

Abstract
Motor proteins move cargos along microtubules, and transport them to specific sub-cellular locations. Because altered transport is suggested to underlie a variety of neurodegenerative diseases, understanding microtubule based motor transport and its regulation will likely ultimately lead to improved therapeutic approaches. Kinesin-1 is a eukaryotic motor protein which moves in an anterograde (plus-end) direction along microtubules (MTs), powered by ATP hydrolysis. Here we report a detailed purification protocol to isolate active full length kinesin from Drosophila embryos, thus allowing the combination of Drosophila genetics with single-molecule biophysical studies. Starting with approximately 50 laying cups, with approximately 1000 females per cup, we carried out overnight collections. This provided approximately 10 ml of packed embryos. The embryos were bleach dechorionated (yielding approximately 9 grams of embryos), and then homogenized. After disruption, the homogenate was clarified using a low speed spin followed by a high speed centrifugation. The clarified supernatant was treated with GTP and taxol to polymerize MTs. Kinesin was immobilized on polymerized MTs by adding the ATP analog, 5'-adenylyl imidodiphosphate at room temperature. After kinesin binding, microtubules were sedimented via high speed centrifugation through a sucrose cushion. The microtubule pellet was then re-suspended, and this process was repeated. Finally, ATP was added to release the kinesin from the MTs. High speed centrifugation then spun down the MTs, leaving the kinesin in the supernatant. This kinesin was subjected to a centrifugal filtration using a 100 KD cut off filter for further purification, aliquoted, snap frozen in liquid nitrogen, and stored at -80 °C. SDS gel electrophoresis and western blotting was performed using the purified sample. The motor activity of purified samples before and after the final centrifugal filtration step was evaluated using an in vitro single molecule microtubule assay. The kinesin fractions before and after the centrifugal filtration showed processivity as previously reported in literature. Further experiments are underway to evaluate the interaction between kinesin and other transport related proteins.
Keywords: Developmental Biology, Issue 62, Drosophila, Kinesin, clarification, polymerization, sedimentation, microtubule
Protocol
Strains of flies can be purchased and amplified in the lab. Depending on the amount of Drosophila culture vials received, one need to frequently 'flip' the culture vials, once the vials has reached maximum capacity. The amplification continues until approximately 50 Drosophila culture vials are filled. One then makes 'fly cups' with 100 ml tricorn beakers (Fly cup making is discussed in the next section). After 24 hours, use embryos from these cups to seed new Drosophila culture vials with affixed fly food at the bottom. At room temperature, the growth time for Drosophila embryos from overnight embryos to full grown flies are about 8.5 days. Seeded embryos hatch after 12-15 hours into the first larvae stage. The larvae grow for about 4 days molting twice into the second and third larvae stage, at 24 and 48 hours after hatching. The larvae then encapsulate into pupariums and submit to a 4 day metamorphosis until emerging from their pupal cases 1. Allow for more growth for about two to three days in the Drosophila culture vials to increase the amount of flies in each. When 400 vials are filled, there will be a sufficient amount of flies for fifty egg laying cups (About 1,000 female flies per cup). Flies need time to adjust to their new environment. It usually takes about two days for them to be ready for collection, after being transferred to the cups. Replacing agar plates daily will keep the fly cups clean, which allow flies to live longer. (For further understanding of Drosophila care and amplification, please refer to D. B. Roberts' Drosophila: A Practical Approach 2).
A good source to obtain large amounts of Drosophila embryos are population cages 3, which are commercially available. The assembly and maintenance of population cages can be seen on chapter 5 and 7 in the book Drosophila Melanogaster: Practical Uses in Cell and Molecular Biology4. However, population cages are expensive and are hard to maintain. Since population cages can hold an abundant amount of flies, the use of carbon dioxide is necessary for transferring and feeding flies. As stated before, carbon dioxide decreases the health of the flies, causing them not to lay as well. On a small scale, 100 ml tricorn beakers can be used. With fifty 100 ml beakers, 9 grams of dechorionated embryos may be attained. In order remove the dead flies, unused yeast paste and other impurities we have created a double layered sieve catcher with different mesh sizes. One sieve traps unwanted materials like dead flies while allowing embryos to pass through (350 micron pore size). The second part contains a mesh (120 microns pore size), which is meant to catch Drosophila embryos and other impurities will pass through the mesh.
1. Fly Cup Preparation and Embryo Collection
Flip the amplified flies from multiple vials into an empty plastic vial, until the amount of flies have reached one inch of the vial's height. Then, transfer those flies into a 100 ml egg collection cup (tricon beaker with nylon mesh windows). Keep tapping the cup on a hard surface to prevent the escape of flies when transferring flies. Cover cup with agar plate containing yeast paste and allow 24-48 hrs of stabilization time.
Collect overnight agar plates from fifty fly cups and wash the contents of the plates to the sieve catcher using a clean paint brush and running water. Flesh the embryos in the sieve with plenty of water until all the yeast paste is washed away.
Dechorionate the cleaned embryos by immersing them in 50% bleach for 3 min.
Rinse with distilled water extensively until embryos loose the bleach odor. Then dry the mesh with embryos by placing it on a c fold towel multiple times. Transfer the embryos to a clean vial and record the weight.
2. Embryo Homogenization and Clarification
Place dechorionated embryos in Dounce homogenizer with 1.5 x volume of ice-cold extraction buffer.
Do 5 strokes with the loose pestle. Aliquot the homogenate into clean centrifuge tubes. Centrifuge at 15,000 g for 40 min. at 4 °C.
Carefully collect the clear supernatant liquid without the upper white lipid layer or the lower pellet.
Transfer supernatant to clean centrifuge tubes and centrifuge at 50,000 g for 30 min. at 4 °C
Resulting high-speed supernatant collected as explained above can be used immediately or can be snap frozen and stored at -80 °C for months.
3. Microtubule Polymerization and Kinesin Binding
Thaw the frozen high-speed supernatant to room temperature. To polymerize microtubules, add GTP (0.3 mM) and taxol (20 μM) and agitate gently at room temperature for 20 min in a rotating shaker.
To bind kinesin, add 2.5 mM nonhydrolyzable ATP analog 5'-adenylyl imidodiphosphate (AMPPNP), followed by a 10 min agitation at room temperature in a rotating shaker.
4. Differential Sedimentation of Microtubules and Kinesin
Sediment through an equal volume sucrose cushion (20% sucrose and 10 μM taxol in extraction buffer) by centrifugation at 23,000 g (sw41ti rotor, TLS-55) for 30 min. at 4 °C.
Wash pellet by resuspension in 10% of original homogenate volume in extraction buffer containing 10 μM taxol and 75 mM NaCl.
Repeat the sedimentation with another equal volume sucrose cushion as in step 4.1.
Re-suspend pellet from salt wash in 5% extraction buffer with 20 μM taxol, 75 mM NaCl, 10 mM MgSO4, and 10 mM ATP.
Sediment at 120,000 g (sw41ti rotor: 31,000 rpm, TLS-55: 42,000 rpm) for 20 mins at 4 °C. Collect supernatant (kinesin fraction).
Perform a centrifugal filtration at 14,000 g for 15 min at 4 °C. Recover concentrated sample and repeat filtration with a new filter as above for 30 minutes and collect the concentrated sample.
Perform a protein assay to determine the concentration. Briefly, varying concentrations of a known protein (bovine serum albumin; BSA) was mixed with a known volume of Bradford reagent and measured the optical density at wavelength 595nm. Then an optical density versus concentration standard curve was established to calculate the unknown concentration of purified kinesin fraction using the optical density of this sample measured under similar conditions as that of the standard BSA samples. Gel electrophoresis as well as western blotting was also performed using a known amount of the purified kinesin.
Aliquot kinesin fraction into small volumes of desired concentration and snap freeze in liquid nitrogen to store at -80 °C.
5. Representative Results
Full-length functional Kinesin was purified from Drosophila embryos. Figure 1 depicts the silver stained gel showing the different fractions during purification. Lane 1 is a sample of the high speed supernatant after clarification (step 2.5), lane 2 is a sample of the pellet after clarification, lane 3 is a sample of the supernatant after the first sedimentation with sucrose cushion (step 4.1), lane 4 is a sample of the pellet after the first sedimentation, lane 5 is a sample of the supernatant after the second sedimentation (step 4.3), lane 6 is a sample of the pellet after the second sedimentation, lane 7 is a sample of the pellet of the final sedimentation (step 4.5), lane 8 is the purified kinesin sample, lane 9 is the filtered kinesin sample, and lane 10 is the marker. The concentrated band in lane 8 and 9 at around 115 kD is kinesin heavy-chain 1, which is consistent with Figure 1, lane 8 of the Saxton paper published in 19885. Figure 2 represents the c western blot of the purified kinesin fraction detected using the kinesin heavy chain antibody. The antibody AKINO1-A does not significantly cross-react with other kinesin family members 6. Note that although this protocol results in a good source of functional kinesin, while the fraction is enriched for kinesin-1, there are likely contaminants, including potentially other kinesins and other motor proteins. We removed a variety of smaller contaminants by filtration. As far as removing other motors which cannot be done by size alone, the purification is similar to what was done by others to study kinesin 7, and we believe that the majority of the active motor is Kinesin-1, but more sophisticated purification would allow removal of potential motor contaminants, such as was done by Cole et al8 and Saxton et al.9
The processivity of kinesin was evaluated by in vitro single molecule microtubule binding assay, as described in more detail in the book by Scholey entitled "Motility Assay for Motor Proteins"10. Briefly, polystyrene beads with a single active motor were brought into contact with the microtubule, in the presence of saturating (1 mm) ATP. The motor attached to the microtubule, and started walking away from the center of the laser trap. At a pre-defined displacement of the bead from the trap center (100 nm) the laser beam power was automatically turned off, allowing the motor to walk along the MT under no load. The movie shows a 500 nm diameter bead with single kinesin walking along MT. The length of the video screen corresponds to 20 μm. Figure 3 represents the measured distribution of run-lengths for single full-length Drosophila kinesin molecules, purified according to the protocol presented here. The exponential fit to the distribution provides the average runlength of single kinesin 1.55 ± 0.1 μm and 1.28 ± 0.12 μm for the filtered and unfiltered sample respectively.
 Figure 1. Silver stained gel of the purified fractions: Samples from each fraction were run in a 10% gel. 10 μg of protein was loaded in each lane. Lane 1 is a sample of the high speed supernatant after clarification (step 2.5), lane 2 is a sample of the pellet after clarification, lane 3 is a sample of the supernatant after the first sedimentation with sucrose cushion (step 4.1), lane 4 is a sample of the pellet after the first sedimentation, lane 5 is a sample of the supernatant after the second sedimentation (step 4.3), lane 6 is a sample of the pellet after the second sedimentation, lane 7 is a sample of the pellet of the final sedimentation (step 4.5), lane 8 is the purified kinesin sample, lane 9 is the filtered kinesin using a 100 kD cut-off Amicon ultra 0.5 ml centrifugal filter (Millipore, USA). The blue arrows indicate the proteins whose amounts were decreased and the red arrow indicate the concentration of kinesin due to the centrifugal filtration step used in this protocol.
Figure 1. Silver stained gel of the purified fractions: Samples from each fraction were run in a 10% gel. 10 μg of protein was loaded in each lane. Lane 1 is a sample of the high speed supernatant after clarification (step 2.5), lane 2 is a sample of the pellet after clarification, lane 3 is a sample of the supernatant after the first sedimentation with sucrose cushion (step 4.1), lane 4 is a sample of the pellet after the first sedimentation, lane 5 is a sample of the supernatant after the second sedimentation (step 4.3), lane 6 is a sample of the pellet after the second sedimentation, lane 7 is a sample of the pellet of the final sedimentation (step 4.5), lane 8 is the purified kinesin sample, lane 9 is the filtered kinesin using a 100 kD cut-off Amicon ultra 0.5 ml centrifugal filter (Millipore, USA). The blue arrows indicate the proteins whose amounts were decreased and the red arrow indicate the concentration of kinesin due to the centrifugal filtration step used in this protocol.
![]() Figure 2. Purified KHC fraction blotted against anti-kinesin antibody: Purified kinesin sample was subjected to gel electrophoresis and blotted (in nitrocellulose membrane) against primary antibody AKINO1-A (1:1,000 in TBST ) for 1 hour at room temperature followed by incubation in Donkey –anti-rabbit antibody (1:10,000 in TBST) for an hour at room temperature. An ECL (chemiluminescent) kit was used to detect the kinesin signal shown above.
Figure 2. Purified KHC fraction blotted against anti-kinesin antibody: Purified kinesin sample was subjected to gel electrophoresis and blotted (in nitrocellulose membrane) against primary antibody AKINO1-A (1:1,000 in TBST ) for 1 hour at room temperature followed by incubation in Donkey –anti-rabbit antibody (1:10,000 in TBST) for an hour at room temperature. An ECL (chemiluminescent) kit was used to detect the kinesin signal shown above.
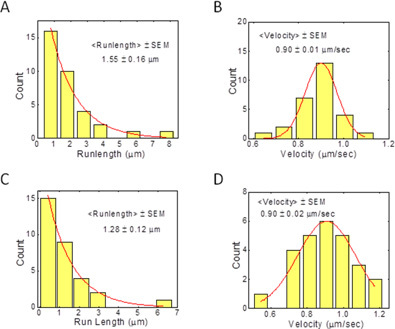 Figure 3. Runlength and Velocity of single Drosophila full length kinesin: (A) and (B) Runlength and Velocity of filtered kinesin sample. (C) and (D) Runlength and velocity of unfiltered kinesin sample.
Figure 3. Runlength and Velocity of single Drosophila full length kinesin: (A) and (B) Runlength and Velocity of filtered kinesin sample. (C) and (D) Runlength and velocity of unfiltered kinesin sample.
Figure 4. Video of Kinesin motility. Click here to view video.
Discussion
Bovine brain is the most widely used starting material 11 to purify full-length kinesin, though murine brain has been used as well 12. A large disadvantage of using bovine brain as a kinesin source is the availability of fresh starting material: slaughterhouses are typically inaccessible, and the brain must be extremely fresh in order to obtain active kinesin. Further, only the brains of young cows are effective. Finally, genetic manipulation of cows is currently not a viable option, so only 'wild-type' protein can be studied.
Mice are more accessible, but harvesting murine brain is somewhat difficult and time consuming as purification can require more than 50 brains. Further, maintaining a mouse colony can be quite expensive.
In contrast to Bovine or Murine sources, Drosophila has a number of advantages. First, the flies can easily be grown in the laboratory with minimal capital outlay, and are thus readily accessible. Drosophila lay prolific embryos, which are easily harvested as described here. Because the Drosophila genome can be manipulated, and indeed many mutant flies are easily obtained, both wild-type and mutant proteins can be purified, and due to the combination of genetic manipulations and a short generation span, more mutants can be isolated. However, it should be noted that, because complete loss of kinesin function is lethal, and protein in the embryos is provided predominantly maternally, it is not possible to purify pure kinesin mutants that are dramatically impaired. Nonetheless, it is possible to purify protein from heterozygous embryos (kin-mut/+), and characterize function of the mixed population, and then potentially relate such changes in function to phenotypes in the animal.
Alteration or impairment of transport appears to underlie many neurodegenerative diseases; however, the mechanistic link between disease and alteration of single-molecule function (due either to mutations or an altered signaling environment) is unclear. The protein purification assay developed here should be an important tool in pursuing this understanding, because single-molecule assays can be used to determine the motors' function. By combining such studies with in vivo characterization of phenotypes, and aided by modeling, this allows direct determination of the relationship between altering specific single molecular properties and disease development/progression (as an example, see the link between altered single-molecule dynein function and impaired neuronal transport, in 13).
Disclosures
We have nothing to disclose.
Acknowledgments
Special thanks to Kris Ngai and Jason Del Rio for their support and assistance in this project. This work was supported by RO1 grant GM070676 to SPG.
References
- Ashburner M, Thompson JN. The laboratory culture of Drosophila 2A. In: Ashburner M, Wright TRF, editors. The genetics and biology of Drosophila. Academic Press; 1978. pp. 1–81. [Google Scholar]
- Roberts DB. Drosophila: A Practical Approach. Oxford University Press; 1998. [Google Scholar]
- Kunert N, Brehm A. Mass production of Drosophila Embryosand Chromatographic Purification of Native Protein Complexes. Methods in Molecular Biology. 2008;420:359. doi: 10.1007/978-1-59745-583-1_23. [DOI] [PubMed] [Google Scholar]
- Goldstein LSB, Fyrberg EA, Wilson L, Matsudaira PT, Eds Drosophila melanogaster: Practical Uses in Cell and Molecular Biology. Methods in Cell Biology. 1994;44 [Google Scholar]
- Saxton WM. Drosophila kinesin: Characterization of microtubule motility and ATPase. Proceedings of the National Academy of Science. 1988;85:1109. doi: 10.1073/pnas.85.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubeita G. Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell. 2008;135:1098–1098. doi: 10.1016/j.cell.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Block SM. Force and Velocity Measured for Single Kinesin Molecules. Cell. 1994;77:773. doi: 10.1016/0092-8674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Cole DG, Saxton WM, Sheehan KB, Scholey JM. A "Slow" Homotetrameric Kinesin-related Motor Protein Purified from Drosophila embryos. J. Biol. Chem. 1994;269:22917–22917. [PMC free article] [PubMed] [Google Scholar]
- Saxton WM. Isolation and Analysis of Microtuble Motor Proteins. Drosophila Melanogaster. Bloomington: Academic. 1994;44:279. doi: 10.1016/s0091-679x(08)60919-x. [DOI] [PubMed] [Google Scholar]
- Scholey J. Motility Assay for Motor Proteins. Methods in Cell Biology. 1993;39:138. [Google Scholar]
- Wagner MC, Pfister K, Brody S, Bloom G. Purification of kinesin from bovine brain and assay of microtubule-stimulated ATPase activity. Methods in Enzymology. 1991;196:157. doi: 10.1016/0076-6879(91)96016-k. [DOI] [PubMed] [Google Scholar]
- Aizawa H. Kinesin Family in Murine Nerwous System. The Journal of Cell Biology. 1992;119:1287–1287. doi: 10.1083/jcb.119.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori-McKenney KM, Xu J, Gross SP, Vallee RB. A cytoplasmic dynein tail mutation impairs motor processivity. Nature Cell Biology. 2010;12:1228–1228. doi: 10.1038/ncb2127. [DOI] [PMC free article] [PubMed] [Google Scholar]