

Abstract
Genomes are organized into three-dimensional structures, adopting higher-order conformations inside the micron-sized nuclear spaces 7, 2, 12. Such architectures are not random and involve interactions between gene promoters and regulatory elements 13. The binding of transcription factors to specific regulatory sequences brings about a network of transcription regulation and coordination 1, 14.
Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET) was developed to identify these higher-order chromatin structures 5,6. Cells are fixed and interacting loci are captured by covalent DNA-protein cross-links. To minimize non-specific noise and reduce complexity, as well as to increase the specificity of the chromatin interaction analysis, chromatin immunoprecipitation (ChIP) is used against specific protein factors to enrich chromatin fragments of interest before proximity ligation. Ligation involving half-linkers subsequently forms covalent links between pairs of DNA fragments tethered together within individual chromatin complexes. The flanking MmeI restriction enzyme sites in the half-linkers allow extraction of paired end tag-linker-tag constructs (PETs) upon MmeI digestion. As the half-linkers are biotinylated, these PET constructs are purified using streptavidin-magnetic beads. The purified PETs are ligated with next-generation sequencing adaptors and a catalog of interacting fragments is generated via next-generation sequencers such as the Illumina Genome Analyzer. Mapping and bioinformatics analysis is then performed to identify ChIP-enriched binding sites and ChIP-enriched chromatin interactions 8.
We have produced a video to demonstrate critical aspects of the ChIA-PET protocol, especially the preparation of ChIP as the quality of ChIP plays a major role in the outcome of a ChIA-PET library. As the protocols are very long, only the critical steps are shown in the video.
Keywords: Genetics, Issue 62, ChIP, ChIA-PET, Chromatin Interactions, Genomics, Next-Generation Sequencing
Protocol
A. Chromatin Immunoprecipitation (ChIP) (see Figure 1)
1. Dual Crosslinking of Chromatin-Bound Proteins and Cell Harvesting
(at 02:10 of the video)
Chromatin immunoprecipitation (ChIP) is the first critical step involved in constructing a ChIA-PET library. This step is important to reduce the level of complexity, background noise and add specificity. The preparation of ChIP will need to be optimized for cell type and factor of interest. This protocol is based on the RNA Polymerase II ChIA-PET library prepared from MCF-7 cells 9 constructed in our laboratory. To ensure that the resulting library is of sufficient complexity, we recommend using 1 x 108 cells to prepare the ChIP material. Depending on the target and cell line, the yield obtained can be between 100 ng to 300 ng. We recommend that a minimum of 100 ng ChIP should be used to construct a ChIA-PET library with fewer than 20 PCR cycles to minimize redundancy during sequencing. Higher amounts of ChIP material will allow for further reductions in redundancy, improving the quality of the libraries.
Wash 1 x 108 MCF-7 cells (equivalent to five 500 cm2 square plates of 2 x 107 cells) twice with Phosphate Buffered Saline (PBS) pre-warmed at 37 °C.
Crosslink MCF-7 cells with 1.5 mM freshly prepared EthylGlycol bis(SuccinimidylSuccinate) (EGS)) for 45 minutes at room temperature (22 °C) with rotation in a chemical fume hood.
Add 37% formaldehyde to a final concentration of 1% for 20 minutes at room temperature (22 °C) with rotation in a chemical fume hood.
![]()
Quench the crosslinkers by adding 2 M glycine to a final concentration of 200 mM. Incubate for 10 minutes at room temperature (22 °C) with rotation.
![]()
Discard quenched crosslinkers into a waste flask and wash cells twice with cold PBS.
Discard PBS and harvest cells by scraping. Keep harvested cells on ice. Rinse the plate with 5ml PBS (with Protease Inhibitors ("+PI") added according to manufacturer's instructions) to maximize collection of cells.
Pellet the cells at 1,800 x g (3,000 rpm) for 10 minutes at 4 °C. Discard supernatant and proceed to cell lysis. Alternatively, store the pellet at -80 °C.
2. Cell Lysis
(at 04:15 of the video)
Resuspend pellet in 15 ml 0.1% SDS Lysis Buffer (50 mM HEPES pH7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 0.1% SDS + PI) thoroughly by pipetting. Rotate for 15 minutes at 4 °C.
Pellet lysed cells at 800 x g (2,000 rpm) for 10 minutes at 4 °C.
Discard supernatant and repeat cell lysis once as per step 2.1 and 2.2.
The isolated nuclear pellet is ready for nuclear lysis. Alternatively, store pellet at -80 °C.
3. Nuclear Lysis
(at 05:00 of the video)
Nuclear lysis is performed to release crosslinked chromatin before chromatin fragmentation. In the absence of nuclear membrane, the crosslinked chromatin can be sonicated using gentler conditions. Sometimes, sonication strength may not be sufficient to break up the nuclear membrane in which case, less chromatin will be obtained because the intact nuclei will be discarded after centrifugation. However, different cell types may require different conditions.
Resuspend isolated nuclei pellet in 15 ml 1% SDS Lysis Buffer (50 mM HEPES pH7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 1% SDS + PI). Transfer nuclei suspension to a high speed-centrifuge tube (Oak Ridge Centrifuge Tube (Polypropylene)) and rotate for 15 minutes at 4 °C.
Pellet lysed nuclei pellet at 47,810 x g (20,000 rpm) for 30 minutes at 4 °C.
Decant supernatant and break pellet with a p1000 pipette tip.
Wash pellet with 30 ml 0.1% SDS Lysis Buffer (+PI).
Rotate for 15 minutes at 4 °C.
Pellet chromatin at 47,810 x g (20,000 rpm) for 30 minutes at 4 °C.
Discard supernatant and repeat wash once as per step 3.4 to 3.6 before proceeding to chromatin fragmentation. Alternatively, store chromatin pellet at -80 °C.
4. Fragmentation of Chromatin
(at 07:03 of the video)
Transfer chromatin pellet to a 14 ml polystyrene round bottom test tube.
Add 1 ml 0.1% SDS Lysis Buffer (+PI) to chromatin pellet. Ensure there are no bubbles in the mixture as this will affect sonication efficiency.
Shear chromatin-DNA to a size of 200 to 600 base pairs with a Branson Digital Sonifer Cell Disrupter (amplitude 35%, 9 minutes (30 seconds on, 30 seconds off)) and keep samples cold at all time to prevent overheating by performing the sonication in a cold room (see Figure 2).
- Reverse-crosslink an aliquot of chromatin and check fragmentation efficiency with the following steps. (The following steps are not shown in the video).
- Aliquot 10 μl of chromatin after sonication.
- Centrifuge sonicated chromatin at 16,110 x g (13,200 rpm) for 5 minutes at 4 °C.
- Transfer supernatant to a new tube and add 2 μl of Proteinase K solution.
- Incubate for 30 minutes at 50 °C.
- Resolve reverse-crosslinked chromatin on a 1.5% agarose gel.
- Repeat sonication if the sizes of DNA are larger than expected.
Centrifuge remaining lysate at 16,110 x g (13,200 rpm) for 30 minutes at 4 °C and transfer sonicated chromatin (supernatant) into a new tube before preclearing with magnetic beads. Alternatively, store sonicated chromatin at -80 °C until sufficient chromatin is collected to start a ChIP.
5. Washing, Preclearing and Coating of Antibody to Beads
(at 08:17 of the video)
- Preclearing of Chromatin
- Use 300 μl of magnetic protein G beads for one IP.
- Wash beads thrice with 5 ml Beads Wash Buffer (PBS, 0.1% Triton X-100). This and future washes involving magnetic protein G beads consist of the following steps. Ensure magnetic beads do not dry out.
- Reclaim beads using a magnetic particle concentrator.
- Discard supernatant.
- Resuspend beads with buffer.
- Rotate for 5 minutes at 4 °C.
- Centrifuge at 129 x g (800 rpm) for 1 minute at 4 °C.
- Reclaim beads using a magnetic particle concentrator.
- Discard supernatant.
- Combine sonicated chromatin with prewashed beads, to remove background binding of chromatin to beads.
- Keep 10 μl of sonicated chromatin as "input" for subsequent enrichment check by quantitative PCR (qPCR). Store at 4 °C.
- Rotate overnight at 4 °C.
- Centrifuge sonicated chromatin at 129 x g (800 rpm) for 1 minute at 4 °C.
- Place tube on the magnetic particle concentrator.
- Coating Antibody to Magnetic Beads
- Aliquot 300 μl of magnetic protein G beads to a fresh tube.
- Wash beads thrice with 5ml Beads Wash Buffer (PBS, 0.1% Triton X-100).
- Add an equivalent volume (as step 5.1.3) of Beads Wash Buffer.
- Add 35 μg of RNA Polymerase II (8WG16) monoclonal antibody.
- Rotate overnight at 4 °C.
- Wash antibody-coated beads twice with 5 ml Beads Wash Buffer.
- Reclaim antibody-coated beads using a magnetic particle concentrator.
6. Chromatin Immunoprecipitation
(at 10:03 of the video)
To reduce the level of complexity and background noise, antibodies against specific protein factors are used to enrich specific chromatin fragments of interest before proximity ligation 6.
Here, we used a mouse RNA polymerase II monoclonal antibody (8WG16) that recognized the initiation form of the protein. By enriching DNA fragments that are associated with RNA polymerase II, specificity of the library can be increased, allowing identification of long-range chromatin interactions between active promoters and their corresponding regulatory regions 9.
Discard wash buffer from antibody-coated beads with the help of a magnetic particle concentrator.
Transfer sonicated chromatin (supernatant from step 5.1.6) with the help of the magnetic particle concentrator to antibody-coated beads (from step 5.2.7).
Rotate overnight at 4 °C.
7. Washing and Elution of Immunoprecipitated DNA-Protein Complexes
(at 11:13 of the video)
Wash chromatin-immunoprecipitated beads thrice with 5 ml 0.1% SDS Lysis Buffer.
Wash beads once with 5 ml High Salt Wash Buffer (50 mM HEPES pH 7.5, 350 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 0.1% SDS).
Wash beads once with 5 ml Lithium Chloride Wash Buffer (10 mM Tris pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% Nonidet P-40, 0.5% Sodium Deoxycholate).
Discard wash buffer and resuspend washed beads with 1 ml TE Buffer.
- Rotate for 5 minutes at 4 °C. The sample is ready for quantitation and ChIP enrichment check. Once sufficient ChIP material has been collected, the sample is ready to be constructed into a ChIA-PET library. This step is critical, and must be done to ensure successful ChIA-PET library construction. ChIP-enriched beads may be stored for up to 2 weeks at 4 °C while undergoing quantitation, ChIP enrichment checks, and accumulation of sufficient material.
- Elute and reverse-crosslink "input" DNA and ChIP-enriched beads with the following steps. (The following steps are not shown in the video.)
- Elute 20% of ChIP-enriched beads with 200 μl ChIP Elution Buffer (50 mM Tris pH 8.0, 10 mM EDTA, 1% SDS). Rotate for 30 minutes at 37 °C. Centrifuge eluted beads at 6,100 x g (800 rpm) for 1 minute at 4 °C. Transfer eluted ChIP complexes (supernatant) to a new tube with the help of the magnetic particle concentrator.
- Reverse-crosslink "input" and eluted ChIP complexes with 2 μl Proteinase K (final concentration of 0.2 g/ml) for 2 hours at 50 °C.
- Transfer reverse-crosslinked "input" DNA and eluted ChIP DNA into a 2 ml MaXtract High Density (respectively). Add 200 μl 25:24:1 Phenol-Chloroform-Isoamyl Alcohol pH 7.9 to the tubes in a chemical fume hood and invert to mix well. Spin the tubes for 5 minutes at 16,110 x g (13,200 rpm) at room temperature.
- Transfer upper aqueous phase to a new tube and precipitate reverse crosslinked DNA with 20 μl 3 M Sodium Acetate pH 5.5, 200 μl isopropanol and 1 μl 15 mg/ml Glycoblue. Incubate at -80 °C for 30 minutes and pellet DNA for 30 minutes at 16,110 x g (13,200 rpm) at 4 °C.
- After centrifugation of isopropanol precipitated DNA, wash DNA pellet with 75% ice-cold ethanol twice and resuspend DNA pellet in 20 μl TE buffer.
- Quantitate "input" DNA (from step 5.1.3) and ChIP DNA by picogreen assay 10.
- Perform an enrichment check via qualitative PCR (qPCR).
B. Chromatin Interaction Analysis using Paired-End Tag Sequencing (ChIA-PET)
The second half of the video will highlight the key steps in the construction of a ChIA-PET library.
1. End-Blunting of ChIP DNA Fragments
This chapter of the video highlights two main points, a step-by-step procedure of washing magnetic beads to remove enzymes and buffering salt of the previous reaction (Step 1.1, 12:22 to 12:54 of the video) and the procedure of setting up enzymatic reactions involving magnetic beads in the reaction mixture (Step 1.2, 12:54 to 13:25 of video).
- Wash ChIP-enriched beads once with ice-cold TE Buffer. This and future washes involving magnetic beads consist of the following steps.
- Centrifuge at 6,100 x g (800 rpm) for 1 minute at 4 °C.
- Reclaim beads using a magnetic particle concentrator.
- Discard supernatant.
- Add buffer to beads.
- Mix beads with buffer by flicking action.
- Centrifuge at 6,100 x g (800 rpm) for 1 minute at 4 °C.
- Reclaim beads using a magnetic particle concentrator.
- Discard supernatant.
- To fill-in sonicated overhangs, prepare a master-mix with 70 μl 10 x Buffer for T4 DNA polymerase, 7 μl 10 mM dNTPs and 615.8 μl nuclease-free water. Mix thoroughly and resuspend washed beads with master-mix. Add 7.2 μl 9.7 U/μl T4 DNA polymerase to a final volume of 700 μl. Mix and incubate for 40 minutes at 37 °C with rotation (using Palico Biotech Intelli-Mixer RM-2L, F8, 30 rpm, U=50, u=60). Perform all subsequent enzymatic reactions with the following steps.
- Dilute reaction buffers with nuclease-free water.
- Add all other reagents (except enzymes) to diluted buffer.
- Mix thoroughly. Resuspend beads/pellet with reaction mix.
- Add enzyme to resuspended beads/pellet (Keep enzymes on benchtop cooler box at all times to ensure maximum enzymatic activity).
- Seal the tube with parafilm.
- Ensure that beads are well-mixed throughout the entire incubation.
Discard reaction mixture and wash away residual buffer salts and enzymes thrice with ice-cold wash buffer (10 mM Tris-Cl pH 7.5, 1 mM EDTA, 500 mM NaCl).
2. Ligation of Biotinylated Half-Linkers to ChIP DNA
This chapter of the video highlights the characteristics of the half-linker oligonucleotides used in the construction of ChIA-PET and the use of nucleotide barcode composition to distinguish between non-specific and specific ligation products (13:28 to 14:24) of the video).The chapter also shows the step-by-step procedure of setting up a half-linker ligation reaction (Step 2.2, 14:24 to 15:54 of the video).
Two types of biotinylated half-linkers are introduced in this protocol and are designed with an internal barcode of four nucleotides (TAAG or ATGT) and a recognition site for the type IIS restriction enzyme MmeI (TCCAAC). After ChIP enrichment, sonicated chromatin fragments are divided equally into two aliquots and are first ligated with an excess of either half-linker A or half-linker B 5,9.
Divide ChIP-enriched beads into two aliquots for DNA half-linker ligation by biotinylated half-linkers A or half-linkers B respectively. These two half-linkers have the same nucleotide sequences, except four nucleotides in the middle which serve (Half-linker A with TAAG; half-linker B with ATGT) as nucleotide barcodes. When combined during proximity ligation, random and non-specific ligations between two different ChIP complexes can be identified from the population of sequences with heterodimer AB linkers, as compared with specific ligations which can be identified from the population of sequences with homodimer AA or BB linkers.
Ligate ChIP-enriched beads with 140 μl 5 x T4 DNA ligase buffer with PEG, 3.5 μl 200 ng/μl biotinylated half-linkers A or B, 553.5 μl nuclease-free water and 3 μl 30 U/μl T4 DNA ligase (Keep T4 DNA ligase on a benchtop cooler box at all times as ligases are unstable even on ice). Seal the tube with parafilm and incubate overnight at 16 °C with rotation.
3. Elution and Proximity Ligation of ChIP DNA Fragments
This chapter of the video explains the role of Buffer EB, SDS and Triton X-100 during the elution of chromatin complexes from beads and also shows the step-by-step procedure of setting up a circularization reaction (Step 3.9, 15:54 to 17:10 of the video).
After ligating the half-linkers to the chromatin fragments, both fractions are combined and eluted off the beads. Interacting DNA fragments will then be connected by a complete linker sequence during proximity ligation.
Using the nucleotide barcode composition, the sequences can be classified into three categories, namely sequences with heterodimer AB linkers (barcode ATGT / TAAG) and sequences with homodimer AA or BB linkers (barcode TAAG / TAAG or ATGT / ATGT) to distinguish non-specific and specific ligation products respectively 9.
Hence, the use of two half-linkers with different nucleotide barcodes allows specification of different experiments or replicates, as well as to monitoring of non-specific chimeric ligation rate between different ChIP complexes 5.
Wash half-linker-ligated beads thrice with ice-cold Wash Buffer (10 mM Tris-Cl pH 7.5, 1 mM EDTA, 500 mM NaCl).
- Combine both tubes of half-linker-ligated beads in the following manner.
- Reclaim one of the tubes of half-linker-ligated beads using a magnetic particle concentrator ("Tube A").
- Keep the other tube of half-linker-ligated beads on ice ("Tube B").
- Discard Wash Buffer (from step 3.2.1) from Tube A and transfer half-linker-ligated beads (from step 3.2.2) from Tube B into Tube A while Tube A is still on the magnetic particle collector.
- Add 700 μl ice-cold Wash Buffer to Tube B to collect any residual half-linker-ligated beads and ensure both tubes of half-linker-ligated beads are combined properly. Transfer the wash buffer into Tube A. Discard the empty Tube B.
Phosphorylate half-linker-ligated beads with 70 μl 10 x T4 DNA ligase buffer (NEB), 616 μl nuclease-free water and 14 μl 10 U/μl of T4 DNA Polynucleotide Kinase. Incubate for 50 minutes at 37 °C with rotation.
Reclaim phosphorylated half-linker-ligated beads using a magnetic particle concentrator and discard reaction mix.
Elute half-linker-ligated chromatin-DNA complex with 200 μl freshly prepared Elution Buffer (TE Buffer, 1% SDS). Incubate for 30 minutes at room temperature (22 °C) with rotation.
Transfer eluate to a fresh tube and rinse beads with 900 μl Buffer EB. Transfer supernatant to the same tube to combine the elutions.
Transfer collected eluate to the filter cups of two centrifuge tube filters and centrifuge at 16,110 x g (13,200 rpm) for 1 minute at room temperature (22 °C).
Sequester SDS by adding 90 μl 20% Triton X-100 and invert to mix well. Incubate for 1 hour at 37 °C.
Circularization is performed under extremely dilute conditions in order to favor ligation events within individual cross-linked chromatin complexes while minimizing ligation events between different chromatin complexes which would give rise to undesirable chimeric DNA molecules. Working on ice, transfer quenched eluate to a 50 ml falcon tube and add 7776 μl nuclease-free water, 1000 μl 10 x T4 DNA Ligase Buffer (NEB) and 33.33 μl 30 U/μl T4 DNA ligase. Incubate overnight at 16 °C.
4. Reverse-Crosslinking and DNA Purification
This chapter of the video shows the step-by-step procedure of phenol:chloroform extraction (Step 4.3, 17:11 to 17:59) and the close-up of the pelleted DNA after centrifugation in Step 4.4.
Reverse-crosslink and degrade protein by adding 100 μl 20 mg/ml Proteinase K solution. Incubate for 2 hours at 50 °C.
Transfer chromatin DNA into a 50 ml MaXtract High Density and add 9ml nuclease-free water to obtain a final volume of 19 ml.
Add 19 ml 25:24:1 Phenol-Chloroform-Isoamyl Alcohol pH 7.9 to the tube in a chemical fume hood and invert to mix well. Spin the tubes for 5 minutes at 1,800 x g (3,000 rpm) at room temperature.
Transfer upper aqueous phase to a 50 ml Oak Ridge Centrifuge Tubes (Teflon FEP) and precipitate chromatin DNA with 1.9 ml 3 M Sodium Acetate pH 5.5, 19 ml isopropanol and 5 μl Glycoblue. Glycoblue is added to enhance visibility of the pellet.
Incubate at -80 °C for at least an hour. Allow the frozen solution to thaw before centrifuging at 38,720 x g (18,000 rpm) for 30 minutes at 4 °C.
After centrifugation of isopropanol precipitated DNA, wash DNA pellet with 75% ice-cold ethanol twice and resuspend DNA pellet in 34 μl Buffer EB. Transfer DNA mixture to a 1.7 ml DNA LoBind tube.
Resuspend DNA pellet in 34 μl Buffer EB.
- (RNase treatment is optional as subsequent washing and purification steps in the ChIA-PET protocol serve to remove contaminating RNA and manual inspections of the ChIA-PET sequences from our in-house libraries have displayed no traces of contaminating RNA).
- Perform RNase digestion by adding 10 μl RNase ONE 10 x Reaction Buffer, 55 μl nuclease-free water and 1 μl 10 U/μl RNase ONE Ribonuclease. Incubate at 37 °C for an hour.
- Transfer chromatin DNA into a 2 ml MaXtract High Density. Add 100 μl 25:24:1 Phenol-Chloroform-Isoamyl Alcohol pH 7.9 to the tube in a chemical fume hood and invert to mix well. Spin the tube for 5 minutes at 16,110 x g (13,200 rpm) at room temperature (22 °C).
- Transfer upper aqueous phase to a new tube and precipitate reverse-crosslinked DNA with 10 μl 3 M Sodium Acetate pH 5.5 and 100 μl isopropanol. Incubate at -80 °C for 30 minutes and pellet DNA for 30 minutes at 16,110 x g (13,200 rpm) at 4 °C.
- After centrifugation of isopropanol precipitated DNA, wash the DNA pellet with 75% ice-cold ethanol twice and resuspend DNA pellet in 34 μl Buffer EB.
5. Immobilization of ChIA-PET DNA to Streptavidin Beads
The introduction of this chapter talks about the presence of the MmeI recognition site and the biotinylated T present in the half-linker oligonucleotides to facilitate the extraction of tag-linker-tag constructs ("PETs", 18:02 to 19:10 of the video). In addition, this chapter of the video also gives a quick overall view of Step 5.2, the step-by-step procedure of setting up a PCR reaction (Step 6.1, 19:10 to 20:00 of the video) and excising a successful ChIA-PET DNA (Step 6.9, 20:00 to 20:30).
Half-linkers A and B contain flanking MmeI recognition sites, allowing this type IIS restriction enzyme to cut 18/20 base pairs downstream of their target binding sites to generate short "tags" of the chromatin fragment, producing paired tag-linker-tag constructs ("PETs").
To enable the purification of PET constructs by streptavidin-coated magnetic beads, both half-linker A and B are modified with biotin, allowing capture and purification of the ChIA-PET constructs.
Release captured ChIA-PET DNA by adding 5 μl 10 x NEBuffer 4, 5 μl freshly prepared 500 μM (10 x) S-adenosylmethionine (SAM) and 1 μl 2 U/μl MmeI to resuspended DNA. Quench excess MmeI by adding 5 μl non-biotinylated half-linkers to reaction mix for MmeI can be self-inhibitory in excess. Incubate for 2 hours at 37 °C.
To capture released ChIA-PET DNA, transfer 50μl of resuspended M-280 streptavidin beads to a DNA LoBind tube. Wash beads twice with 2 x Binding and Washing Buffer (2 x B&W: 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl). Resuspend beads in 50 μl 2 x B&W Buffer and transfer 50 μl of digestion mix (from step 5.1) to the same tube. Mix well and incubate for 45 minutes at room temperature with rotation.
Reclaim beads using a magnetic particle concentrator and discard supernatant. Wash beads twice with 150 μl 1 x Binding and Washing Buffer (1 x B&W: 5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl).
Ligate 454 GS20 Adaptors to ChIA-PET DNA with 5 μl 10 x T4 DNA Ligase Buffer (Note that the Ligase Buffer used in this step is different from step 3.9, the Ligase Buffer used in this step is from the same company as that which supplies the T4 DNA Ligase), 4 μl 200 ng/μl 454 GS20 Adaptor A, 4 μl 200 ng/μl 454 GS20 Adaptor B, 36 μl nuclease-free water and 1 μl 30 U/μl T4 DNA ligase. Incubate overnight at 16 °C with rotation. ChIA-PET DNA can also be ligated with Illumina-NN adaptors, in which case we recommend amplification of the library using PCR primer PE1.0 and PCR primer PE 2.0 purchased from Illumina.
Reclaim beads using a magnetic particle concentrator and discard supernatant. Wash beads thrice with 150 μl 1 x B&W Buffer.
Nick translate ChIA-PET DNA with 5 μl 10 x NEBuffer 2, 2.5 μl 10 mM dNTPs, 38.5 μl nuclease-free water and 4 μl 10 U/μl Escherichia coli DNA Polymerase I. Incubate overnight at room temperature (22 °C) with rotation.
Reclaim beads using a magnetic particle concentrator and discard supernatant. Wash beads thrice with 150 μl 1 x B&W Buffer.
Resuspend beads with 50 μl Buffer EB.
6. Amplification of ChIA-PET DNA
Set up PCR reactions with 2 μl beads suspension, 21 μl nuclease free water, 1 μl 10 μM Illumina 1-454 primer, 1 μl 10 μM Illumina 2-454 primer and 25 μl Phusion High Fidelity Master Mix.
To determine the number of cycles necessary to generate enough PCR products for sequencing, set up three test PCR reactions with 16, 18 and 20 cycles. Do not exceed 20 cycles as we have found that doing so results in very low library complexity.
| Initial Step | 30 seconds | 98 °C | (denaturation) |
| 18 to 25 cycles | 10 seconds | 98 °C | (denaturation) |
| 30 seconds | 65 °C | (annealing) | |
| 30 seconds | 72 °C | (extension) | |
| Final Step | 5 minutes | 72 °C | (final extension) |
Determine the lowest possible cycle number by running the PCR reactions on a 4-20% gradient TBE gel and staining with SYBR Green I, ensuring the presence of a band of 223 base pairs, which is the length of the ChIA-PET DNA.
Amplify the rest of the ChIA-PET DNA-bound streptavidin beads in a large-scale PCR with as few PCR cycles as possible while still obtaining sufficient yield for sequencing.
Reclaim beads from all PCR reactions using a magnetic particle concentrator and pool supernatant to two fresh DNA LoBind tubes. Precipitate each tube of supernatant by adding 60 μl 3 M Sodium Acetate, 2 μl GlycoBlue and 600 μl isopropanol to pooled reactions.
Incubate at -80 °C for 30 minutes. Centrifuge at 16,110 x g (13,200 rpm) for 30 minutes at 4 °C.
After centrifugation of isopropanol precipitated DNA, wash DNA pellet with 75% ice-cold ethanol twice and resuspend DNA pellet in 100 μl TE buffer and 5 μl 6 x loading dye.
Distribute PCR products evenly into the wells of a 6% TBE gel. Run the gel in 1 x TBE buffer at 200 V for 35 minutes and stain with SYBR Green I. Visualize the gel with a Dark Reader Transilluminator.
- Carefully excise the 223 bp band from the gel and perform "gel crush" DNA extraction protocol as follows:
- Place excised gel fragments in 0.6 ml microcentrifuge tubes that have been pierced at the bottom with a 21-G needle.
- Place each tube inside a 1.5 ml screw cap tube and centrifuge at 16,110 x g (13,200 rpm) for 5 minutes at 4 °C.
- Add 200 μl TE buffer pH 8.0 to each tube and ensure the gel pieces are fully immersed in the buffer.
- Freeze at -80 °C for an hour and incubate at 37 °C overnight.
- Transfer gel pieces together with the buffer in each tube to the filter cup of a centrifuge tube filter and centrifuge at 16,110 x g (13,200 rpm) for 10 minutes at 4 °C.
- Rinse each 1.5 ml tube with 200 μl TE buffer pH 8.0 and transfer rinsing buffer to each filter unit upon completion of the first spin.
- Pool filter-through and precipitate with isopropanol.
- Resuspend DNA pellet with 15 μl TE buffer.
7. Quality Check and Amplification of ChIA-PET DNA
Proceed with quality check using Agilent DNA 1000 Assay or Agilent High Sensitivity DNA Assay on Agilent Bioanalyzer 2100.
The electropherogram of the Agilent DNA assay should display only one peak along with a flat baseline before proceeding to Illumina Genome Analyzer IIx sequencing.
- Prior to Illumina Genome Analyzer IIx sequencing, ChIA-PET DNA ideally should have a minimum concentration of 10 nM. Perform a 4-point quantitative PCR according to Illumina qPCR Quantification Protocol Guide to determine the amount of sample to load onto the flow cell. Alternatively, perform a one-point quantitative PCR as follows:
- Dilute the control template to 100 pM, 10 pM and 1 pM. As described in Illumina qPCR Quantification Protocol, a control template is defined as any library prepared for sequencing on the Illumina platform and it should be as similar as possible in terms of template size, GC content and library type.
- Dilute the ChIA-PET DNA to 10 pM.
- Set up PCR triplicates of each dilution with 0.1 μl qPCR Primer 1.1, 0.1 μl qPCR Primer 2.1, 5 μl LightCycler480 DNA SYBR Green I Master Mix, 3.8 μl nuclease-free water and 1 μl of template with Roche Applied Science LightCycler 480 Real-Time PCR System. Include two negative controls using 1 μl of nuclease-free water as template.
- Initial Denaturation
- 95 °C 5 minutes 4.8 °C/s
- Amplification
- 95 °C 10 seconds 4.8 °C/s
- 60 °C 1 minute 2.5 °C/s
- 72 °C 30 seconds 4.8 °C/s
- Melting Curve
- 95 °C 5 seconds 4.8 °C/s
- 65 °C 1 minute 2.5 °C/s
- 95 °C 5 acquisitions per °C
- Cooling
- 40 °C 10 seconds 2.0 °C/s
- Ensure that the negative controls show no amplification and that the standard deviation for the Ct values is less than 0.1 before calculating the concentration of the library templates based on the standard curve generated from the control template dilutions.
Generate clusters on the surface of Illumina flow cells by loading 1.5pM into Illumina cBot Cluster Generation System according to onscreen instructions.
- Proceed to sequence ChIA-PET DNA on Illumina Genome Analyzer IIx using Illumina 3-454 sequencing primer and Illumina 4-454 sequencing primer in a single lane to see the quality of the library. Store remaining ChIA-PET DNA at -20 °C. If the library has good data, prepare the sample for additional runs of 4 to 8 lanes as needed based on the sample lane results to obtain 18 to 20 million unique reads.
- The amount of ChIA-PET DNA loaded on the flow cells depends greatly on the version of the sequencing data analysis software, Illumina's Sequencing Control Studio. For version 2.9, the loading concentration ranges from one-third to half of the maximum capacity of each tile, which equates to approximately 21 million pass filtered reads with about 9.5 million reads of PETs.
- Reduced loading is necessary to ensure correct base-calling for the barcode sequences of the linkers. This error occurs when a high number of similar nucleotides are sequenced, as is the case if the linker is sequenced, to obtain barcoding information. If the barcoding information is not desired, and the linkers are not read, then full loading concentration can be used.
The qSeq files converted by the offline base caller can be further analyzed using the ChIA-PET tool according to the ChIA-PET Tool installation manual (Version 4.1) 8.
C. Representative ChIA-PET Results
We successfully constructed ChIA-PET libraries using RNA Polymerase II antibody (8WG16) in MCF-7 cells (CHM160 and CHM163) 9 using 672 ng of ChIP material as described above. The initial diagnostic gel run of this PCR-amplified library, as mentioned in Step 6.2 of the ChIA-PET protocol, displayed a bright and well-defined band at the expected size of 223 base pairs for all PCR cycling used (see Figure 4).
16 PCR cycles was used to amplify the ChIA-PET library and a total yield of 17.1 ng was obtained. A single, intense electropherogram peak was observed at the expected size of 223 base pairs via Agilent DNA 1000 analysis as mentioned in Step 7.1 of the ChIA-PET protocol (see Figure 5).
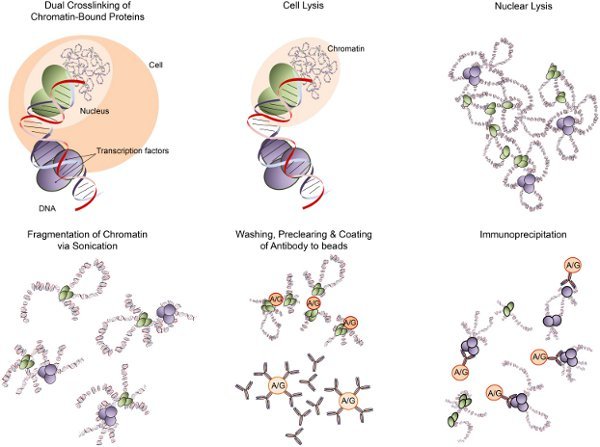 Figure 1. ChIP overview. MCF-7 cells are dual cross-linked with EthylGlycol bis(SuccinimidylSuccinate (EGS) and formaldehyde sequentially, resulting in covalent links between spatially adjacent chromatin. The cross-linked chromatin was obtained from the fixed MCF-7 cells by cell lysis and nuclear lysis. The chromatin was then subjected to fragmentation to a size range of 200-600 base pairs. After pre-clearing the sonicated chromatin with Protein G magnetic beads to remove non-specific DNA, the pre-cleared chromatin was immunoprecipitated overnight with antibody-coated beads to capture chromatin of interest.
Figure 1. ChIP overview. MCF-7 cells are dual cross-linked with EthylGlycol bis(SuccinimidylSuccinate (EGS) and formaldehyde sequentially, resulting in covalent links between spatially adjacent chromatin. The cross-linked chromatin was obtained from the fixed MCF-7 cells by cell lysis and nuclear lysis. The chromatin was then subjected to fragmentation to a size range of 200-600 base pairs. After pre-clearing the sonicated chromatin with Protein G magnetic beads to remove non-specific DNA, the pre-cleared chromatin was immunoprecipitated overnight with antibody-coated beads to capture chromatin of interest.
 Figure 2. Gel analysis of sonicated chromatin fragments. A 100 bp DNA ladder is shown in the first and last lane for size reference. The sonicated chromatin displayed strong intensity between 200 to 600 bp which is ideal to capture long-range chromatin interactions.
Figure 2. Gel analysis of sonicated chromatin fragments. A 100 bp DNA ladder is shown in the first and last lane for size reference. The sonicated chromatin displayed strong intensity between 200 to 600 bp which is ideal to capture long-range chromatin interactions.
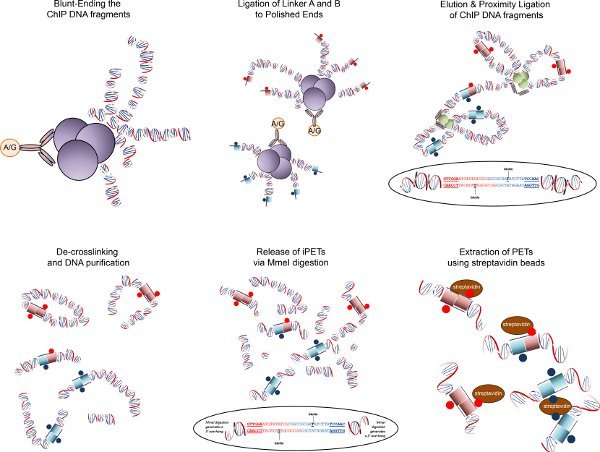 Figure 3. ChIA-PET overview. Fragmented chromatin fragments are end-blunted and ligated to biotinylated half-linkers containing flanking MmeI restriction sites. Intact chromatin complexes are then eluted off the beads and subjected to proximity ligation under extremely dilute conditions, such that interacting DNA fragments are preferentially ligated to one another. After reverse cross-linking to remove DNA-associated proteins, MmeI digestion is carried out to release tag-linker-tag (PET) constructs, which are then purified by selective binding to streptavidin beads. The PET constructs are ligated with adapters for high-throughput sequencing.
Figure 3. ChIA-PET overview. Fragmented chromatin fragments are end-blunted and ligated to biotinylated half-linkers containing flanking MmeI restriction sites. Intact chromatin complexes are then eluted off the beads and subjected to proximity ligation under extremely dilute conditions, such that interacting DNA fragments are preferentially ligated to one another. After reverse cross-linking to remove DNA-associated proteins, MmeI digestion is carried out to release tag-linker-tag (PET) constructs, which are then purified by selective binding to streptavidin beads. The PET constructs are ligated with adapters for high-throughput sequencing.
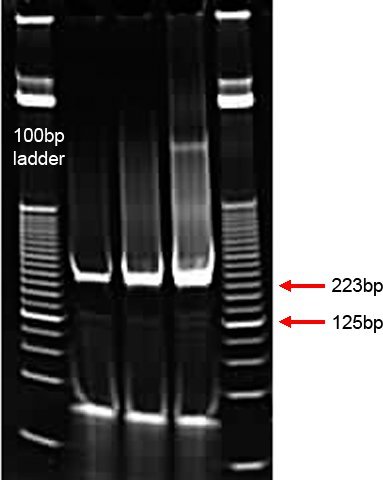 Figure 4. Gel analysis of ChIA-PETs after PCR amplification. A 25 bp DNA ladder is shown in lane 1 and 5 for size reference. Lanes 2 to 4 are PCR products generated after 16, 18 and 20 cycles of PCR amplification from 2 μl of bead-immobilized template, respectively. This is a successful library, as indicated by the bright, well-defined bands at the expected size of 223 bp. The non-specific smear is generated when the number of PCR cycles is increased while the lowest band of each PCR reaction is made up of primer dimers.
Figure 4. Gel analysis of ChIA-PETs after PCR amplification. A 25 bp DNA ladder is shown in lane 1 and 5 for size reference. Lanes 2 to 4 are PCR products generated after 16, 18 and 20 cycles of PCR amplification from 2 μl of bead-immobilized template, respectively. This is a successful library, as indicated by the bright, well-defined bands at the expected size of 223 bp. The non-specific smear is generated when the number of PCR cycles is increased while the lowest band of each PCR reaction is made up of primer dimers.
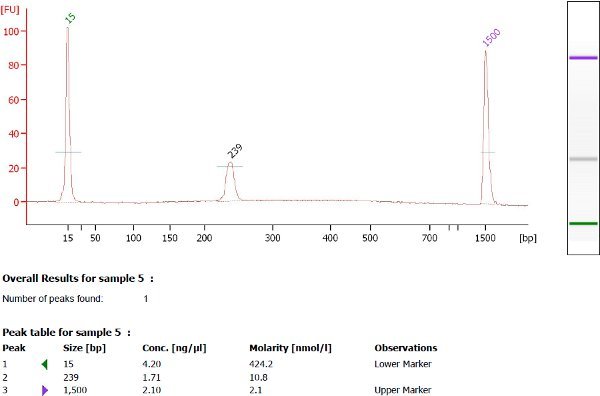 Figure 5. Agilent 2100 Bioanalyzer analysis of purified Illumina-454 adapter-ligated ChIA-PETs. Screen capture of Agilent 2100 Bioanalyzer electropherograms profiling a successful library, with a single intense peak at the expected size of 223 bp. Note that the Agilent Bioanalyzer assay usually reports a slightly higher-than-expected size; in this case, the desired peak is displayed at 237 bp instead of 223 bp. This is within the 10% error range of the Agilent assay.
Figure 5. Agilent 2100 Bioanalyzer analysis of purified Illumina-454 adapter-ligated ChIA-PETs. Screen capture of Agilent 2100 Bioanalyzer electropherograms profiling a successful library, with a single intense peak at the expected size of 223 bp. Note that the Agilent Bioanalyzer assay usually reports a slightly higher-than-expected size; in this case, the desired peak is displayed at 237 bp instead of 223 bp. This is within the 10% error range of the Agilent assay.
Discussion
ChIA-PET is a method developed to identify long-range interactions in transcription regulation. One of the critical factors that determine the quality of a ChIA-PET library is the quality of ChIP material.
The protocol shown in the video incorporates the use of EGS and formaldehyde to cross-link the cells. The use of formaldehyde combined with a second cross-linking reagent bearing a longer spacer arm may help in the binding of proteins which could not be bound by formaldehyde alone 3,11,15. We have constructed libraries with this method which have demonstrated robust binding sites and long-range interactions 9. However, the cross-linking and ChIP conditions should be optimized for each factor of interest, and it is important not to over-crosslink as too much cross-linking will result in difficulties in fragmentation by sonication, and may possibly result in spurious chromatin interactions. Chromatin interactions identified by ChIA-PET should be validated by a different method, such as fluorescence in-situ hybridization 4.
We recommend a minimum of 100 ng of chromatin material. While we have constructed good quality libraries from 50 ng of chromatin material, we have observed that large amounts of starting material allowed construction of ChIA-PET libraries with less than 16 PCR cycles, thereby minimizing amplicons and redundancy of each library. This lower redundancy correlated with higher unique mapped tags and also a high percentage of usable data, thereby enabling a more comprehensive chromatin interaction map with fewer lanes of sequencing. The final packed volume of beads in each tube should be 50 μl and 100 μl for magnetic and Sepharose beads respectively. If the packed bead volume is less than stated, bring to the minimum packed volume with similarly pre-cleared blank magnetic or Sepharose beads to minimize loss of DNA-bearing beads in subsequent steps. Sawed-off tips or large-core tips should be used for pipetting Sepharose beads.
The following modifications were incorporated following the previously published ChIA-PET protocol 5. Firstly, magnetic G beads were used to minimize sample loss during washes. In addition, we identified non-specific bands with approximate sizes of 100 bp and 138 bp to be amplicons of self-ligated half-linkers or/and adaptors. Hence, we reduced the concentration of biotinylated half-linkers and 454 GS20 adaptors to minimize non-specific bands during PCR amplification. The proximity ligation volume was reduced from 50 ml to 10 ml to minimize sample loss during subsequent purification steps and also save on reagent costs. We also increased the incubation time to immobilize ChIA-PET DNA to beads to ensure maximal capture of ChIA-PET DNA on the streptavidin beads.
During the proximity ligation step, chimeric ligations that do not represent true in vivo chromatin interactions are inevitably generated in a non-specific and random manner. Hence, to evaluate the quality of data from any ChIA-PET experiment, the rate of chimerism is estimated from the use of two different half-linkers with specific nucleotide barcodes TAAG and ATGT 5. After high-throughput sequencing, the ChIA-PET sequences are first analyzed for linker barcode composition and sequences derived from specific ligation products and non-specific ligation products can be distinguished 8. The percentage of known chimeras (i.e. heterodimers AB linkers) present in our in-house MCF-7 RNA Polymerase II ChIA-PET libraries is less than 15%.
ChIA-PET sequences are subsequently classified into two categories, namely self-ligation PETs and inter-ligation PETs. Self-ligation PETs are obtained from self-circularization ligation of the chromatin fragments while inter-ligation PETs are derived from inter-ligation between two different DNA fragments. The latter is then sub-divided into three different categories based on the genomic distance of each tag on the same chromosome (intrachromosomal inter-ligation PETs) or that both tags are mapped to two different chromosomes (interchromosomal inter-ligation PETs). We have developed a ChIA-PET tool software package to sort out the different categories 8. This will be based on the DNA fragments that are in the library. Generally, smaller ChIP fragments will give a higher resolution and the cut off for these RNA Polymerase II ChIA-PET libraries is about 4 kb.
In addition, real chromatin interactions can be distinguished from random noise by counting the number of inter-ligation PETs in an interaction cluster; in other words, a cluster of high PET count is said to have a higher probability of being a real chromatin interaction 8.
To filter our false positives arise from highly enriched anchors which can form inter-ligation PETs by random chance, a statistical analysis framework has also been formulated to account for random formation of any inter-ligation PETs between two anchors 8.
In conclusion, the ChIA-PET technique allows mapping chromatin interaction networks on a global scale. The implementation of ChIP in ChIA-PET allows the reduction of library complexity and background noise. In addition, ChIP adds specificity to chromatin interactions, enabling the examination of specific chromatin interactions associated with particular transcription factors 5.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors are supported by A*STAR of Singapore. In addition, M.J.F. is supported by an A*STAR National Science Scholarship, a L'Oreal For Women in Science National Fellowship and a Lee Kuan Yew Post-Doctoral Fellowship. Y.R. is supported by NIH ENCODE grants (R01 HG004456-01 and R01 HG003521-01). The authors also acknowledge the videography team of 8 Pixels Productions, Singapore, in particular Mr. Kelvin Issey, Mr. Chang Kai Xiang and Mr. Sherwin Gan for filming the scenes, Ms. Siti Rahim for video editing and Ms. Michelle Teo for the voice-over.
References
- Cai S, Lee CC. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat. Genet. 2006;38:1278–1288. doi: 10.1038/ng1913. [DOI] [PubMed] [Google Scholar]
- Cook PR. The organization of replication and transcription. Science. 1999;284:1790–1795. doi: 10.1126/science.284.5421.1790. [DOI] [PubMed] [Google Scholar]
- Das PM, Ramachandran K. Chromatin immunoprecipitation assay. Biotechniques. 2004;37:961–969. doi: 10.2144/04376RV01. [DOI] [PubMed] [Google Scholar]
- Deng W, Blobel GA. Do chromatin loops provide epigenetic gene expression states. Curr. Opin. Genet. Dev. 2010;20:548–554. doi: 10.1016/j.gde.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Han Y. Chromatin interaction analysis using paired-end tag sequencing. Curr. Protoc. Mol. Biol. 2010;Chapter 21:21–25. doi: 10.1002/0471142727.mb2115s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Hassan AB. Visualization of focal sites of transcription within human nuclei. EMBO J. 1993;12:1059–1065. doi: 10.1002/j.1460-2075.1993.tb05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Fullwood MJ. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010;11:R22. doi: 10.1186/gb-2010-11-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ruan X. Extensive Promoter-Centered Chromatin Interactions Provide a Topological Basis for Transcription Regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P, Wei CL. Paired-end diTagging for transcriptome and genome analysis. Curr. Protoc. Mol. Biol. Chapter. 2007;Chapter 21:12. doi: 10.1002/0471142727.mb2112s79. [DOI] [PubMed] [Google Scholar]
- Nowak DE, Tian B. Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques. 2005;39:715–725. doi: 10.2144/000112014. [DOI] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- Rippe K, von Hippel PH. Action at a distance: DNA-looping and initiation of transcription. Trends Biochem. Sci. 1995;20:500–506. doi: 10.1016/s0968-0004(00)89117-3. [DOI] [PubMed] [Google Scholar]
- Schoenfelder S, Sexton T. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng PY, Vakoc CR. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques. 2006;41:694–698. doi: 10.2144/000112297. [DOI] [PubMed] [Google Scholar]