

Abstract
Tumor heterogeneity represents a fundamental feature supporting tumor robustness and presents a central obstacle to the development of therapeutic strategies1. To overcome the issue of tumor heterogeneity, it is essential to develop assays and tools enabling phenotypic, (epi)genetic and functional identification and characterization of tumor subpopulations that drive specific disease pathologies and represent clinically relevant targets. It is now well established that tumors exhibit distinct sub-fractions of cells with different frequencies of cell division, and that the functional criteria of being slow cycling is positively associated with tumor formation ability in several cancers including those of the brain, breast, skin and pancreas as well as leukemia2-8. The fluorescent dye carboxyfluorescein succinimidyl ester (CFSE) has been used for tracking the division frequency of cells in vitro and in vivo in blood-borne tumors and solid tumors such as glioblastoma2,7,8. The cell-permeant non-fluorescent pro-drug of CFSE is converted by intracellular esterases into a fluorescent compound, which is retained within cells by covalently binding to proteins through reaction of its succinimidyl moiety with intracellular amine groups to form stable amide bonds9. The fluorescent dye is equally distributed between daughter cells upon divisions, leading to the halving of the fluorescence intensity with every cell division. This enables tracking of cell cycle frequency up to eight to ten rounds of division10. CFSE retention capacity was used with brain tumor cells to identify and isolate a slow cycling subpopulation (top 5% dye-retaining cells) demonstrated to be enriched in cancer stem cell activity2.
This protocol describes the technique of staining cells with CFSE and the isolation of individual populations within a culture of human glioblastoma (GBM)-derived cells possessing differing division rates using flow cytometry2. The technique has served to identify and isolate a brain tumor slow-cycling population of cells by virtue of their ability to retain the CFSE labeling.
Keywords: Medicine, Issue 62, Label-Retaining Cells, Slow-Dividing Cells, Cancer Stem Cells, Glioblastoma, CFSE
Protocol
1. Preparing Glioblastoma Single Cell Suspension
Gliomasphere culture is established and maintained using the neurosphere assay (NSA) as previously described2,11,12.
At the appropriate time for passaging the gliomaspheres, the medium containing the spheres is removed, placed in an appropriate size sterile tissue culture tube, and centrifuged at 800 rpm (110 g) for 5 min at room temperature.
The supernatant is discarded and the pellet of spheres is resuspended in 1 ml of 0.05% trypsin-EDTA and incubated at 37 °C in a water bath for 3-5 min.
An equal volume of soybean trypsin inhibitor is then used to stop the trypsin activity.
The cell suspension is gently pipetted up and down to ensure homogeneity and complete neutralization of the trypsin activity.
The cell suspension is again centrifuged at 800 rpm for 5 min. The supernatant is removed and the cells are resuspended in 1ml of NeuroCult NSC Basal Medium.
10μl of the single-cell suspension is mixed with 90μl of trypan blue to perform a cell count.
2. Staining with Cell Trace Carboxyfluorescein Succinimidyl Ester (CFSE) Green Fluorescent Dye
For every 1 million cells to be stained, 1 ml of staining reagent is prepared by adding 1 μl of 5 mM Cell Trace CFSE dye (Molecular Probes, Invitrogen) to 1 ml of NeuroCult NSC Basal Medium to give a final staining concentration of 5 μM.
The staining reagent containing the cells is mixed well until homogeneity and incubated at room temperature for 10 min.
During the incubation, ice-cold NeuroCult NSC Basal Medium is prepared as the quenching solution.
To stop the staining process, approximately 5-10 volumes of ice-cold quenching solution is added to the CFSE-loaded cells.
The solution is then centrifuged at 800 rpm for 5 min and the supernatant is discarded.
At this point, there should appear a pellet of cells at the bottom of the tube with a bright-green tint of color, indicating the staining was successful.
The cells are washed by resuspending the pellet in 1-2 ml of fresh NeuroCult NSC Basal Medium, centrifuging at 800 rpm for 5 min and discarding the supernatant. This procedure is repeated for a total of three washes.
After the third wash, the cells are resuspended in 1 ml of NeuroCult NSC Basal Medium and a cell count is performed.
The CFSE stained cells are then plated in sterile tissue culture flasks at an appropriate density [50,000 cells/ml] and placed in a 37 °C / 5% CO2 humidified incubator.
3. CFSE Loading Verification
Cells plated in NSA culture can be monitored under a fluorescent microscope to verify CFSE staining. At this stage, all of the cells exhibit bright green color (Figure 1). Alternatively, a drop of the cell suspension from CFSE stained cells could be placed on a microscope slide and covered with a coverslip to visualize cells under the fluorescent microscope.
Successful CFSE labeling is further confirmed via flow cytometry. As many as 1-5 x 105 cells from each group is sufficient for CFSE staining verification. The CFSE loaded cells and unloaded negative control cells are resuspended respectively in 1 x -PBS or NeuroCult NSC Basal Medium for a total volume of 500 μl and placed in separate FACS tubes.
The flow cytometer is adjusted based on its user manual instructions for related parameters. CFSE has a wavelength excitation maximum of 492 nm and emission maximum of 517 nm.
First, the unloaded control cells are run and the acquired events are recorded in forward and side scatter (FSC vs. SSC) and histogram plots, and the main cell population is gated (Figure 2A).
Similarly, the CFSE loaded cells are analyzed using the same parameters used for control cells (Figure 2B).
4. Isolation of the Slow-dividing [i.e. CFSE Retaining] Cell Population Using Fluorescent Activated Cell Sorting (FACS)
Upon division, CFSE intensity is decreased and hGBM cells form gliomaspheres that are composed of cells exhibiting different levels of green fluorescence intensity (Figure 3). When the gliomaspheres have reached an average size of approximately 150-200 μm in diameter (approximately 5 to 10 days post CFSE loading depending on the cell line growth rate), the medium containing the spheres is removed, placed in an appropriate size sterile tissue culture tube, and centrifuged at 800 rpm for 5 min at room temperature.
A single-cell suspension is prepared as described in prior steps and the cells are resuspended in PBS for a cell count in order to prepare a cell suspension with a density up to 107 cells per milliliter.
After resuspending the cells in an appropriate volume of PBS, Propidium Iodide (PI, Sigma, 500 μg/ml) is added at a concentration of 1 μl/ml of cell suspension to exclude dead cells when sorted/analyzed by flow cytometry.
Two 15 ml sterile tissue culture tubes each containing 1ml of complete NeuroCult NSC Medium is used to collect the sorted slow-dividing cells (CFSE retaining cells) and the fast dividing cells (CFSE diluting cells).
First, the cell suspension from the unloaded cell group is run through the flow cytometer.
The cells (events) are plotted based on forward scatter (FSC) versus side scatter (SSC) and voltage parameters for both parameters are adjusted so that the majority of the cells are included in the flow plot.
To exclude dead or damaged cells, the cells are plotted based on SSC versus PI reactivity and the single live cell population (PI negative) is gated.
Then, a homogeneous cell population is selected based on forward scatter (FSC) versus side scatter (SSC).
The homogeneous single live cell population is then plotted in a histogram based on cell frequency versus CFSE intensity set on a logarithmic scale. The CFSE laser intensity is adjusted in order to place the negative signal between 0 and 100.
Using the same parameters and a similar gating strategy, the CFSE labeled cells are run through the flow cytometer and analyzed (Figure 4C).
Subsequently, a slow dividing population of cells and an overall population (faster dividing cells) are identified based on their ability to retain CFSE (CFSEhigh-top 5% and CFSElow-bottom 85%, respectively).
These different cell populations are then sorted into separate 15 ml sterile tissue culture tubes containing 1ml of complete NSC medium.
Sorted cells are then centrifuged at 800 rpm for 5 min.
Supernatant is discarded and the pellet is resuspended in an appropriate volume of medium (depending on the number of isolated events or pellet size) and a cell count is performed.
Cells from the different sorted populations are then plated at 50,000 cells per milliliter in an appropriate sterile tissue culture flask and placed in a 37 °C / 5% CO2 humidified incubator for 5-10 days before being serially passaged or used for downstream experiments.
5. Representative Results
After CFSE loading, all the cells present an intense green fluorescence as visualized under the microscope (Figure 1). This green-tinted color can be seen even with the naked eye (see the video). Analyzing these cells with flow cytometry also shows highly intense green fluorescence compared to control cells (Figure 2). Upon plating CFSE loaded cells in neurosphere culture, these cells proliferate and form 150-200 μm gliomaspheres in 5-10 days. As the cells proliferate, the fluorescent dye is equally distributed between daughter cells upon divisions, leading to the halving of the fluorescence intensity with every cell division. Glioblastomas are functionally heterogeneous and are composed of cells proliferating on different time scales. The method described in this protocol enables one to identify and isolate fast and slow dividing cells by virtue of their ability to dilute or retain CFSE, respectively (Figure 3, also see the video).
Flow cytometry analysis of dissociated gliomaspheres generated from CFSE-loaded cells in comparison to unloaded cells demonstrates diverse CFSE retention properties (Figure 4). Fast dividing cells (bottom 85%) or slow dividing cells (the top 5% - CFSEhigh) exhibit gliomapshere forming ability when cultured in the neurosphere assay conditions (Figure 5).
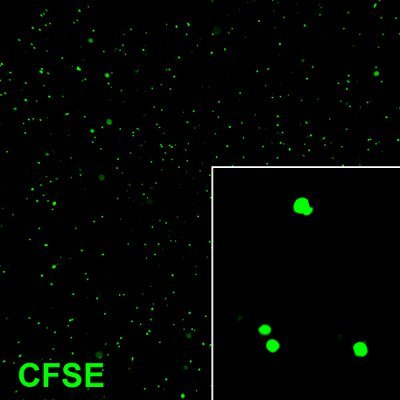 Figure 1. Dissociated tumor cells immediately after loading with CFSE dye. The cells exhibit bright green color demonstrating successful labeling. Original magnification; 10 x.
Figure 1. Dissociated tumor cells immediately after loading with CFSE dye. The cells exhibit bright green color demonstrating successful labeling. Original magnification; 10 x.
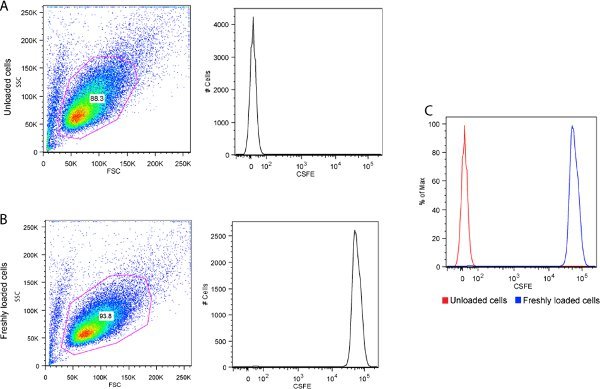 Figure 2. CFSE loading confirmation using flow cytometry. A) Unloaded cells, B) CFSE loaded cells and C) Comparing CFSE fluorescence intensity in loaded versus unloaded cells. Note that there is several orders of magnitude difference in the fluorescence intensity between the labeled and unlabeled cells.
Figure 2. CFSE loading confirmation using flow cytometry. A) Unloaded cells, B) CFSE loaded cells and C) Comparing CFSE fluorescence intensity in loaded versus unloaded cells. Note that there is several orders of magnitude difference in the fluorescence intensity between the labeled and unlabeled cells.
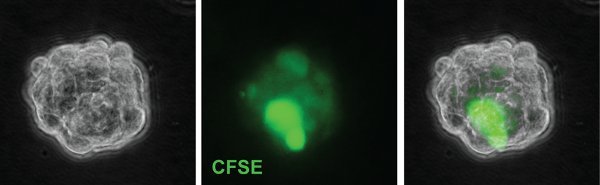 Figure 3. A representative gliomasphere derived from a CFSE-loaded cell 6 days after plating. Some cells exhibit very bright CFSE staining. Original magnification; 63 x.
Figure 3. A representative gliomasphere derived from a CFSE-loaded cell 6 days after plating. Some cells exhibit very bright CFSE staining. Original magnification; 63 x.
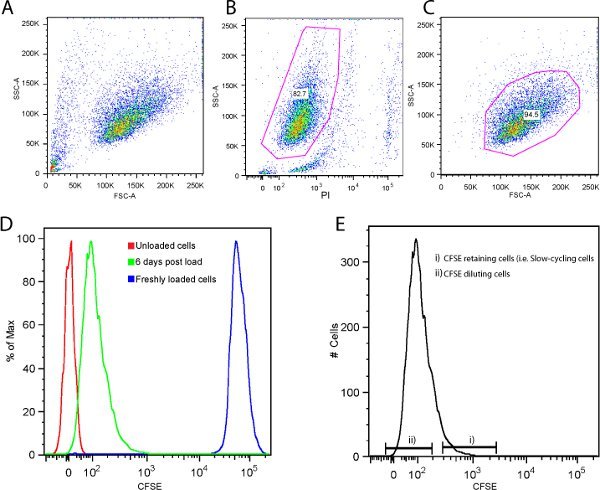 Figure 4. Representative flow cytometry plots/histograms for isolation of fast and slow dividing cells 6 days after CFSE staining. A) FSC vs. SSC to show the entire cell population, B) SSC vs. PI reactivity to exclude dead or damaged cells, C) FSC vs. SSC to gate a homogenous live cell population, D) Demonstrates the presence of CFSE retaining cells 6 days after CFSE labeling as evidenced by a greater fluorescence intensity compared to the negative control. The histogram also shows that the CFSE intensity decayed over time. E) Histogram displaying the gating strategy to identify CFSE retaining cells (top 5%) and CFSE diluting cells (bottom 85%).
Figure 4. Representative flow cytometry plots/histograms for isolation of fast and slow dividing cells 6 days after CFSE staining. A) FSC vs. SSC to show the entire cell population, B) SSC vs. PI reactivity to exclude dead or damaged cells, C) FSC vs. SSC to gate a homogenous live cell population, D) Demonstrates the presence of CFSE retaining cells 6 days after CFSE labeling as evidenced by a greater fluorescence intensity compared to the negative control. The histogram also shows that the CFSE intensity decayed over time. E) Histogram displaying the gating strategy to identify CFSE retaining cells (top 5%) and CFSE diluting cells (bottom 85%).
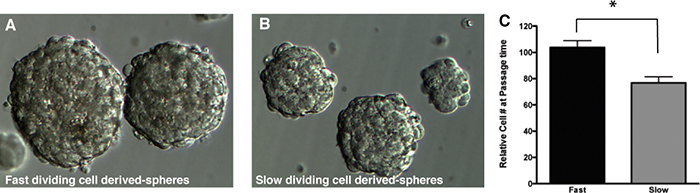 Figure 5. Representative gliomaspheres generated from A) fast-dividing cells (CFSElow) and B) slow-dividing cells (CFSEhigh). Original magnification: 10x and 20x respectively. C) Representative cell count obtained from fast or slow dividing cell-derived culture at passage time. P<0.05, t-test. Click here to view larger figure.
Figure 5. Representative gliomaspheres generated from A) fast-dividing cells (CFSElow) and B) slow-dividing cells (CFSEhigh). Original magnification: 10x and 20x respectively. C) Representative cell count obtained from fast or slow dividing cell-derived culture at passage time. P<0.05, t-test. Click here to view larger figure.
Discussion
An increasing number of studies have demonstrated the importance of a slow-dividing sub-compartment of cells in cancer which contribute to tumor formation and treatment resistance2-8. Therefore it is critical to describe and standardize experimental methods enabling the study of this specific subpopulation of cells or more generally to compare subpopulations with different growth rates. Using CFSE to identify and isolate sub-fractions of cells based on their rate of cell division is a starting point to further characterizing these specific cellular fractions, helping to advance our understanding of the heterogeneity described in tumors. In this protocol we gave the example of identifying and isolating a slow-dividing population within glioblastoma, but this protocol can also be applied to other tumor cells, including and not limited to breast, pancreatic and skin cancers. Downstream analysis of these populations can be performed and can include functional studies comparing proliferative potential2, tumor formation ability2, treatment sensitivity, and (epi)genetic comparisons. This methodology can also be used with non-cancerous systems and can be applied, for example, to somatic stem/precursor cells.
Critical technical points within the procedure involved adequate dissociation of cells with trypsin, and sufficient resuspension of cells in the staining media to establish a proper density and ensure homogenous staining. A small sample of freshly stained cells should be checked by flow cytometry to ensure homogenous labeling characterized by a Gaussian-like narrow profile (see Figure 2B). In the case of heterogeneous initial labeling, a pre-sort can be performed in order to improve peak resolution with narrower range of CFSE fluorescence. However it should be ensured that this pre-sort does not select for specific sizes. The use of propidium iodide (PI) labeling is important for identifying live cells and for dead/damaged population removal from the working gate, due to the bias that dead cells can introduce in downstream applications. It is worth noting the particularly high fluorescence intensity of the CFSE labeling and its potential to bleed into other channels when using neighboring fluorochromes in the spectrum, such as the fluorochrome phycoerythrin (PE). It is crucial to note that multiplex labeling experiments with CFSE and fluorochrome-conjugated antibodies may require a compensation process at the acquisition or analysis time. Be sure to have all the necessary controls that include unstained cells (providing the level of autofluorescence), singly stained cells and your specific combination. Multiple-color labeling in combination with CFSE works best with fluorochromes such as Pacific Blue or Allophycocyanin (APC). If the purpose of the experiment is to quantify the absolute number of cell divisions over a defined period of time, the CFSE Mean Fluorescence Intensity (MFI) has to be measured at a minimum of two different time points2 with the first one at least 24 hours post CFSE loading. CFSE intensity drops drastically within the first 24 hours in the absence of cell division but then stabilizes and decreases in relation to cell division. It is also necessary to include a control with non-proliferative CFSE loaded cells providing the baseline for CFSE intensity decay not related to cell division.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by the Florida Center for Brain Tumor Research, the Preston A. Wells Jr. Center for Brain Tumor Therapy and the National Institutes of Health (1R21CA141020-01).
References
- Tian T, Olson S, Whitacre JM, Harding A. The origins of cancer robustness and evolvability. Integr. Biol. (Camb) 2011;3:17–30. doi: 10.1039/c0ib00046a. [DOI] [PubMed] [Google Scholar]
- Deleyrolle LP. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain. 2011;134 (Pt. 5):1331–1343. doi: 10.1093/brain/awr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy K, Wang G, Rokhfeld D, Bieberich E. Deoxycholate promotes survival of breast cancer cells by reducing the level of pro-apoptotic ceramide. Breast Cancer Res. 2008;10:106–106. doi: 10.1186/bcr2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Roesch A. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis. 2009;26:611–623. doi: 10.1007/s10585-009-9260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham SM. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- Holyoake TL. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood. 2001;97:720–728. doi: 10.1182/blood.v97.3.720. [DOI] [PubMed] [Google Scholar]
- Parish CR. Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol. Cell Biol. 1999;77:499–508. doi: 10.1046/j.1440-1711.1999.00877.x. [DOI] [PubMed] [Google Scholar]
- Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J. Immunol. Methods. 2000;243:147–154. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- Azari H. Isolation and Expansion of Human Glioblastoma Multiforme Tumor Cells Using the Neurosphere Assay. J. Vis. Exp. 2011. pp. e3633–e3633. [DOI] [PMC free article] [PubMed]
- Azari H. Isolation and Expansion of the Adult Mouse Neural Stem Cells Using the Neurosphere Assay. J. Vis. Exp. 2010. pp. e2393–e2393. [DOI] [PMC free article] [PubMed]