

Abstract
This protocol describes a fluorescence microscope-based screening of Arabidopsis seedlings and describes how to map recessive mutations that alter the subcellular distribution of a specific tagged fluorescent marker in the secretory pathway. Arabidopsis is a powerful biological model for genetic studies because of its genome size, generation time, and conservation of molecular mechanisms among kingdoms. The array genotyping as an approach to map the mutation in alternative to the traditional method based on molecular markers is advantageous because it is relatively faster and may allow the mapping of several mutants in a really short time frame. This method allows the identification of proteins that can influence the integrity of any organelle in plants. Here, as an example, we propose a screen to map genes important for the integrity of the endoplasmic reticulum (ER). Our approach, however, can be easily extended to other plant cell organelles (for example see1,2), and thus represents an important step toward understanding the molecular basis governing other subcellular structures.
Keywords: Genetics, Issue 62, EMS mutagenesis, secretory pathway, mapping, confocal screening
Protocol
1. EMS Treatment
Arabidopsis thaliana seeds are mutagenized using as mutagen agent ethyl methane sulfonate (EMS)3,4, which induces into the genome C-to-T changes resulting in C/G to T/A mutations5-7.
Weigh 0.8 g Arabidopsis seeds (~40,000 seeds) carrying the organelle fluorescent marker (specifically, in this study ssGFPHDEL (signal sequence-GFP-HDEL tetrapeptide) has been used as an ER marker).
Transfer the seeds in a 50-ml falcon tube, then add 25 ml distilled water.
Add 0.2% (v/v) ethyl methane sulfonate.
Incubate for 16 hours on nutating mixer at low speed.
Aspirate the liquid and discard it into a flask containing 1.0 M NaOH to inactivate the EMS.
Add 25 ml water to the Falcon tube containing the seeds, close, and invert 5 times to wash the seeds; wait until all the seeds have settled, then aspirate the water and discard into the 1.0 M NaOH flask.
Repeat the washing step up to 10 times.
After the final wash, re-suspend the seeds in a minimal amount of water.
Proceed to seed sterilization adding 25 mL of 10 % bleach, shake vigorously for 30 s. Let seeds settle to bottom, then pour off bleach and rinse with 25 mL of sterile water. Pour off the sterile water and add 25 mL 70 % ethanol. Shake tubes for 30 s. Let seeds settle to bottom. Pour off ethanol and rinse with 25 mL of sterile water. Repeat twice the wash with sterile water, then pour seeds on a plastic Petri dish containing 3 MM filter paper and let dry under the hood. Store at 4 °C for 2 days.
Plate the seeds on ½ MS phytagel (half concentration of Murashige and Skoog medium), 150-mm Petri dish (~250-300 seeds for plate).
Grow M1 seeds on plate for 2 weeks, then transplant into soil.
Collect M2 seeds from individual M1 plants to generate M2 lines (1000 independent lines).
2. Confocal Screening of M2 and M3 Populations
In this section we describe the observation of seedlings with a confocal or fluorescence microscope as described earlier8.
Sixty seeds from each M2 line are grown for 7 to 10 days on plastic Petri dishes, ½ MS phytagel; on the same plate are also grown 5 seedlings EMS-untreated (control).
Five to ten cotyledons are mounted with the abaxial side toward the lens (40X) on a microscope slide and enclosed with a coverslip.
Each cotyledon is observed, from the cortical to the medial region, under fluorescence for any altered subcellular distribution of the organelle marker.
Positive plants are transplanted into soil and their seeds are collected and screened again to confirm the mutant phenotype in the M3 generation.
- Remove the contaminating background mutations through backcrossing at least three times to a wild-type genome possibly containing the desired organelle fluorescent marker.
- From the mother plant, using fine scissors or forceps remove mature siliques, and open flowers.
- Remove the buds that are too small from the meristem.
- Insert the tip of one pair of forceps between petals and sepals to open one flower bud and remove all anthers.
- Using one pair of forceps take an open mature flower from the father plant and rub the anthers on the stigma of the emasculated plant.
3. Mapping
This section describes essentially how to map a recessive mutation using a modified protocol from Borevitz9 which is faster if compared to traditional mapping methods10,11 . This approach use high-density oligonucleotide arrays with the ability to detect numerous single feature polymorphisms (SFPs) in a single assay12. Using the Affymetrix Arabidopsis ATH1 GeneChip array is possible to analyze approximately 24,000 genes. A pool of F2 individuals showing the mutation is compared to a pool of wild-type F2 plants collected within the same segregating population. Then, the mutation will be mapped in the region where the mutant pool is enriched for mutant genotype alleles and consequently in the same region the wild-type pool will result enriched for the wild-type parent alleles13.
Genomic DNA (3 μg) is extracted from the homozygous mutant Columbia (M3) using a Qiagen DNeasy Plant Mini Kit and is submitted for Illumina Genome Analyzer II (GA II)14 sequencing.
The homozygous mutant Columbia (M3) is crossed with Landsberg erecta to generate a mapping population.
The mapping is performed on 70 up to 100 F2 individuals showing the aberrant phenotype and the same number of F2 plants with a wild-type phenotype.
Collect from each plant a leaf disc (0.60 mm) using a hole punch. The leaf disc should be collected from leaves of the same age to be sure to have similar amounts of DNA. The samples can be processed for genomic extraction separately or in groups. In this case, it is possible to collect 5-10 samples for each Eppendorf tube so that in the end you will have fewer eppendorf tubes from which genomic DNA has to be extracted.
MasterPure Plant Leaf DNA Purification Kit (Epicentre) is used to extract the genomic DNA. The genomic DNA obtained from each sample is quantified with a nanodrop.
The same quantity of genomic DNA from each sample is then put together up to a total of 300 ng (~30 μL) of plant DNAs; add 60 μL 2.5X random primers solution [125 mM Tris-HCl (pH 6.8), 12.5 mM MgCl2, 25 mM 2-mercaptoethanol, 750 μg/ml oligodeoxyribonucleotide primers (random octamers)] (Bioprime kit) and 42 μL water (final volume 132 μL).
Denature DNAs at >95 °C for 5 to 10 min.
Cool on ice.
To each denatured DNA add 15 μL 10X dNTPs mix with Biotin dCTP [1 mM biotin-14-dCTP, 1 mM dCTP, 2 mM dATP, 2 mM dGTP, 2 mM dTTP in 10 mM Tris-HCl (pH 7.5), 1 mM Na2EDTA] (Bioprime kit), and complete with 3 μL Klenow polymerase (Bioprime kit).
Incubate overnight at 25 °C.
On the following day, add 15 μL 3 M NaOAc and 400 μL cold 100% EtOH; mix.
Incubate at -80 °C for 1 hour, then spin at 20,500x g for 15 min, remove supernatant, and wash with 500 μL cold 75% EtOH.
Spin at 20,500x g for 10 min.
Dry DNA pellets at 37 °C for 10 min and resuspend in 100 μL water.
Use 5 μL to check yield and quality on a gel (Figure 2).
Send 95 μL of labeling reaction for the wild-type and mutant for GeneChip Arabidopsis ATH1 Genome Array Hybridization.
The arrays are scanned and the .CEL files obtained are analyzed using R software (http://www.r-project.org/).
Install R software, then open the program and paste the following string: source("http://bioconductor.org/biocLite.R") biocLite() Then press return key-this will install the standard Bioconductor packages (http://www.bioconductor.org).
In a new folder on your desktop, download from the following website (http://www.naturalvariation.org/methods) the files: readcel.R SFP.R Map.R ath1V5.RData ColLerCEL.zip The file ColLerCEL.zip should be unpacked and the resulting files pasted into your new folder.
Rename the data obtained from your GeneChip experiment into wildtype.CEL and mutant.CEL.
Copy now your GeneChip data (wildtype.CEL and mutant.CEL) into your folder.
Open R, a) for Mac: click "Misc," and then click "Change working directory." b) for PC: click "File" then "Change directory"
a) for Mac: Select the folder containing the files. Click "Workspace," "Load workspace file" and select ath1V5.RData, b) for PC: Select the folder containing the files above, click "File," then "Load workspace" and select ath1V5.RData.
Open readCEL.R using Notepad and copy the whole text to R.
Open SFP.R using Notepad and copy the whole text to R.
Open Map.R using Notepad and copy the whole text to R.
In the Console window you will see the following message: Chromosome x limits Mb y z Search genes at TAIR paste in this link3http://arabidopsis.org/servlets/sv?action=download&chr=x&start=y&end=z
Copy and paste the link into your internet browser. A window will appear and ask to open or save the file; you should select save. Choose a file name and attach the extension ".xls," then click Save.
Open the file ".xls" where you can find the coordinate for your mutant (the result is graphically represented in Figure 3).
Within the mapped area in the assembled Illumina reads of the mutant genome identify specific EMS transitions 4 among the single nucleotide polymorphisms. Complement the mutant phenotype by transformation with wild type gene(s) using standard procedures15.
4. Representative Results
Figure 1 shows the approach used for identification of a mutant of the secretory pathway using confocal microscopy screening. Figure 2 shows a typical preparation of labeled genomic DNA for array hybridization. In figure 3, a typical result expected after analyzing the data obtained from the GeneChip Arabidopsis ATH1 Genome Array is presented.
 Figure 1. Arabidopsis transgenic plants expressing ssGFPHDEL (ER marker) (1) were cultivated to produce enough seeds for EMS treatment (2). Seeds treated with EMS were then sown to generate M1 plants (3). Each M1 plant represents a different line and seed were collected separately from each of them. M2 seed were plated on ½ MS substrate (4) and then screened for defects in ER morphology by confocal microscopy (5). During the screening we found plants that conserved the wild-type ER morphology (6) and plants that show defective ER phenotypes (7). These plants were grown to obtain the M3 generation and to confirm the phenotype (8). Genomic DNA from M3 plant was used for Solexa Illumina sequencing (a). The same plant was also used for crosses with Ler-wt to obtain the F2 mapping population (b).
Figure 1. Arabidopsis transgenic plants expressing ssGFPHDEL (ER marker) (1) were cultivated to produce enough seeds for EMS treatment (2). Seeds treated with EMS were then sown to generate M1 plants (3). Each M1 plant represents a different line and seed were collected separately from each of them. M2 seed were plated on ½ MS substrate (4) and then screened for defects in ER morphology by confocal microscopy (5). During the screening we found plants that conserved the wild-type ER morphology (6) and plants that show defective ER phenotypes (7). These plants were grown to obtain the M3 generation and to confirm the phenotype (8). Genomic DNA from M3 plant was used for Solexa Illumina sequencing (a). The same plant was also used for crosses with Ler-wt to obtain the F2 mapping population (b).
 Figure 2. Bioprime random labeling reactions (5 μL of 100 μL) were loaded on agarose gel 1%. Lane a is from a pool of wild-type F2 plants, and lane b is from a pool of mutant F2 plants. (Marker is 1 Kb DNA ladder-N3232, from NEB).
Figure 2. Bioprime random labeling reactions (5 μL of 100 μL) were loaded on agarose gel 1%. Lane a is from a pool of wild-type F2 plants, and lane b is from a pool of mutant F2 plants. (Marker is 1 Kb DNA ladder-N3232, from NEB).
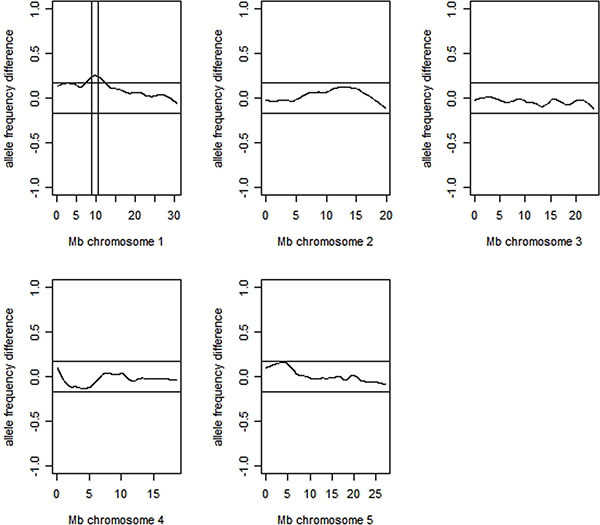 Figure 3. The figure represents an example of mapping of Col-0 mutation using GeneChip Arabidopsis ATH1 Genome Array Hybridization. The mutation in this example is located on chromosome 1, delimited by vertical bars. The horizontal bars represent the thresholds for detection.
Figure 3. The figure represents an example of mapping of Col-0 mutation using GeneChip Arabidopsis ATH1 Genome Array Hybridization. The mutation in this example is located on chromosome 1, delimited by vertical bars. The horizontal bars represent the thresholds for detection.
Discussion
Here we described a confocal microscopy-based screening for the identification of endomembrane mutants. This approach can be easily extended to other organelles of the cell for which specific fluorescent protein markers are available. The screen is based on the identification of mutants that show an aberrant distribution of the fluorescent marker either in the target organelle or to organelles that are not supposed to contain the marker. Respectively, these mutants represent populations in which either the ability of the organelle to subcompartimentalize the marker is compromised or mutants that cannot properly translocate the marker among organelles. During the screening we noticed that some mutants showed a phenotype in early development stages as early as 7 days after germination; however, others showed the phenotype only in a later stage of development. While this may be linked to a number of reasons, including development-dependent expression of the mutated allele(s), this suggests that plants should be examined at different growth stages (at least 7 to 14 days) to guarantee that potentially interesting mutants are not discarded.
The approach described in this protocol allows us to map a gene in a relatively short time compared to the classical mapping method. In fact, collecting a sufficient number of individuals of the F2 population used for the GeneChip Array requires a relatively small amount time compared to F2 plants required for the classical fine-mapping procedure. However, there are critical steps that should be observed. Selection of F2 population samples showing or not showing the phenotype should be carried out carefully, since the addition of even a small number of plants with the wrong phenotype by mistake can introduce errors into the final result. This is mostly linked to the fact that the F2 population used for the rough mapping is very small. For this reason, the selection of plants that either show or do not show the phenotype should be conducted every time at the same day after germination. This is because, as we mentioned above, the appearance of the phenotype may be linked to growth. Another important point to consider is to look carefully at the next generation data postalignment, which is generated by programs that give us important information like the percentage of reads containing the variant. The theoretical value expected for a heterozygous variant is around 50%, this means that 50% of the sequences would contain the variant while for a homozygous variant, is circa 100%. Unfortunately the situation after a postalignment can be far from the theoretical value, in fact the percentage of reads containing the variant for heterozygous can vary from the 20 to 80% while from 60 to 100% for homozygous16. Therefore, in order not to miss the key single nucleotide polymorphisms (SNP), it is important to not to discard mutants with less than 100% percentage of reads for the variant.
Disclosures
We have nothing to disclose.
Acknowledgments
We acknowledge support by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, US Department of Energy (award number DE-FG02- 91ER20021) and National Science Foundation (MCB 0948584) (F.B.). We are thankful to Ms Karen Bird for editing the manuscript.
References
- Marti L. A missense mutation in the vacuolar protein GOLD36 causes organizational defects in the ER and aberrant protein trafficking in the plant secretory pathway. The Plant journal : for cell and molecular biology. 2010;63:901–913. doi: 10.1111/j.1365-313X.2010.04296.x. [DOI] [PubMed] [Google Scholar]
- Stefano G, Renna L, Moss T, McNew J, Brandizzi F. Arabidopsis the spatial and dynamic organization of the endoplasmic reticulum and Golgi apparatus is influenced by the integrity of the c-terminal domain of RHD3, a non-essential GTPase. The Plant Journal. 2011. Forthcoming. [DOI] [PubMed]
- Kim Y, Schumaker KS, Zhu JK. EMS mutagenesis of Arabidopsis. Methods in molecular biology. 2006;323:101–103. doi: 10.1385/1-59745-003-0:101. [DOI] [PubMed] [Google Scholar]
- Maple J, Moller SG. Mutagenesis in Arabidopsis. Methods in molecular biology. 2007;362:197–206. doi: 10.1007/978-1-59745-257-1_14. [DOI] [PubMed] [Google Scholar]
- Greene EA. Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics. 2003;164:731–740. doi: 10.1093/genetics/164.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg DR. Ethyl methanesulfonate-induced reversion of bacteriophage T4rII mutants. Genetics. 1963;48:561–580. doi: 10.1093/genetics/48.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk I, Kovalchuk O, Hohn B. Genome-wide variation of the somatic mutation frequency in transgenic plants. The EMBO journal. 2000;19:4431–4438. doi: 10.1093/emboj/19.17.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulaflous A, Faso C, Brandizzi F. Deciphering the Golgi apparatus: from imaging to genes. Traffic. 2008;9:1613–1617. doi: 10.1111/j.1600-0854.2008.00769.x. [DOI] [PubMed] [Google Scholar]
- Borevitz J. Genotyping and mapping with high-density oligonucleotide arrays. Methods in molecular biology. 2006;323:137–145. doi: 10.1385/1-59745-003-0:137. [DOI] [PubMed] [Google Scholar]
- Konieczny A, Ausubel FM. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. The Plant journal : for cell and molecular biology. 1993;4:403–410. doi: 10.1046/j.1365-313x.1993.04020403.x. [DOI] [PubMed] [Google Scholar]
- Bell CJ, Ecker JR. Assignment of 30 microsatellite loci to the linkage map of Arabidopsis. Genomics. 1994;19:137–144. doi: 10.1006/geno.1994.1023. [DOI] [PubMed] [Google Scholar]
- Hazen SP, Kay SA. Gene arrays are not just for measuring gene expression. Trends in Plant Science. 2003;8:413–416. doi: 10.1016/S1360-1385(03)00186-9. [DOI] [PubMed] [Google Scholar]
- Hazen SP. Rapid array mapping of circadian clock and developmental mutations in Arabidopsis. Plant Physiology. 2005;138:990–997. doi: 10.1104/pp.105.061408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel D, Glazebrook J. A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2002. Arabidopsis. [Google Scholar]
- Voelkerding KV, Dames S, Durtschi JD. Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology. J. Mol. Diagn. 2010;12:539–551. doi: 10.2353/jmoldx.2010.100043. [DOI] [PMC free article] [PubMed] [Google Scholar]