

Abstract
Herpes simplex virus type-1 (HSV-1) establishes a life-long latent infection in peripheral neurons. This latent reservoir is the source of recurrent reactivation events that ensure transmission and contribute to clinical disease. Current antivirals do not impact the latent reservoir and there are no vaccines. While the molecular details of lytic replication are well-characterized, mechanisms controlling latency in neurons remain elusive. Our present understanding of latency is derived from in vivo studies using small animal models, which have been indispensable for defining viral gene requirements and the role of immune responses. However, it is impossible to distinguish specific effects on the virus-neuron relationship from more general consequences of infection mediated by immune or non-neuronal support cells in live animals. In addition, animal experimentation is costly, time-consuming, and limited in terms of available options for manipulating host processes. To overcome these limitations, a neuron-only system is desperately needed that reproduces the in vivo characteristics of latency and reactivation but offers the benefits of tissue culture in terms of homogeneity and accessibility.
Here we present an in vitro model utilizing cultured primary sympathetic neurons from rat superior cervical ganglia (SCG) (Figure 1) to study HSV-1 latency and reactivation that fits most if not all of the desired criteria. After eliminating non-neuronal cells, near-homogeneous TrkA+ neuron cultures are infected with HSV-1 in the presence of acyclovir (ACV) to suppress lytic replication. Following ACV removal, non-productive HSV-1 infections that faithfully exhibit accepted hallmarks of latency are efficiently established. Notably, lytic mRNAs, proteins, and infectious virus become undetectable, even in the absence of selection, but latency-associated transcript (LAT) expression persists in neuronal nuclei. Viral genomes are maintained at an average copy number of 25 per neuron and can be induced to productively replicate by interfering with PI3-Kinase / Akt signaling or the simple withdrawal of nerve growth factor1. A recombinant HSV-1 encoding EGFP fused to the viral lytic protein Us11 provides a functional, real-time marker for replication resulting from reactivation that is readily quantified. In addition to chemical treatments, genetic methodologies such as RNA-interference or gene delivery via lentiviral vectors can be successfully applied to the system permitting mechanistic studies that are very difficult, if not impossible, in animals. In summary, the SCG-based HSV-1 latency / reactivation system provides a powerful, necessary tool to unravel the molecular mechanisms controlling HSV1 latency and reactivation in neurons, a long standing puzzle in virology whose solution may offer fresh insights into developing new therapies that target the latent herpesvirus reservoir.
Keywords: Immunology, Issue 62, neuron cell culture, Herpes Simplex Virus (HSV), molecular biology, virology
Protocol
1. Isolation and Culture of SCG Neurons from Rat Embryos
To provide a useful context for understanding this protocol, and for a comprehensive discussion of earlier literature that established methods of SCG neuron culture, including the basis for SCG in vitro culture, plate-coating substrates, and the components of serum-free media, the reader is referred to references2-4.
The use of rats as a source for SCG neurons was conducted in accordance with NIH guidelines under an active protocol approved by the Institutional Animal Care & Use Committee (IACUC).
Before commencing the dissection, prepare collagen and laminin coated 96 well tissue culture dishes. Using a multi-channel pipetting device, fill all 96 wells with a solution containing 0.66 mg / ml rat tail collagen. Immediately remove the collagen, which can be recovered and used for up to 8 dissections. After removing the collagen, it is very important to let the wells dry under a laminar flow hood. The amount of time it takes to dry depends upon the number of wells in the dish. For example, it typically takes approximately 5-10 min. for wells in a 96 well dish to dry, but can take up to 30 - 40 min if a larger format 24 well dish is used. Failure to properly dry the wells results in poor SCG attachment. Then repeat the procedure using a solution of 2 μg / ml laminin. Incubate the laminin solution of at least 2 hr at 37 °C in a humidified CO2 incubator until you are ready to plate your neurons (step 1.14).
Commercially obtained pregnant female rats are euthanized using CO2. After spraying the cadaver with 70% ethanol, a U-shaped incision is made around the abdomen. After peeling back the skin, a second u-shaped incision is made through the abdominal muscle wall. The uterus is visible upon lifting up the abdominal muscle layer. Remove the uterus and place in a 15 cm dish. Carefully open the uterus using a blunt scissor to avoid damaging the pups within. Each pup must be released from its embryonic sac, the umbilical cord severed, and the pup wiped clean with 70% ethanol and Kimwipes.
Working at a dissection hood, sacrifice unborn E21 rat pups by shearing the head from the torso. Aim the scissors at the base of the neck, just above the shoulders. To expose the ganglia, pin down the head (neck-side up) using 23 G needles in three locations: i) spinal cord; ii) anterior skin pulled up over the nose and pinned; and iii) esophagus / trachea must be pinned away from the base of the neck.
Look for the two SCG* positioned on either side of the carotid artery bifurcation (two SCGs per embryo). The SCG is located just underneath the branch. Separate the SCG from the arteries by pulling the bifurcation apart. Place the ganglia into 12 ml of L15-media (supplemented with 0.4% D(+)-glucose) in 15 ml conical tube on ice.
*The SCG is translucent and colorless compared to the opaque, yellowish adipose tissue that surrounds the bifurcation. Try to remove as much of the residual adipose tissue and blood vessels, before collecting the ganglia.
Repeat 1.4 and 1.5 until all SCGs are harvested from the embryos.
Gently centrifuge the ganglia for 1 min at 600 rpm and aspirate the excess media.
Resuspend the ganglia in 1 ml of L15-media containing trypsin (0.25%, without EDTA) and collagenase (1 mg/ml). Incubate for 30 min at 37 °C, agitate every 10 min.
Add 10 ml of C-media (1x MEM, 0.4% D(+)-glucose, 2 mM L-glutamine, 10% FBS) to inactivate the trypsin and centrifuge for 1 min at 600 rpm.
Remove as much of the trypsin/collagenase media as possible* and wash the ganglia with 10ml of C-media. Centrifuge for 1 min at 600 rpm. Repeat this step once.
*Residual collagenase will interfere with the collagen substrate required for cell attachment on the culture plate.
Remove the wash media and re-suspend the ganglia in 1 ml of C-media and triturate using 21 G needle attached to a 5 ml syringe to dissociate large clumps of cells*. Finish triturating with 23 G needle until the visible clumps are dissociated.
*Be careful not to over-triturate, as this will damage the cells. If the trypsin and collagenase treatment was successful, a maximum of fifteen up and down cycles with the 21 G needle; and three cycles with the 23 G needle should be sufficient.
Filter the dissociated neurons through a 70 μm nylon filter [BD Biosciences cell strainer] into a 50 ml conical tube to discard any remaining clumps.
Withdraw a 10 μl aliquot of the filtered cell suspension and mix with trypan blue to determine the number of live cells. Count the number of cells that actively exclude the dye using a hemocytometer.
Remove the laminin solution from the 96 well dishes waiting in the 37 °C incubator (see STEP 1.2). Do not dry the dish, it can be used immediately for plating cells once the laminin is removed. Dispense* 5,000-6,000 total live cells (50 - 100 μl per well) into the collagen and laminin pre-coated 96 well tissue culture dish. Include 50 ng / ml nerve growth factor (NGF) in the C-media.
*It is critical to employ good sterile tissue culture technique because antibiotics are omitted from the growth media. In addition, there can be significant variability among collagen sourced from different suppliers. We have had consistent results across batches using rat tail collagen from Millipore.
At DIV (day in vitro) 1, replace the C-media used to plate the cells with 50 μl NBM (0.4% D(+)- glucose, 2 mM L-glutamine, B-27 supplement containing 50 ng / ml NGF, 5 μM aphidicolin and 20 μM 5-fluorouracil [5-fluoro-2'-deoxyuridine]. Incubate the cultures for 5 days*. At this point, trypsinizing and counting the cells remaining in several wells can determine the number of neurons surviving the anti-mitotic agent treatment. Typically, approximately 1,000 neurons remain per well in a 96 well plate.
*Problems with fungal or bacterial contamination are usually evident within the first 24 hrs of culture. Examine each well carefully for microbial growth prior to replacing plating media. If only a limited number of wells are affected, they can be treated with a dilute bleach solution, rinsed with PBS and dried in order to proceed with the experiment. We recommend repeating any experiment where even a limited number of wells had microbial contamination. If extensive microbial contamination is observed in the 96 well dish, the experiment must be terminated.
2. Infection of SCG Cultures with HSV-1 Us11-EGFP and Establishment of Latency
For useful background on virological techniques, including basic virus propagation, determining virus titer, and multiplicity of infection (MOI), the reader is referred to reference5. For a discussion of herpesvirus biology, the reader is referred to reference6. Finally, the reader is referred to references7-10 for prior examples, other protocols, and additional background regarding alpha- herpesvirus lytic infection of SCG neurons and for comparison with our protocol's adaptations to study latency and reactivation.
At DIV 6, add acyclovir (ACV), final concentration 100 μM to the existing media in each well*. For example, if each well contains 50 μl, add 25 μl of a 300 μM ACV stock. Typically, ACV is added the night before infection, but can also be added 6-8 hr prior to infection.
*It is exceedingly important to minimize unnecessary physical manipulation of the neuron at all times. Simply removing and replacing media or infecting with virus stocks must be done very gently and slowly. Otherwise, the resulting mechanical stress can adversely impact upon neuron viability.
At DIV 7, infect the SCG cultures with HSV-1 Us11-EGFP (described in ref11) at a multiplicity of infection (MOI) between 1 -2 (see important comment below on determining optimal MOI)*. Add the diluted virus directly to the existing media in the well. Include a mock-infected control and a lytic infection positive control in media lacking ACV. Allow the infection to proceed for 2-3 hr at 37 °C.
*MOI is calculated using virus titer determined by performing a plaque assay in Vero cells12 and the number of plated, live cells seeded per well in step 1.14. This calculation is useful only in an operational sense, as the total cell number seeded contains neurons together with contaminating glia, and fibroblasts. As these contaminating dividing cells are killed by treatment with anti-mitotic agents, the effective MOI for the surviving neurons is actually greater. From the initial starting quantity of 5,000 - 6,000 live cells, approximately 1,000 neurons remain after treating with anti-mitotic agents (as assessed by trypsinizing and direct cell counting). The optimal MOI to use when infecting neuronal cultures can vary somewhat from one virus stock preparation to the next. With every new preparation of virus, we recommend testing a limited range of different MOI's from 1 to 2. The goal here is to identify the one that has the least effects upon neuron viability and results in the greatest number of inducible reactivation events (defined in section 3). The Us11-EGFP virus is particularly helpful in rapidly optimizing the conditions but one can also use other readouts such as plaque assay or quantitative PCR. It may help to use virus purified through a sucrose cushion to remove impurities that reduce cell viability, though this is not essential and not routinely performed.
Carefully replace the infection media with fresh NBM containing 50 ng / ml NGF and 100 μM ACV*.
*It is critically important to be EXTREMELY gentle when changing the infection media. Aim the tip of the pipette at the wall of the well rather than at the bottom of the well, and allow the media to gently slide down onto the cells. Even rapid ejection of media from the pipette can generate sufficient force to detach the axons from the substrate and cause the cells to slough off typically as a sheet of cells. If only a limited number of neurons detach (20-30%), the consequences are minimal provided that the cells appear healthy. The detached neurons are likely to reattach over time; however, the axons will no longer be nicely extended and new axons will regrow. While not optimal, the experiment can proceed if only a limited number of wells are affected. If there is extensive detachment of neurons (70-80%), we do not recommend including the affected well(s) in the experiment. If the majority of wells contain 70-80% detached neurons, we recommend terminating the experiment. While the neurons may still reattach, they typically will form large clumps. This complicates the proper assessment of individual reactivated neurons. We recommend repeating any experiment where wells with detached neurons were observed to ensure that reactivation rates were not influenced by differential adherence of neurons to the wells.
In addition, it is critical to always handle the cultures as gently as possibly. Mechanical stress from unnecessary, sudden movements (including repeated opening and closing of an incubator chamber door) or forceful media application could compromise the cultures ability to support HSV-1 latency, possibly resulting in unacceptably high rates of spontaneous reactivation in a given experiment.
Maintain the cultures for 6 days during which time the virus will establish latency. During this period, use a fluorescent microscope to monitor neuron health and Us11-EGFP expression as an indicator of lytic replication. Us11-EGFP should be detected ONLY in the control wells lacking ACV treatment, indicating successful primary infection and productive lytic replication. Neurons may show cytopathic effects even in the absence of productive viral growth. The infected cultures should be closely monitored and compared to mock-infected neurons.
3. Reactivation and Assessment
At DIV 14, carefully replace the ACV-containing media with fresh media lacking ACV. If appropriate, include pharmacological (or biological) variables at this time. Be sure to follow the precautions stated in 2.3.
Over a 5 to 6 day period, monitor the live cultures using a fluorescent microscope to detect neurons undergoing reactivation. Alternatively collect samples for other types of analyses(protein, nucleic acid, plaque assay etc).
Calculate the reactivation frequency by counting the number of wells containing EGFP-positive neurons and express this as a proportion of the total number of sample wells. Note that EGFP expression in an individual well does not distinguish between a primary reactivation event and subsequent infection due to viral spread. Since infectious spread is contained to a single culture well, one or more EGFP-positive neurons in a well is operationally defined as one reactivation event under these conditions.
*Multiple reactivation events can occur in a culture, as described in1. To distinguish a primary reactivation event from viral spread resulting from reactivation, the encapsidation inhibitor WAY-150138 can be added. By blocking encapsidation, infectious virus is not produced and spread resulting from reactivation is not possible. Thus, the EGFP signal detected in the presence of WAY-150138 reflects the number of independent reactivation events within a singe culture well1,13-15. Note that WAY-150138 is only effective against the HSV-1 Patton strain and not commercially available. As an alternative to WAY-150138, consider using any of the following: i) a mutant virus impaired in its ability to spread to neighboring cells can be utilized; ii) a reporter virus where EGFP is expressed from an IE promoter in the continuous presence of ACV; iii) treatment with PAA or ACV to prevent late gene expression (and infectious virus production) followed by immunofluorescence using antibodies directed against an IE gene product.
4. Alternative Methods to Assess Reactivation using the Assay
Results from an individual, single experiment should be obtained from a single preparation of SCG. Replicate experiments can be performed with independent SCG preparations as long as each replicate was conducted using a single SCG batch.
Assessment of HSV-1 lytic transcripts. Collect RNA at desired times after ACV removal in the presence or absence of different experimental inducers of reactivation. When using 96 well culture plates, we recommend pooling at least 20 wells together for each sample (roughly 105 cells per RNA sample). Alternatively, SCGs can be plated in larger wells (e.g. for a 24 well plate, use 4-5 x104 cells/well). While it is certainly possible to detect RNA from less than 20 wells, the small volumes involved result in unwarranted sample-to-sample variability.
Detection of HSV-1 lytic proteins. Collect lysates at a desired time after induction of reactivation. Add 7.5 μl of lysis buffer to each of 10 wells in a 96 well plate and analyze by SDS-PAGE and immunoblotting. Alternatively, viral proteins can be detected by indirect immunofluorescence microscopy. The initial plating must be done on a mountable substrate for imaging such as a sterile glass coverslip that has been pre-coated with poly-D-lysine (0.2 mg/ml, Sigma) prior to the application of collagen and laminin (see 1.10).
Detection of infectious particles. Collect culture supernatant upon reactivation. Perform a plaque assay on monolayers of Vero cells using serial dilutions of the supernatant. Additionally, plaque assays can be performed using lysates of freeze-thawed cultures to include cell-associated particles to determine titer.
5. Representative Results
Figure 2A illustrates an example where 1 μM trichostatin A (TSA), a known inducer of reactivation16-18, is applied to SCG cultures latently infected with the wild-type HSV1 EGFP-Us11 reporter strain. With TSA, reactivation reaches 50% of the maximum within 2 days, and by 5 days the reactivation plateaus at around 60% of the wells. Base line ('spontaneous') reactivation is approximately 10% in this experiment and typically ranges from 10-20% using this in vitro system. Maximum reactivation levels vary depending on which reactivation inducer is utilized. Many reactivation inducers can be applied for the duration of the experiment as they do not interfere with productive viral replication, but this needs to be determined empirically. Other inducers, including any reagent that affects the viability of the neurons or hinders completion of the viral productive life cycle, may require a transient pulse application to provoke reactivation.
While the accumulation of EGFP-Us11 in each well of the experiment depicted in Figure 2A is indicative of reactivation from latency, it does not distinguish whether the Us11-EGFP signal in individual neurons is derived from an independent reactivation event, or the spread of a reactivated virus through the culture. To evaluate the number of neurons undergoing independent reactivation events in each well, cultures were pre-treated with WAY-150138, a compound that specifically blocks viral spread by preventing encapsidation of the viral DNA genome13-15. Infected sympathetic neuron cultures were treated with WAY-150138 and reactivation induced with TSA. Significant numbers of EGFP-positive neurons were only detected in TSA-treated wells, compared to DMSO-treated control cultures, demonstrating that a number of independent reactivation events occur per individual culture (Figure 2B).
In our assay, the Us11-EGFP reporter is expressed from the endogenous Us11 promoter11. Since Us11 is a "true-late" or γ2 viral lytic gene, viral DNA replication is required for robust expression. This is illustrated in Figure 3, as EGFP-Us11 accumulation is impaired by the viral DNA polymerase inhibitor phosphonoacetic acid (PAA) in cultures induced to reactivate with the PI3-kinase inhibitor LY294002.
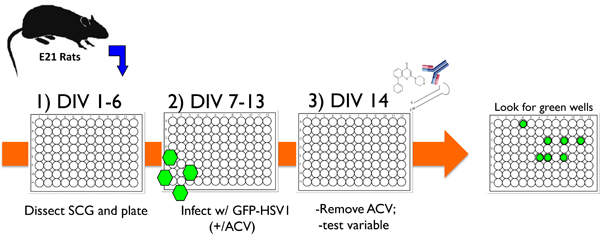 Figure 1. Schematic illustrating a typical experimental protocol using the cultured neuron in vitro system to study HSV-1 latency and reactivation. 1) After dissecting superior cervical ganglia (SCG) from E21 rats, dissociated neurons are plated in 96 well dishes and treated with anti-mitotic agents to remove non-neuronal cells. 2) After 6 days in vitro (DIV) the cultures are infected with recombinant HSV-1 that contains EGFP fused to the virus-encoded Us11 late gene (EGFP-HSV-1) in the presence acyclovir (ACV), an antiviral drug that blocks lytic replication. 3) By day 14 (DIV 14), the acyclovir is removed and EGFP-expression is not detected. Cultures can be stably maintained in this manner for 1 month or more, or a test variable can be added to evaluate its ability to provoke reactivation from latency. The variable can be in the form of a small molecule chemical inhibitor, an antibody against a soluble neurotrophin or cell surface protein, or a lentivirus expressing either a gene-specific shRNA or an ectopically expressed protein. Reactivation is then monitored by scoring EGFP-positive wells in real-time.
Figure 1. Schematic illustrating a typical experimental protocol using the cultured neuron in vitro system to study HSV-1 latency and reactivation. 1) After dissecting superior cervical ganglia (SCG) from E21 rats, dissociated neurons are plated in 96 well dishes and treated with anti-mitotic agents to remove non-neuronal cells. 2) After 6 days in vitro (DIV) the cultures are infected with recombinant HSV-1 that contains EGFP fused to the virus-encoded Us11 late gene (EGFP-HSV-1) in the presence acyclovir (ACV), an antiviral drug that blocks lytic replication. 3) By day 14 (DIV 14), the acyclovir is removed and EGFP-expression is not detected. Cultures can be stably maintained in this manner for 1 month or more, or a test variable can be added to evaluate its ability to provoke reactivation from latency. The variable can be in the form of a small molecule chemical inhibitor, an antibody against a soluble neurotrophin or cell surface protein, or a lentivirus expressing either a gene-specific shRNA or an ectopically expressed protein. Reactivation is then monitored by scoring EGFP-positive wells in real-time.
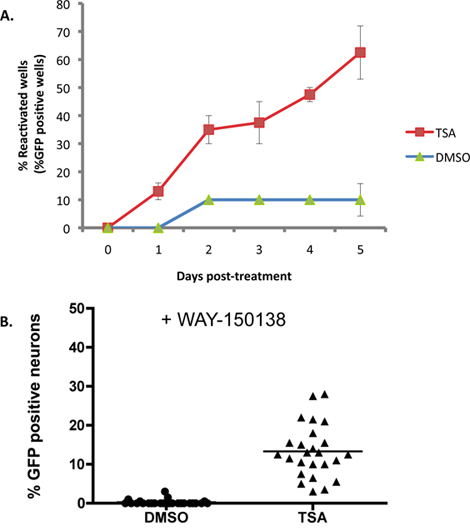 Figure 2. TSA reactivates HSV-1 in SCG cultures. SCG cultures were latently infected as described in Figure 1. At DIV 14, ACV was removed and replaced with media containing 1 μM trichostatin A (TSA). (A) The cultures were visualized and scored using fluorescence microscopy each day after drug treatment for a period of 5 days. The percentage of wells undergoing reactivation over the course of the experiment is shown compared to DMSO control-treated cultures. Percentage of reactivation was calculated by the number of EGFP-positive wells out of 20 wells of a 96 well culture plate. Error bars indicate standard error of mean. (B) Latently infected cultures were treated with TSA and an inhibitor of viral DNA encapsidation, WAY-150138 (20 μg/ml). The number of EGFP+ neurons was compared 48 hrs after treatment with the drugs to the control treated with DMSO and WAY-150138. Each data point represents the ratio of EGFP+ neurons out of 1,000 neurons in a culture well. Bar shows the average percentage of EGFP+ neurons in a well.
Figure 2. TSA reactivates HSV-1 in SCG cultures. SCG cultures were latently infected as described in Figure 1. At DIV 14, ACV was removed and replaced with media containing 1 μM trichostatin A (TSA). (A) The cultures were visualized and scored using fluorescence microscopy each day after drug treatment for a period of 5 days. The percentage of wells undergoing reactivation over the course of the experiment is shown compared to DMSO control-treated cultures. Percentage of reactivation was calculated by the number of EGFP-positive wells out of 20 wells of a 96 well culture plate. Error bars indicate standard error of mean. (B) Latently infected cultures were treated with TSA and an inhibitor of viral DNA encapsidation, WAY-150138 (20 μg/ml). The number of EGFP+ neurons was compared 48 hrs after treatment with the drugs to the control treated with DMSO and WAY-150138. Each data point represents the ratio of EGFP+ neurons out of 1,000 neurons in a culture well. Bar shows the average percentage of EGFP+ neurons in a well.
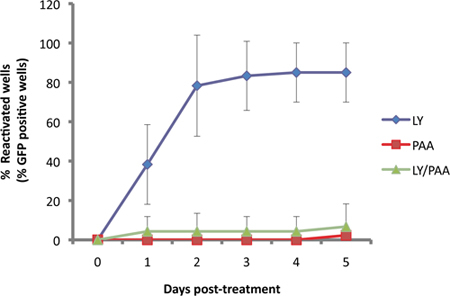 Figure 3. EGFP detecion is dependent on viral DNA replication. SCG cultures were latently infected then treated with 10 μM of LY29004, a known inducer of reactivation 1. Reactivation induced by LY (blue) was compared to those treated with viral DNA synthesis inhibitor, phophonoacetic acid, PAA (300μg/ml, red) and the two compounds together (green).
Figure 3. EGFP detecion is dependent on viral DNA replication. SCG cultures were latently infected then treated with 10 μM of LY29004, a known inducer of reactivation 1. Reactivation induced by LY (blue) was compared to those treated with viral DNA synthesis inhibitor, phophonoacetic acid, PAA (300μg/ml, red) and the two compounds together (green).
Discussion
This primary neuron culture and infection system provides a simple and effective method to explore the molecular mechanisms underlying HSV-1 latency and reactivation. The system faithfully recapitulates the accepted hallmarks of latency defined in both human infections and in live-animal models. When the virus is latent in the SCG cultures, infectious particles and viral lytic gene products cannot be detected. The only viral gene product detected in latently-infected neurons is the non-coding LAT transcript, which accumulates within nuclei. Reactivation coincides with expression of viral lytic genes and culminates in the production of infectious particles. Moreover, this primary neuron cell culture model is responsive to accepted reactivation inducers such as TSA, which effectively induces lytic viral replication.
Different chemical inducers stimulate reactivation to varying extents and with characteristic kinetics. For example, TSA reproducibly induces reactivation in 60-70% of the cultures over a 5 d observation period. Similar levels of TSA-induced reactivation have been reported using latently-infected, differentiated rat pheochromocytoma PC12 cells 17 or murine trigeminal neuron cultures 18. The kinetics of reactivation we observe and the inability of TSA to reactivate 100% of the cultures could reflect heterogeneity in the capacity of the latent genomes to be derepressed. Consistent with this proposal, the efficacy of TSA-induced reactivation reportedly decreases as a function of time in culture 18. In contrast to TSA, LY29004 induces reactivation in 80-90% of the cultures within 2-3 d. Differences in establishing latency or MOI are not likely to account for differences in inducible reactivation between TSA and LY29004, as our conditions for establishing latency reproducibly result in approximately 20 % of neurons that express the virus-encoded latency associated transcript LAT 1. However, we have not yet measured the fraction of viral genome-positive cells, raising the possibility that LY29004 induces reactivation in a greater number of neurons than TSA. While the reasons underlying the differential capacity of TSA and LY20004 to induce reactivation remain to be determined, it is clear different inducing agents can promote reactivation with characteristic efficiencies and kinetics.
A powerful aspect of the system is the ability to use wild type virus or reporter strains that behave indistinguishably from a wild-type virus. Unlike earlier cell culture systems to study HSV-1 latency, which relied on mutant HSV variants that were unable to replicate productively 16,18,19, reviewed in 20, our model system incorporates an EGFP-reporter strain that is indistinguishable from wild-type virus in terms of lytic replication efficiency in cultured cells 11. The use of an EGFP reporter virus allows for a sensitive and straightforward visual assessment of latency and reactivation. In contrast to other reporters such as β-galactosidase, which requires fixation and lysis of cells for analysis, EGFP allows the researcher to monitor the viral state throughout the entire course of an experiment, providing an easy, real-time assessment of spontaneous and induced reactivation. Other methods, like quantitative PCR, can also be utilized to look at viral gene expression patterns that precede EGFP-expression, since EGFP is expressed as a "true-late" gene, or for virus strains that lack a reporter gene. Alternatively, other reporter viruses where EGFP is fused to immediate-early (IE) or early (E) genes could be developed. An advantage of using the "true-late" EGFP-reporter described is that EGFP-expression is stringently dependent upon viral DNA synthesis, indicating that the virus lifecycle has proceeded into the final gene expression phase associated with lytic growth. Indeed, "true-late" EGFP expression to levels detectable by visual inspection correlates well with infectious virus production, a biological indicator that productive reactivation has occurred. However, it remains to be seen if each neuron that commences reactivation, as measured by productive cycle (lytic) gene expression, is capable of progressing through the complete lytic program to produce infectious virus. Perhaps viruses have ample opportunity to abort the lytic program and reestablish latency. Thus, reporter viruses expressing EGFP fused to IE or E proteins may result in less reliable reactivation readouts. From this perspective, comparing the efficiency of reactivation measured using EGFP-reporters expressed from IE, E, or late promoters, or even a combined reporter with different fluorescent reporter proteins fused to a gene product from each kinetic class (IE vs. E vs. late) would be useful. In addition, our neuronal cell culture system can also be used with different HSV-1 strains, regardless of their neuroinvasive potential or ability to regulate the host immune response 21-24. Finally, the in vitro culture system described here can be applied to other neuronal cell types and ex vivo peripheral ganglia cultures obtained from higher-order species, including humans 25-27.
By using a pure population of neurons, detailed studies of the interactions between HSV1 and its neuronal host at the molecular level are now possible. Significantly, this model system avoids the many complex issues associated with studying latency and reactivation in immunocompetent animals, where additional levels of control are layered on top of fundamental virus-neuron interactions. Moreover, work in this system clearly demonstrates that there is indeed a stable relationship between virus and host neuron that can be established and maintained without assistance from an acquired immune response. While innate and adaptive immunity clearly play important roles in the biology of latent HSV infections, the virus has the intrinsic capacity i) to establish a non-productive infection resembling latency in neurons; and ii) to switch to a lytic, productive replication program in response to environmental and neuronal cues applied to neurons alone. Importantly, the molecular basis for this unique relationship between HSV and the neuron can now be dissected using cell biological, pharmacological, gene silencing, and gene delivery tools 1. Studies of this sort that are focused on the neuron and virus in isolation are not possible in animal models where several cell types occupy the ganglial compartment. We hope that by using this model system, a comprehensive understanding of the intricate molecular circuitry regulating HSV-1 latency in neurons can finally be achieved. Whenever possible, principles gleaned from this cultured neuron system can then be tested in vivo using established animal models, illustrating the complementary nature of these two approaches to the overall study of HSV-1 latency.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank the reviewers for their thoughtful suggestions that helped to improve this manuscript. This work was supported by grants to MVC (NS21072, HD23315), ACW (GM61139, S10RR017970) and IM (AI073898, GM056927) from the NIH. MK was supported in part by an NIH training grant (5T32 AI007180).
References
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host & Microbe. 2010;8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MI. Primary cultures of sympathetic ganglia. In: Fedoroff S, Richardson A, editors. Protocols for Neural Cell Culture. 3rd Ed. Totowa, N.J: Humana Press, Inc; 2001. pp. 71–94. [Google Scholar]
- Letourneau PC. Preparation of substrata for in vitro culture of neurons. In: Fedoroff S, Richardson A, editors. Protocols for Neural Cell Culture. 3rd Ed. Totowa, N.J: Humana Press, Inc; 2001. pp. 245–254. [Google Scholar]
- Price JP, Brewer GJ. Serum-free media for neural cell cultures. In: Fedoroff S, Richardson A, editors. Protocols for Neural Cell Culture. 3rd Ed. Totowa, N.J: Humana Press, Inc; 2001. pp. 255–264. [Google Scholar]
- Flint SJ, Enquist LW, Racaniello VR, Skalka AM. Principles of virology. 3rd Ed. ASM Press; 2008. [Google Scholar]
- Roizman B, Pellett PE. The family Herpesviridae: A brief introduction. In: Knipe DM, Howley PM, editors. Fields Virology. Vol. 2. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 2381–2397. [Google Scholar]
- Price RW, Rubenstein R, Khan A. Herpes simplex virus infection of isolated autonomic neurons in culture: viral replication and spread in a neuronal network. Arch. Virol. 1982;71:127–140. doi: 10.1007/BF01314882. [DOI] [PubMed] [Google Scholar]
- Tomishima MJ, Enquist LW. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 2001;154:741–752. doi: 10.1083/jcb.200011146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Flood EA, Enquist LW. Culturing primary and transformed neuronal cells for studying pseudorabies virus infection. Methods Mol. Biol. 2005;292:299–316. doi: 10.1385/1-59259-848-x:299. [DOI] [PubMed] [Google Scholar]
- Wang F, Tang W, McGraw HM, Bennett J, Enquist LW, Friedman HM. Herpes simplex virus type 1 glycoprotein E is required for axonal localization of capsid, tegument, and membrane glycoproteins. J. Virol. 2005;79:13362–13372. doi: 10.1128/JVI.79.21.13362-13372.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benboudjema L, Mulvey M, Gao Y, Pimplikar SW, Mohr I. Association of the herpes simplex virus type 1 us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003;77:9192–9203. doi: 10.1128/JVI.77.17.9192-9203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho J, Morton ER, Yedowitz JC. Herpes simplex virus: propagation, quantification and storage. Curr. Protoc. Microbiol. 2005;Chapter 14:Unit 14E.1. doi: 10.1002/9780471729259.mc14e01s00. [DOI] [PubMed] [Google Scholar]
- Van Zeijl M, Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld BL, O'Hara BL, Bloom JD, Johann SV. Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J. Virol. 2000;74:9054–9061. doi: 10.1128/jvi.74.19.9054-9061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb WW, Brown JC. Inhibition of herpes simplex virus replication by WAY-150138: assembly of capsids depleted of the portal and terminase proteins involved in DNA encapsidation. J. Virol. 2002;76:10084–10088. doi: 10.1128/JVI.76.19.10084-10088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesola JM, Zhu J, Knipe DM, Coen DM. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J. Virol. 2005;79:4516–14525. doi: 10.1128/JVI.79.23.14516-14525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JL, Scarpini CG, Connor V, Lachmann RH, Tolkovsky AM, Efstathiou S. Herpes simplex virus type 1 promoter activity during latency establishment, maintenance and reactivation in primary dorsal root neurons in vitro. J. Virol. 2001;75:3885–3895. doi: 10.1128/JVI.75.8.3885-3895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher RJ, Jacob RJ, Steiner MR, Allen WR, Hill JM, Miller CS. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript- independent manner in neuronal cells. J. Neurovirol. 2005;11:306–317. doi: 10.1080/13550280590952817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry-Allison T, Smith CA, DeLuca NA. Relaxed repression of herpes simplex virus type 1 genomes in murine trigenminal neurons. J. Virol. 2007;71:12394–12405. doi: 10.1128/JVI.01068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Preston CM. Establishment of latency in vitro by the herpes virus type 1 mutant in1918. J. Gen. Virol. 1991;72:907–913. doi: 10.1099/0022-1317-72-4-907. [DOI] [PubMed] [Google Scholar]
- Wagner EK, Bloom DC. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 1997;10:419–443. doi: 10.1128/cmr.10.3.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow LI, Laycock KA, Jun PY, Rader KA, Brady RH, Miller JK, Pepose JS, Leib DA. A structural and functional comparison of the latency-associated transcript promoters of herpes simplex virus type 1 strains KOS and McKrae. J. Gen Virol. 1994;75:2475–2480. doi: 10.1099/0022-1317-75-9-2475. [DOI] [PubMed] [Google Scholar]
- Stroop WG, Banks MC. Herpes simplex virus type 1 strain KOS-63 does not cause acute or recurrent ocular disease and does not reactivate ganglionic latency in vivo. Acta Neuropathol. 1994;87:14–22. doi: 10.1007/BF00386250. [DOI] [PubMed] [Google Scholar]
- Sawtell NM, Poon DK, Tansky CS, Thompson RL. The latent herpes simplex virus type 1 genome copy number in individual neurons is virus strain specific and correlates with reactivation. J. Virol. 1998;72:5343–5350. doi: 10.1128/jvi.72.7.5343-5350.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Cook ML, Devi-Rao G, Wagner EK, Stevens JG. Functional and molecular analysis of the avirulent wild-type herpes simplex virus type 1 strain KOS. J. Virol. 1986;58:203–211. doi: 10.1128/jvi.58.1.203-211.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Smith RL, Freed CR, Johnson EM., Jr Nerve growth factor-dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J. Neurosci. 1990;10:1268–1275. doi: 10.1523/JNEUROSCI.10-04-01268.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehm PC, Camarena V, Gardner JB, Wilson AC, Mohr I, Chao MV. Cultured vestibular ganglion neurons demonstrate latent herpes simplex type I reactivation. Laryngoscope. 2011;121:2268–2275. doi: 10.1002/lary.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn MA, Nayak S, Camarena V, Gardner J, Wilson A, Mohr I, Chao MV, Roehm PC. A cell culture model of facial palsy resulting from reactivation of latent herpes simplex virus type 1. Otology & Neurotology. 2012. Forthcoming. [DOI] [PMC free article] [PubMed]