

Abstract
Neural stem cells (NSCs) can be isolated and expanded in large-scale, using the neurosphere assay and differentiated into the three major cell types of the central nervous system (CNS); namely, astrocytes, oligodendrocytes and neurons. These characteristics make neural stem and progenitor cells an invaluable renewable source of cells for in vitro studies such as drug screening, neurotoxicology and electrophysiology and also for cell replacement therapy in many neurological diseases. In practice, however, heterogeneity of NSC progeny, low production of neurons and oligodendrocytes, and predominance of astrocytes following differentiation limit their clinical applications. Here, we describe a novel methodology for the generation and subsequent purification of immature neurons from murine NSC progeny using fluorescence activated cell sorting (FACS) technology. Using this methodology, a highly enriched neuronal progenitor cell population can be achieved without any noticeable astrocyte and bona fide NSC contamination. The procedure includes differentiation of NSC progeny isolated and expanded from E14 mouse ganglionic eminences using the neurosphere assay, followed by isolation and enrichment of immature neuronal cells based on their physical (size and internal complexity) and fluorescent properties using flow cytometry technology. Overall, it takes 5-7 days to generate neurospheres and 6-8 days to differentiate NSC progeny and isolate highly purified immature neuronal cells.
Keywords: Neuroscience, Issue 62, neural Stem Cell, Neuronal Progenitor Cells, Flow Cytometry, Isolation, Enrichment
Protocol
1. Basic Set Up Before Proceeding to Cell Culture
NeuroCult NSC Basal Medium and NeuroCult NSC Proliferation Supplements are mixed at a 9:1 ratio, respectively, to make complete NSC medium.
A 37 °C water bath is used to warm up the medium.
Appropriate amount of heat inactivated fetal calf serum (FCS) is thawed.
Appropriate amount of stock growth factor solutions including epidermal growth factor (EGF at 10 μg/ml), basic fibroblastic growth factor (b-FGF at 10 μg/ml) and 0.2% heparin solution in phosphate buffered saline (PBS) are thawed.
Tissue culture flasks (T25, T75 or &T175) are needed for NSC expansion and differentiation.
Tissue culture treated 96 and 24 well plates and glass coverslips are needed for plating sorted cells.
2. Isolation, and Expansion (Passaging) of NSCs
Neural stem and progenitors are isolated and expanded from mouse brain as described in separate protocols 1, 2 3.
When the neurospheres (either primary or passaged neurospheres) are ready for subculture (150-200 μm in diameter), the medium containing spheres is transferred to an appropriate size sterile tissue culture tube, and centrifuged at 800 rpm (110 g) for 5 min at room temperature.
The supernatant medium is removed and the spheres are re-suspended in 1 ml of pre-warmed 0.05 % trypsin-EDTA.
After incubation in a 37 °C water bath for 2-3 min, an equal volume of soybean trypsin inhibitor is added to the cell suspension to stop the trypsin activity.
To ensure that the trypsin has been completely inactivated, the cell suspension is gently pipetted up and down 3-5 times.
After centrifugation at 800 rpm (110 g) for 5 min, the supernatant is removed and the cells are re-suspended in 1 ml of complete NSC medium.
10μl of the cell suspension is mixed with 90 μl of trypan blue to perform a cell count. Note: Other appropriate cell dilutions can also be used.
The cells are then used for subculture 1, 2 or for differentiation as described below.
3. Differentiation of Neural Stem and Progenitor Cells, The Neuroblast Assay (NBA)
Cells resulting from dissociated neurospheres are plated at a concentration of 2-3 x 105 cells/ml in the complete NSC medium supplemented with 20 ng/ml EGF, 10 ng/ml bFGF, 1 μl/ml of 0.2% heparin and 5% heat inactivated FCS in an appropriate size tissue culture flask. 5 ml medium is used for T25, 20 ml for T75 and 40 ml for T175 flasks. Note: FCS will cause the cells to firmly attach to the substrate and there is no need for coating the flasks for NBA culture.
Culture flasks are then incubated in a 37 °C humidified incubator with 5% CO2 for 3-4 days. This period is called proliferation stage, during which NSC progeny attach to the substrate and proliferate as a monolayer of cells.
When the culture becomes 90-95% confluent, the medium of each flask is switched to the complete NSC medium supplemented with 5% FCS without any growth factors.
Culture flasks are again incubated in a 37 ° C humidified incubator with 5% CO2 for another 3-4 days. This period is called differentiation stage, during which neuronal progenitors appear on top of an astrocytic monolayer and proliferate to make colonies of immature neuronal cells. On day 4-5 of differentiation stage, the culture is ready for isolation of the neuroblast cells using flow cytometry technology.
4. Cell Preparation for Flow Cytometry
Trypsinization
The culture of differentiating NSC progeny on day 4-5 of differentiation stage is washed once with an appropriate amount of PBS (2 ml for T25, 4 ml for T75 and 8 ml for T175 flask) to remove FCS from the culture. Note: FCS blocks Trypsin-EDTA activity.
Then, a sufficient amount of pre-warmed 0.05% Trypsin-EDTA (2 ml for T25, 4 ml for T75 and 8 ml for T175 flask) is added to each flask to cover the cell monolayer and incubated for 1-2 min in a 37 °C humidified incubator.
The flask is checked under the microscope to evaluate the effect of Trypsin-EDTA on the culture. The flask is then held with one hand and struck firmly with the other hand to dislodge the cells from the bottom of the flask.
Equal volume of soybean trypsin inhibitor is added to the flask to quench trypsin activity. The cell suspension is gently pipetted up and down to ensure trypsin inactivation and to achieve a homogenous single cell suspension. Note: Alternatively, medium supplemented with 5% FCS could be used to quench trypsin activity.
To remove non-dissociated clumps, the cell suspension is passed through a 40-μm-size mesh filter.
The cell suspension is then centrifuged at 800 rpm (110g) for 5 min, the supernatant is removed and the cells are re-suspended in an appropriate volume of complete NSC medium.
The final volume of cell suspension is adjusted so that the cell density does not exceed 2-3x106-cells/ml. High cell density might cause stream blockage in the flow cytometry machine.
If you would like to isolate neuronal cells just based on their physical properties, now you can proceed to FACS sorting as described in part 4.
Before sorting, Propidium Iodide (PI, Sigma, 500 μg/ml in PBS) is added at a concentration of 1 μl/ ml of cell suspension to exclude dead cells.
Live cell immunolabeling
To achieve a higher purity of neuronal cells, harvested single cell from the NBA culture can be stained for PSA-NCAM (a marker of early immature neuronal progenitors) and then the positive cells from the neuronal population can be isolated using the FACS machine.
- After performing a cell count, the single cell suspension from the NBA culture is divided into four groups:
- Cells alone group, serves as a control to adjust parameters and gates (0.5-1 x 106 cells).
- Cells plus PI, serves as a control to adjust parameters and gates (0.5-10 x 106 cells). PI negative cells from this group can also be sorted based on the cells physical characteristics.
- Secondary alone (Isotype control) group that also serves as a control to adjust parameters and gates (1-2 x 106cells).
- Immunolabeling group (8-10 x 106 cells) that is stained for PSA-NCAM to achieve further purification of the immature neuronal cells. To exclude dead cells upon sorting, PI is added to this group as well.
To start immunolabeling, cells are centrifuged at 800 rpm (110 g) for 5 min. The supernatant is removed and the cells are re-suspended in 100 μl of complete NSC medium.
Phycoerythrin (PE) conjugated anti-PSA-NCAM antibody is added at a ratio of 10 μl / 5-10 x 106 cells to the cell suspension to be stained for PSA-NCAM. In the same way, a PE conjugated mouse IgG1 isotype control antibody is added to the isotype control cell suspension. Follow the instructions on the antibody specification sheet regarding the concentration of antibody to be used. The cell suspension is then mixed gently and incubated at room temperature for 10-15 min in dark.
The cell suspensions (PSA-NCAM and Isotype control tubes) are then centrifuged at 800 rpm (110 g) for 5 min, the supernatant is removed and the cells are re-suspended in 1 ml of complete NSC medium.
To remove excess un-bounded antibodies from the samples, step 4 is repeated 2-3 times.
After re-suspending cells in an appropriate volume of medium, Propidium Iodide (PI, Sigma, 500 μg/ml in PBS) is added at a concentration of 1 μl/ml of the cell suspensions (cells plus PI, isotype control and PSA-NCAM-stained samples) to exclude dead cells when analyzed by flow cytometry.
15 ml sterile falcon tubes containing 1 ml of complete medium are prepared to collect sorted cells from different cell populations.
5. Fluorescence-activated Cell Sorting (FACS) of Immature Neurons
The FACS machine is set up based on its user manual instruction via the flow cytometry facility expert technician.
Phosphate buffered saline (PBS) or any other appropriate solution depending on manufacturer instructions, could be used as the sheath fluid at 28 PSI through a 90 μm nozzle.
Differential pressure on the system is adjusted such that the sort trigger rate does not exceed 2500 events/second.
The cell suspension from the cells alone group is run through the FACS machine.
First, the cells (events) are plotted based on their size (Forward Scatter (FSC) light) versus internal complexity (Side Scatter (SSC) light) to distinguish different cell populations. To do so, voltage parameters for FSC and SSC should be adjusted so that the cells can be seen in the flow cytometry plot.
To exclude cell clumps and doublets, the cells are first plotted based on forward scatter area (FSC-A) versus forward scatter pulse width (FSC-W) and the single cell population is gated as population 1 (P1). Then the P1 cells are plotted based on side scatter area (SSC-A) versus side scatter pulse width (FSC-W) and this time the single cells are gated as population 2 (P2). Having established these two consecutive gates, clumps or doublets (high FSC pulse width and high SSC pulse width) are being excluded.
To exclude dead or damaged cells, the single cells (P2) are plotted based on PI reactivity versus FSC-A and the single live cell population is gated.
Single live cells are plotted based on FSC versus SSC to distinguish different cell populations based on cell size and internal granularity. At this stage two main cell populations are gated as FSClow SSClow (P3) and FSChigh SSChigh (P4) cell populations.
To sort PSA-NCAM immuno-reactive immature neuronal cells from the FSClow SSClow population, first the FSClow SSClow population from control (cells plus PI and isotype control) groups is plotted based on PE immunoreactivity versus FSC-A and the negative cells are gated (P5), and then positive cells from PSA-NCAM stained group are gated as population 6 (P6).
After adjusting all needed gates, cells of interests are sorted into 15 ml sterile tissue culture tubes containing 1ml NSC medium.
Sorted cells are then centrifuged at 1200 rpm (240 g) for 5 min.
Supernatant is discarded and the pellet is re-suspended in an appropriate volume of medium (depending on the pellet size) and a cell count is performed.
Cells are then plated in Poly-L- Ornithine coated 96-well plates at a density of 20-30 x 103 cells /well in 200-250 μl of complete NSC medium with 5% FCS and 20 ng/ml human recombinant BMP4 5.
Note: to coat 96 wells, 100 μl of Poly-L- Ornithine working solution (1.5 ml Poly-O and 8.5 ml of sterile PBS) is added to each well and the plate is incubated in a 37 °C incubator for at least one hour. Then, the Poly-O is removed and each well is washed three times with sterile PBS (let the PBS remain in the well for 10 min each time). Use 500 μl of Poly-L- Ornithine working solution / well if 24-well plates are used.
Cells are then fixed at different time points after sort using cold 4% paraformaldehyde and immunostained for appropriate neuronal and glial cell markers to assess the phenotype of sorted cells from different cell populations.
6. Representative Results
In the neuroblast assay (NBA) differentiation culture, plated cells attach to the tissue culture substrate and grow as a monolayer. Depending on initial cell plating density, the culture will turn into a 90-95% confluent culture after 3-4 days. At this stage, a monolayer of mainly astrocytic cells can be visualized underneath with small round neuronal progenitor cells on top4,5 (Figure 1). After switching the medium to a growth factor free medium, neuronal progenitor cells start dividing rapidly and generate clusters of immature neuroblast cells on top of the astrocytic monolayer in 4-5 days (Figure 2). Flow cytometry analysis of dissociated NBA culture on day 4-5 of differentiation stage, shows two main cell populations; FSClow SSClow and FSChigh SSChigh based on cell size (FSC) and internal granularity (SSC) (Figure 3). Immuno-fluorescent analysis of the sorted cells shows that the neuronal cells are mainly located in the FSClow SSClow populations (with more than 75% of them expressing β-III tubulin), while sorted cells from the FSChigh SSChigh population are mostly GFAP immunoreactive astrocytes (Figure 4). Further purification of immature neuronal cells up to near homogeneity (97%) could be achieved with positive selection of PSA-NCAM IR cells from the FSClow SSClow population (Figure 3, 4). Enriched neuronal cells generated from the NSCs isolated from E14 mouse ganglionic eminences acquire a GABAergic phenotype and express DARPP-32, a marker of medium spiny neurons, upon differentiation (Figure 5).
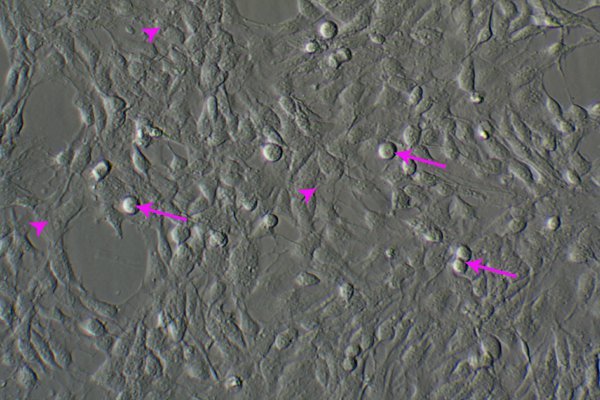 Figure 1. Neuroblast assay culture from E14 mouse neural stem cells on day 4 of proliferation stage. This monolayer culture consists of flat astrocytic cells (arrowheads) underneath and round neuronal progenitor cells (arrows) on top. Original magnification; 20 x.
Figure 1. Neuroblast assay culture from E14 mouse neural stem cells on day 4 of proliferation stage. This monolayer culture consists of flat astrocytic cells (arrowheads) underneath and round neuronal progenitor cells (arrows) on top. Original magnification; 20 x.
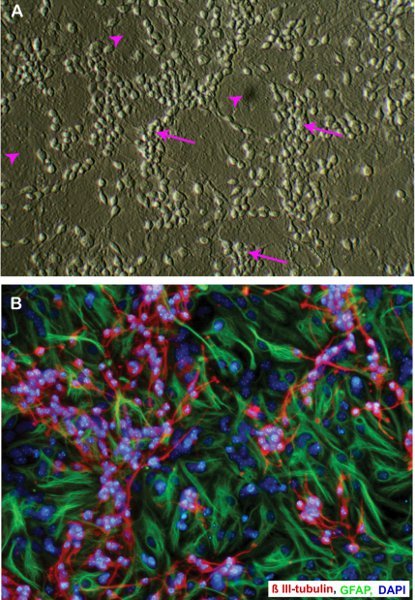 Figure 2. Neuroblast assay culture from E14 mouse neural stem cells on day 4 of differentiation stage: A) Phase contrast, B) Immunofluorescent staining. Note the cluster of immature neuronal cells (arrows in A and β III tubulin staining in B) on top of the astrocytic monolayer (arrow heads in A, GFAP staining in B). Original magnification; 20 x.
Figure 2. Neuroblast assay culture from E14 mouse neural stem cells on day 4 of differentiation stage: A) Phase contrast, B) Immunofluorescent staining. Note the cluster of immature neuronal cells (arrows in A and β III tubulin staining in B) on top of the astrocytic monolayer (arrow heads in A, GFAP staining in B). Original magnification; 20 x.
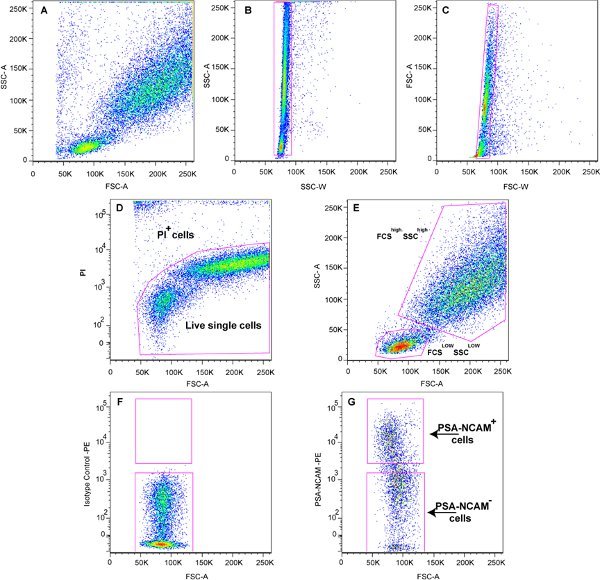 Figure 3. Representative sort plots: A). FSC vs. SSC sort plot before exclusion of doublets and dead cells. B, C). Sort plots for exclusion of doublets based on FSC and SSC pulse width. D). Exclusion of dead and damaged cells based on Propidium Iodide immunoreactivity. E). FSC vs. SSC plot after exclusion of doublets and dead cells, showing the two main populations labeled as the FSClow SSClow and the FSChigh SSChigh populations. F, G). Sort plots to isolate PSA-NCAM IR immature cells from the FSClow SSClow population.
Figure 3. Representative sort plots: A). FSC vs. SSC sort plot before exclusion of doublets and dead cells. B, C). Sort plots for exclusion of doublets based on FSC and SSC pulse width. D). Exclusion of dead and damaged cells based on Propidium Iodide immunoreactivity. E). FSC vs. SSC plot after exclusion of doublets and dead cells, showing the two main populations labeled as the FSClow SSClow and the FSChigh SSChigh populations. F, G). Sort plots to isolate PSA-NCAM IR immature cells from the FSClow SSClow population.
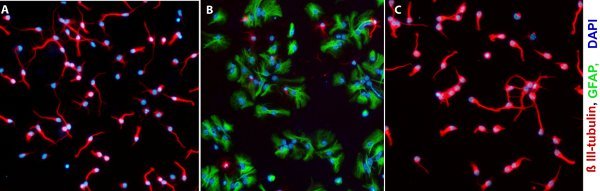 Figure 4. Representative micrographs from cells sorted from different populations one day after plating. A) FSClow SSClow population (more than 75% of these cells are neurons). B) FSChigh SSChigh population (the majority of these cells are astrocytes). C) PSA-NCAM+ sorted cells from FSClow SSClow population (almost all of the sorted cells are neurons). Original Magnification; 20 x.
Figure 4. Representative micrographs from cells sorted from different populations one day after plating. A) FSClow SSClow population (more than 75% of these cells are neurons). B) FSChigh SSChigh population (the majority of these cells are astrocytes). C) PSA-NCAM+ sorted cells from FSClow SSClow population (almost all of the sorted cells are neurons). Original Magnification; 20 x.
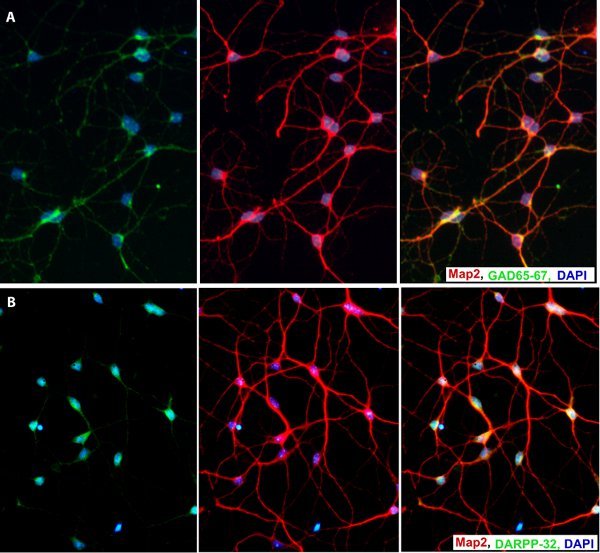 Figure 5. Sorted immature neurons differentiate into GABAergic neurons (A) and express DARPP-32 (B), a marker for medium spiny neurons.
Figure 5. Sorted immature neurons differentiate into GABAergic neurons (A) and express DARPP-32 (B), a marker for medium spiny neurons.
Discussion
The ability to isolate defined neural cell populations (i.e. neurons, astrocytes and oligodendrocytes) might solve some of the impediments associated with the clinical application of neural stem cells (NSCs). First, it enables us to fully characterize the starting population of donor cells. Second, it allows us to use a particular cell type or a combination of different cell types with a specific ratio, depending on the nature or stage of the disease. Lastly, using highly enriched pre-differentiated neural cell progenitors can dramatically decrease possible uncontrolled proliferation and tumor formation associated with using heterogeneous neural precursors containing bona fide NSC6. Generally differentiation of neural stem cells will result in 5-10% neuronal cells and more than 80% astrocytes. Using the NBA method of differentiation, the percentage of neuronal cells will increase up to 20-30% but still there are lots of astrocytes and other stem and progenitor cells in the culture, which may not be desirable if one would intend to study pure neuronal cells in vitro or to study their therapeutic effect in animal models of neurological diseases. In this protocol, we took advantage of inherent differences in the physical and fluorescent properties of differentiating NSC progeny to purify immature neuronal cells5. Our flow cytometry purification methodology increases the percentage of neuronal cells from 20-30% to 75-97% with no detectable astrocytes and un-differentiated bona fide neural stem and progenitor cells.
Application of this methodology to human NSCs might benefit neuronal cell replacement therapy in neurological disease. This approach could also be useful for in vitro studies that need highly purified neuronal progenitor cells such as drug screening, neurotoxicology, developmental studies and electrophysiology.
To be able to consistently generate high quality immature neurons from NSCs generated from E14 mouse ganglionic eminences, we recommend:
Not to let the spheres grow too large. Large neurospheres are associated with more cell death and less neurogenic abilities.
Not to trypsinize the spheres for more than 2-3 minutes. Leaving trypsin for more than 3 minutes causes damage to the cells and decreases their neurogenic efficiency.
Not to let the proliferating monolayer become over-confluent. This may interfere with their normal differentiation process. Always, switch medium when the culture reaches about 90% confluency.
To give the culture a medium change on the day before sort. This conditioned medium can be collected on the day of sort and used for plating cells. This medium contains a lot of unidentified soluble factors from the astrocytic cells that will help the sorted immature neuronal cells to survive and acquire a more mature phenotype.
As drawbacks to this technology, passing cell suspension though flow cytometry machine could be associated with some risks including shearing force that might damage the cells and cause cell death upon sort and also fungal or bacterial contamination. To avoid damage by shearing force, we recommend sorting the cell at an appropriate speed, and using right sheath fluid (PBS is recommended) and right size nozzles (90-100 μm) not to let the sort trigger rate exceed 2500 events/second. To avoid contamination, make sure the instrument has been cleaned properly using disinfectant reagents before sort and also use antibiotics in your collecting medium.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by funding from the Overstreet Foundation.
References
- Azari H, Rahman M, Sharififar S, Reynolds B. Isolation and Expansion of the Adult Mouse Neural Stem Cells Using the Neurosphere Assay. J. Vis. Exp. 2010. p. e2393. [DOI] [PMC free article] [PubMed]
- Azari H, Sharififar S, Rahman M, Ansari S, Reynolds B. Establishing Embryonic Mouse Neural Stem Cell Culture Using the Neurosphere Assay. J. Vis. Exp. 2011. p. e2457. [DOI] [PMC free article] [PubMed]
- Siebzehnrubl FA, Vedam-Mai V, Azari H, Reynolds BA, Deleyrolle LP. Isolation and Characterization of Adult Neural Stem Cells. Methods. Mol. Biol. 2011;750:61–77. doi: 10.1007/978-1-61779-145-1_4. [DOI] [PubMed] [Google Scholar]
- Scheffler B. Phenotypic and functional characterization of adult brain neuropoiesis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9353–9358. doi: 10.1073/pnas.0503965102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azari H. Purification of immature neuronal cells from neural stem cell progeny. PLoS One. 2011;6:e20941–e20941. doi: 10.1371/journal.pone.0020941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amariglio N. Donor-Derived Brain Tumor Following Neural Stem Cell Transplantation in an Ataxia Telangiectasia Patient. Plos Medicine. 2009;6:221–231. doi: 10.1371/journal.pmed.1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]