

Abstract
Calmodulin (CaM), by mediating the stimulation of the activity of two adenylyl cyclases (ACs), plays a key role in integrating the cAMP and Ca2+ signaling systems. These ACs, AC1 and AC8, by decoding discrete Ca2+ signals can contribute to fine-tuning intracellular cAMP dynamics, particularly in neurons where they predominate. CaM comprises an α-helical linker separating two globular regions at the N-terminus and the C-terminus that each bind two Ca2+ ions. These two lobes have differing affinities for Ca2+, and they can interact with target proteins independently. This study explores previous indications that the two lobes of CaM can regulate AC1 and AC8 differently and thereby yield different responses to cellular transitions in [Ca2+]i. We first compared by glutathione S-transferase pull-down assays and offline nanoelectrospray ionization mass spectrometry the interaction of CaM and Ca2+-binding deficient mutants of CaM with the internal CaM binding domain (CaMBD) of AC1 and the two terminal CaMBDs of AC8. We then examined the influence of these three CaMBDs on Ca2+ binding by native and mutated CaM in stopped-flow experiments to quantify their interactions. The three CaMBDs show quite distinct interactions with the two lobes of CaM. These findings establish the critical kinetic differences between the mechanisms of Ca2+-CaM activation of AC1 and AC8, which may underpin their different physiological roles.
Adenylyl cyclase (AC) synthesizes the ubiquitous second messenger, cAMP, which is fundamental for numerous physiological processes in the cell. Nine membrane-bound AC isoforms have been cloned and characterized in mammals. They are all activated by Gsα; however, each isoform is also differentially regulated by other signals, such as Ca2+ and protein kinases.1 Type 1 and 8 ACs (AC1 and AC8, respectively) are the two isoforms that are directly stimulated by physiological concentrations of Ca2+ in a mechanism that requires the mediation of calmodulin (CaM). AC1 and AC8 thus play central roles in imparting sophisticated outcomes to the activation of Ca2+ signaling. Despite their similarities in terms of Ca2+-CaM stimulation, the kinetics of these two enzymes in response to a modest Ca2+ entry differed significantly.2 The cAMP increase in cells expressing AC1 was slower than in cells expressing AC8, although AC1 is more sensitive to Ca2+ than AC8 (EC50 values of AC1 and AC8 in vitro of 150 and 560 nM, respectively). Moreover, AC8 gave rise to cAMP oscillations in response to CCh-evoked Ca2+ oscillation,3 whereas AC1 activity yielded only a steady cAMP increase.2 These kinetic differences are thought to be at least partially due to the distinct CaM regulation of these two enzymes.
CaM is a small (16.7 kDa) ubiquitous protein, which is widely distributed and involved in the regulation of more than 300 Ca2+-dependent processes in eukaryotic cells,4 including shaping levels of second messengers and components of the actin cytoskeleton. CaM comprises globular N- and C-terminal lobes (N-lobe and C-lobe, respectively) separated by a long flexible α-helical linker. Each lobe contains a pair of EF-hands: EF1 and EF2 in the N-lobe and EF3 and EF4 in the C-lobe.5 The introduction of mutations into each EF-hand to impair Ca2+ binding has provided valuable research tools for investigating the interaction of CaM with its targets. Mutation of the first aspartate residue at each EF-hand (D20, D56, D93, and D129) to alanine produces a mutant CaM, CaM1234, which is at least 100-fold less sensitive to Ca2+ than wild-type CaM (CaMWT).6,7 Alanine substitutions in EF1 and EF2 or EF3 and EF4 alone have generated CaM12 and CaM34 to allow separate investigation of the roles of the C- and N-lobe CaM, respectively.8,9
The CaM binding domains (CaMBDs) of AC1 and AC8 are located at relatively different places in the molecules; AC1 has an amphipathic CaM binding domain in the C1b domain,a,10,11 whereas AC8 has an amphipathic CaMBD at the N-terminus and an IQ-like motif (IQlm) in the C2b domain.12 The N-terminal CaMBD of AC8 is not absolutely required for stimulation of catalytic activity by Ca2+ in vitro; CaM activates AC8 via the C2b domain by a disinhibitory mechanism. Removal of the C2b domain makes AC8 insensitive to Ca2+ but superactive.12,13 The N-terminal CaMBD of AC8 appears to act as a CaM tethering site, because exogenous CaM is needed to permit stimulation by Ca2+ when the N-terminus is truncated,14 and even when the critical C-terminal CaMBD is mutated, the consequences are not apparent unless the N-terminus has been either deleted or mutated so that it can no longer bind CaM.13 The activation mechanism of AC1 is distinct from that of AC8; mutagenesis studies identified a key residue within the C1b domain responsible for Ca2+ regulation of AC1,15 although the point mutation of this residue did not prevent CaM from binding to this region of AC1.2
In a previous study using Ca2+-binding mutants of CaM, we discovered lobe-specific regulation by CaM of AC1 and AC8; no CaM mutant could regulate AC1, while partially liganded CaM could regulate AC8.2 The N-lobe mutant of CaM inhibited AC8 activity by competing with endogenous CaM at the N-terminus, and the C-lobe mutant of CaM stimulated AC8 through the C2b domain. The regulation by half-liganded CaM, however, does not signify that CaMBDs of AC8 directly bind CaM with two Ca2+ ions bound, as it could be a consequence of an indirect interaction. Additionally, the lack of regulation of AC1 by half-occupied CaM may possibly be due to an interaction of CaMBD of AC1 with the wrong lobe of CaM, resulting in an unproductive complex that renders catalysis impossible.
This study addressed potential differences in CaM regulatory mechanisms by interrogating lobe-specific interaction of CaM with the CaMBDs of AC1 and AC8. This was achieved using a number of experimental approaches. First, glutathione S-transferase (GST) pull-down assays and offline nanoelectrospray ionization (ESI) mass spectrometry (MS) demonstrated their Ca2+-dependent interactions. Second, offline nano-ESI-MS revealed the relative binding affinities of the CaMBD peptides of AC1 and AC8 for CaM. Lastly, stopped-flow measurements explored the binding of CaM to CaMBDs of AC1 and AC8 and how they influenced the affinity of each lobe of CaM for Ca2+. A previous mutagenesis study of AC8 had allowed the formulation of a model in which CaM tethered at the N-terminal CaMBD might be transferred to the first half of the C-terminal CaMBD of AC8.13 Results from this study help to expand this model and to generate a contrasting model for AC1. Overall, we have revealed yet more distinctions between the mechanisms of the CaM activation of these two enzymes, which illustrates the elaborate complexity of the Ca2+ regulation of ACs.
Experimental Procedures
Materials
ECL Plus Western Blotting Analysis System, Hyperfilm, glutathione-Sepharose 4B, and the pGEX4T-1 vector were obtained from GE Healthcare (Little Chalfont, U.K.). Horseradish peroxidase-conjugated goat anti-mouse IgG and DNase were from Promega (Madison, WI). TALON resin was from BD Clontech (Basingstoke, U.K.). Monoclonal CaM antibody was from Upstate Biotechnology (Dundee, U.K.). M13 was from Cambridge Bioscience (Cambridge, U.K.). Constructs of CaM mutants were gifts from J. H. Caldwell (University of Colorado Health Sciences Center, Denver, CO). Peptides were synthesized by S. Rothemund (IZKF Leipzig, Leipzig, Germany) or JPT Peptide Technologies (Berlin, Germany). All other reagents were purchased from Sigma (Poole, U.K.) unless stated otherwise.
In Vitro Measurement of Adenylyl Cyclase Activity
Determination of AC activity in vitro was performed as described previously16 with some modifications. Crude membranes isolated from HEK293 cells transfected with either AC1 or AC8 were washed twice with buffer containing excess EGTA to remove Ca2+ and CaM prior to the assay. AC activity was measured in the presence of the following components: 12 mM phosphocreatine, 2.5 units of creatine phosphokinase, 100 μM cAMP, 1.4 mM MgCl2, 100 μM ATP, 40 μM GTP, 500 μM phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine, 10 μM forskolin, 0.5 μCi of [α-32P]ATP, 200 μM EGTA, 0.3 or 1 μM free Ca2+, and the indicated concentrations of M13 to remove residual CaM.2 Wild-type CaM (1 μM) was included where indicated. The reaction mixture (final volume, 100 μL) was incubated at 30 °C for 20 min, and the reactions were terminated with 0.5% (w/v) sodium dodecyl sulfate. The [32P]cAMP formed was quantified using a sequential chromatography technique.17 Data points are mean activities ± the standard deviation (SD) of triplicate determinations. IC50 values, from at least three independent experiments, are means ± SD.
Expression and Purification of His-Tagged CaM and GST Fusion Proteins
Wild-type and mutant rat His-CaM were propagated in XL10 Gold cells, and BL21(DE3) cells were used for propagation of GST, GST-8Nt, GST-8C2b, and GST-1C1b. Proteins were expressed overnight at 20 °C via induction with 0.1 mM isopropyl β-d-1-thiogalactopyranoside. Cells were lysed by sonication in lysis buffer [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 10 μM EDTA, 1 mM phenylmethanesulfonyl fluoride, 1 mM benzamidine, 1× protease inhibitor cocktail, and 1 μg of DNAase (pH 7.4)] supplemented with 200 μg/mL lysozyme. Homogenates were centrifuged at 12000 rpm and 4 °C for 15 min. The supernatant fraction was then passed through a base volume of TALON resin (His-CaM) or glutathione-Sepharose 4B resin (GST) chromatography and washed until no protein remained in the eluate (assessed by measurement of the absorbance at 280 nm). A volume of phosphate-buffered saline (PBS) [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 (pH 7.4)] equal to the volume of resin was used to create a 50% slurry, to which 0.02% (v/v) sodium azide was added for storage at 4 °C.
GST Pull-Down Assays
For pull-down assays assessing the binding of CaM to GST, GST-1C1b, GST-8Nt, and GST-8C2b, glutathione-Sepharose 4B supporting the appropriate immobilized GST or GST fusion protein was washed three times in PBS and added to PBS supplemented with 1% (v/v) Triton X-100, 0.5 μM His-CaM, and either 20 μM CaCl2 or 200 μM EGTA. Samples were rotated for 4 h at 4 °C, centrifuged at 2500 rpm and 4 °C for 5 min, and washed three times in PBS. This was followed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis and immunoblotting with CaM (1:1000) and GST (1:50000) antibodies following goat anti-mouse IgG conjugated to horseradish peroxidase (1:20000), as described previously.2
Stopped-Flow Measurement
Stopped-flow fluorescence experiments were performed at 22 °C and 300 V with a drive pressure of 3 bar, using an Applied Photophysics (Leatherhead, U.K.) spectrofluorimeter (model SX.2.0MV-R) with a dead time of 5 ms. All solutions were prepared in MOPS buffer (20 mM, pH 7.4) supplemented with 100 mM KCl and 1 mM MgCl2. The stopped volume was 100 μL after 1:1 mixing, and thus, the final concentrations in the optical chamber are half of the values stated below. CaCl2 (20 μM) and CaM (2 μM) alone, or combined with 2 μM peptide, were mixed with 150 μM quin-2. The concentration of peptide sufficient to saturate CaM was determined experimentally using various peptide concentrations and by examining the relative change in the amplitudes. AC1-C1b is 28 amino acids long and corresponds to residues 495–522 of bovine AC1 (Table 1 of the Supporting Information). AC8-Nt and AC8-C2b are 25 amino acids long and correspond to residues 30–54 and 1187–1211, respectively, of rat AC8 (Table 1 of the Supporting Information). Synthetic peptides were >95% pure, and their C-termini were protected by amidation. Concentrations of peptides were determined spectrophotometrically (at 280 nm) with molar extinction coefficients of 1615 M–1 cm–1 for AC1-C1b, 5500 M–1 cm–1 for AC8-Nt, and 1490 M–1 cm–1 for AC8-C2b. The fluorescence from quin-2 was detected using an excitation wavelength of 334 nm, controlled by a 405 nm cutoff filter (Applied Photophysics). Although the units of fluorescences are arbitrary (au) and the signals varied each day and drifted over time, the rate constants and the changes in amplitude remained consistent. Data from five to eight injections (runs) were averaged and fit with either a single- or double-exponential function. To obtain kinetic parameters, a mean ± SD was generated from at least three independent averages for each type of experiment.
Offline Nano-ESI-MS Measurements
Offline nano-ESI measurements were conducted on an LTQ-OrbitrapXL mass spectrometer (ThermoFisher Scientific, Bremen, Germany) equipped with a nano-ESI source using metal-coated borosilicate emitters (Proxeon, Odense, Denmark). Mass spectra of peptides were acquired in the positive ionization mode in the m/z range of 200–2000, and signals were deconvoluted with Xcalibur version 2.0.7 (ThermoFisher Scientific).
Results
Removal of Endogenous CaM Inhibits AC1 and AC8 Activities
To compare the ability of AC1 and AC8 to compete for CaM, building on a strategy used previously,18 we used a high-affinity 26-mer peptide, “M13” (KRRWKKNFIAVSAANRFKKISSSGAL), derived from rabbit skeletal muscle myosin light chain kinase (Kd ∼ 0.2 nM19) as a “CaM sponge” to remove endogenous CaM from the membranes of HEK cells expressing AC1 and AC8.2 Crude membranes were washed with excess EGTA to remove free and weakly bound Ca2+-CaM prior to the assay, and the activities of AC1 and AC8 were measured in the presence of 10 μM forskolin, Ca2+, and increasing concentrations of M13. The values of [Ca2+]free that maximally stimulated AC1 (0.3 μM) and AC8 (1 μM) were chosen. Data were converted into the percentage of maximal activity in the absence of M13 to directly compare the inhibition of AC1 and AC8. M13 inhibited AC1 and AC8 equally in a concentration-dependent manner, with IC50 values of 0.044 ± 0.005 μM (n = 3) and 0.048 ± 0.010 μM (n = 4), respectively (Figure 1A). Their activities were effectively inhibited by 1 μM M13 in the absence of added CaM (Figure 1A); however, in the presence of 1 μM exogenous CaM, this (IC50) concentration was largely ineffective (Figure 1B). The IC50 values were increased to 1.56 ± 0.16 μM (n = 5) for AC1 and 1.53 ± 0.21 μM (n = 5) for AC8 in the presence of CaM. These results clearly show that AC1 and AC8 compete for CaM equally in vitro at the level of AC activity.
Figure 1.
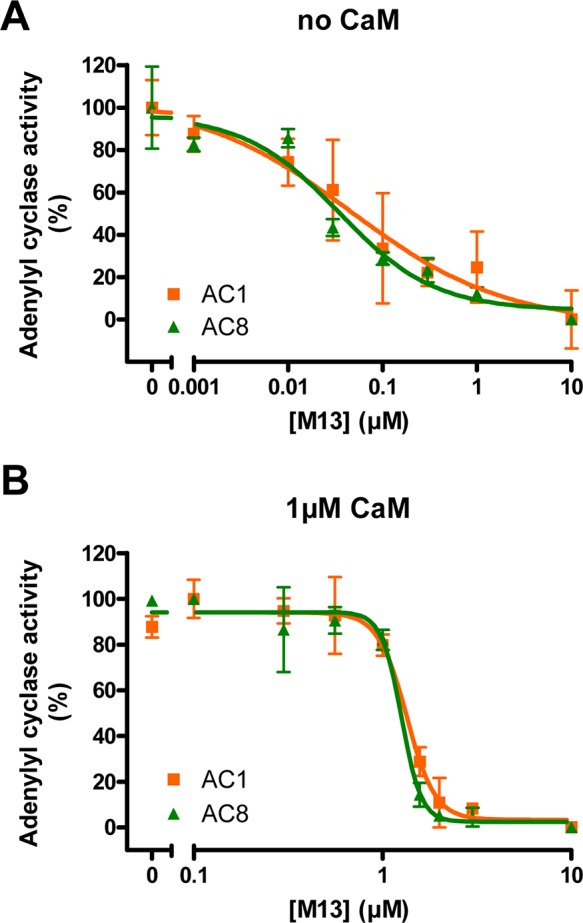
Effect of M13 on Ca2+ regulation of AC1 and AC8 in vitro. Adenylyl cyclase activities of AC1 (orange squares) and AC8 (green triangles) were measured with an increasing concentration of M13 in the absence (A) or presence (B) of 1 μM exogenous CaM. The data were normalized to the maximal activity in the absence of M13 as 100% and the minimal activity with 10 μM M13 as 0%. Data are plotted as means ± SD and are representative of at least three experiments.
The Interaction of CaM with AC1 and AC8 Is Ca2+-Dependent
Direct binding of CaM to the CaMBDs of AC1 and AC8 was first assessed by GST pull-down assays. GST was N-terminally fused to the C1b domain of AC1 (GST-1C1b) or the N-terminus (GST-8Nt) or C2b domain (GST-8C2b) of AC8. GST-1C1b contains 68 amino acid residues from Ser-481 to Leu-548 of bovine AC1 (Table 1 of the Supporting Information). The GST fusion proteins for AC8 previously used comprised residues 1–96 (N-terminal CaMBD) and 1106–1248 (C-terminal CaMBD) of rat AC8.14 To confirm any interactions with a higher level of specificity, shorter GST-8Nt (residues 1–7720) and GST-8C2b (residues 1183–124813), which are similar in length to GST-1C1b, were used (Table 1 of the Supporting Information). Each GST fusion protein was incubated with 0.5 μM CaM in the presence of either 200 μM EGTA or 20 μM Ca2+. Even at high levels, GST alone did not bind CaM, showing the absence of a nonspecific interaction (Figure 2). GST-8Nt, GST-8C2b, and GST-1C1b, on the other hand, pulled down CaM, and their interactions were clearly dependent on Ca2+ (Figure 2).
Figure 2.
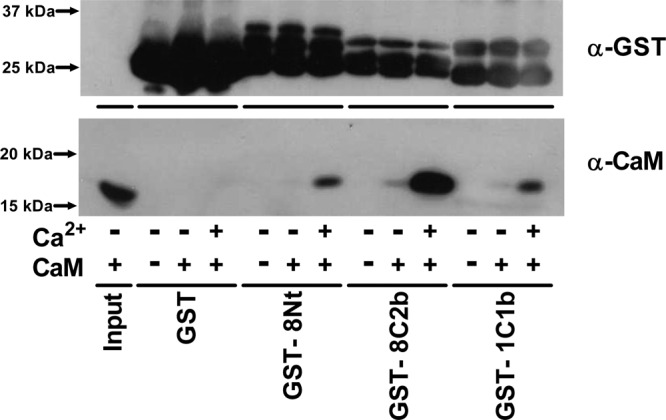
Interaction between CaM and GST-tagged CaMBDs of AC1 and AC8. Binding of CaM to GST (26 kDa), the N-terminus of AC8 (GST-8Nt, 34.5 kDa), the C2b domain of AC8 (GST-8C2b, 33.5 kDa), and the C1b domain of AC1 (GST-1C1b, 33.3 kDa) was assessed by a GST pull-down assay with either 200 μM EGTA (−Ca2+) or 20 μM Ca2+ (+Ca2+). Inclusion of 0.5 μM CaMWT is indicated as +CaM. The horizontal lines indicate CaM input (1%) or different GST fusion proteins used in the assay. The top blot shows the expression of GST, and the bottom blot shows the CaM pulled down. A representative experiment of three is shown.
The Ca2+-dependent interaction between CaM and CaMBDs of AC1 and AC8 was explored more quantitatively by offline nano-ESI-MS measurements. ESI-MS allows the determination of noncovalent interactions between CaM and its target peptides.21−24 Mass spectrometry is not a quantitative technique as the signal intensities depend on the ionization efficiency of the respective molecule. Moreover, absolute signal intensities can vary between different ESI-MS measurements. As such, it is not possible to use absolute MS signal intensities for quantification. Thus, the signal intensities of the CaM·peptide complexes are given as a fraction of the total intensity of all CaM species. Experiments were performed in a solution containing 10 μM CaM and 10 μM peptide (Table 1 of the Supporting Information) with either 200 μM EGTA or 1 mM Ca2+. In the presence of EGTA, all three peptides bound Ca2+-loaded CaM (Figure 3C,E,G). This confirms that the affinity of CaM for Ca2+ becomes higher when a CaMBD peptide binds CaM, as there was no MS signal visible for Ca2+-loaded CaM in the absence of a peptide (Figure 3A). ESI-MS data can also be used to compare the relative affinities of CaM for ligands,25,26 including CaMBD peptides.27 The peak intensities of Ca2+-CaM·peptide complexes were higher for all three peptides in the presence of 1 mM Ca2+ than the peak intensities of CaM (Figure 3D,F,H), compared to those observed in the presence of EGTA (Figure 3C,E,G). For AC1-C1b and AC8-Nt peptides, the signals of non-peptide-bound CaM were not even visible in the presence of Ca2+ (Figure 3D,F). This confirms the higher affinities of all three peptides for CaM in the presence of Ca2+. In the absence of added Ca2+, the peak ratio of the Ca2+-CaM·peptide complex to CaM was the highest with AC1-C1b and the lowest with AC8-C2b (Figure 3C,E,G), showing the order of the relative affinities for partially loaded CaM was as follows: AC1-C1b > AC8-Nt > AC8-C2b. In excess Ca2+, CaM binds four Ca2+ ions, yet we also observed adducts of CaM with more than four Ca2+ ions in ESI-MS experiments (Figure 3B). This is explained by the auxiliary cation binding sites that CaM possesses in addition to the four high-affinity sites,28 as shown by other ESI experiments.21,22,29,30 When the mass spectra were recorded with AC8-C2b and 1 mM Ca2+, there were two signals corresponding to free CaM and peptide-bound CaM with four to eight Ca2+ ions (Figure 3H). In contrast, there was no signal for free CaM when AC1-C1b or AC8-Nt was used (Figure 3D,F), demonstrating that all CaM molecules in the solution formed a complex with these two peptides in the presence of Ca2+. This result illustrated the stronger binding of fully occupied CaM to these two peptides than to AC8-C2b.
Figure 3.

Interaction between CaM and peptides derived from CaMBDs of AC1 and AC8. Offline nano-ESI-MS measurements were performed with 10 μM CaM alone (A and B) or in the presence of 10 μM AC1-C1b (C and D), AC8-Nt (E and F), or AC8-C2b (G and H). The measurements were performed with either 200 μM EGTA (A, C, E, and G) or 1 mM Ca2+ (B, D, F, and H). Deconvoluted mass spectra are presented. The Ca2+-free condition used 200 μM EGTA to mimic the AC assay conditions, which yields negligible concentrations of free Ca2+.19
Lobe-Specific Interaction of CaM with CaMBDs of AC1 and AC8
Offline nano-ESI-MS measurement showed that partially liganded CaM interacted with all three peptides. To understand the lobe-specific interaction of CaM, the ability of CaM mutants to bind the GST fusion proteins was then investigated. None of the CaM species were pulled down by GST alone (Figure 4A), despite the robust expression of GST (Figure 1A of the Supporting Information). GST-1C1b interacted with only CaMWT and not with any CaM mutants (Figure 4B). This result suggests that the absence of regulation by partially liganded CaM2 was due to the inability of the CaM mutants to interact with the CaMBD of AC1. In other words, Ca2+ binding in both lobes of CaM is required for interaction and, by inference, regulation of AC1. In contrast, GST-8Nt and GST-8C2b could pull down partially loaded CaM. GST-8Nt interacted with CaM12 as well as CaMWT in a Ca2+-dependent manner (Figure 4C). The interaction between the C-lobe-occupied CaM and the N-terminus of AC8 mirrored functional data observed previously.2 The result suggests that the preassociation with the N-terminus of AC8 requires at least two Ca2+ ions bound at the C-lobe, although the interaction is enhanced when two more Ca2+ ions bind to the N-lobe. GST-8C2b pulled down CaM34 as well as CaMWT (Figure 4D). The intensity of the CaM34 band was higher than that of the CaMWT band. Hence, CaM requires only two Ca2+ ions bound at the N-lobe to bind to the C-terminal CaMBD of AC8. CaM1234 did not bind to any of the GST fusion proteins, showing that apo-CaM cannot bind to these three CaMBDs.
Figure 4.

Interaction between different CaM species and CaMBDs of AC1 and AC8. The left panels show data for binding of CaM mutants to GST (A), GST-1C1b (B), GST-8Nt (C), and GST-8C2b (D) was assessed by a GST pull-down assay with either 200 μM EGTA (−Ca2+) or 20 μM Ca2+ (+Ca2+). The horizontal lines indicate different species of CaM (0.5 μM) included in the assay or CaM input (1%). The right panels show signal intensities quantified using ImageJ (National Institutes of Health, Bethesda, MD). The level of CaM pulled down was expressed as a band density relative to the GST intensity (Figure 1 of the Supporting Information) and each CaM input and normalized to the level of CaM1234 with EGTA of each blot. Data are plotted as means ± the standard error of the mean and are an average of three experiments.
Ca2+ Dissociation Kinetics of CaM
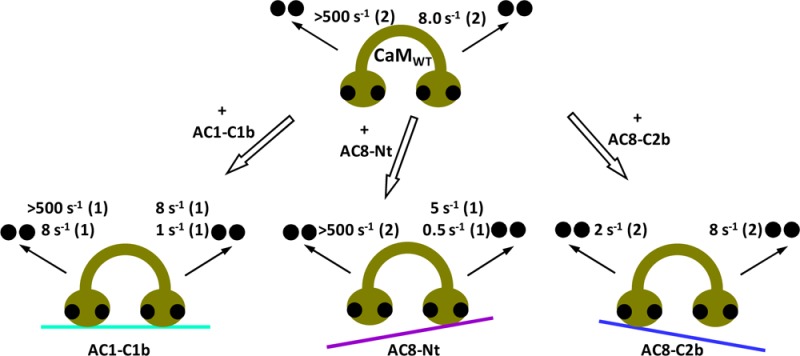
The interaction between each lobe of CaM and the CaMBDs of AC1 and AC8 revealed by the GST pull-down assay supported their lobe-specific regulation revealed previously.2 However, this approach is quite undiscerning, and the effect that each CaMBD exerts on CaM could not be compared. In addition, the association between partially liganded CaM and the C1b domain of AC1 observed using ESI-MS measurement could not be explained. Therefore, stopped-flow kinetic measurement was employed to more precisely explore the lobe-specific interaction of CaM. Ca2+ binding influences the ability of CaM to interact with target proteins.31 Likewise, interaction with target proteins affects the affinity of CaM for Ca2+.32,33 Stopped-flow kinetic studies indicate that the increased affinity of CaM is largely due to a reduction in the Ca2+ dissociation rate constant in Ca2+-CaM·peptide/protein complexes, while the calculated Ca2+ association rate constant is not altered in many cases.34 Accordingly, rates of dissociation of Ca2+ from numerous complexes have been examined to gain insights into their particular interactions. Determining the Ca2+ dissociation rate constants by stopped-flow kinetic studies is especially instructive as Ca2+ ions dissociate from CaM in two steps, one from each lobe. The C-lobe of CaM has a higher affinity for Ca2+ than the N-lobe; subsequently, dissociation of Ca2+ from the C-lobe is slower than that from the N-lobe. In the presence of the CaMBD peptide, dissociation of Ca2+ from CaM not only is slowed but also takes place in two or more steps, from which the important lobe(s) for the target interaction can be inferred.
The rate of dissociation of Ca2+ from the N- and C-lobes of CaM can be measured by monitoring quin-2 fluorescence in stopped-flow measurements.35−37 Because of its high affinity for Ca2+ (Kd ∼ 60 nM38), quin-2 rapidly binds all Ca2+ ions within the dead time of the spectrofluorimeter when micromolar concentrations of quin-2 and Ca2+ are mixed.39 The very fast on rate of quin-2 is ideal, as an increase in fluorescence detected in the stopped-flow apparatus correlates directly with the amount of chelated Ca2+ released from CaM, but not free Ca2+. When mixed with 20 μM Ca2+ and 2 μM CaM1234, quin-2 also chelated Ca2+ within the dead time, and no change in the fluorescence was observed (data not shown), confirming that the aspartate to alanine substitution in each EF-hand Ca2+-binding loop was sufficient to preclude Ca2+ binding. Although CaM-target peptides can restore some Ca2+ binding by CaM mutants,40 stopped-flow measurement did not show the change in the fluorescence in the presence of CaM1234 and CaMBD peptides (data not shown), and thus, we assumed that the Ca2+ dissociations observed under this condition were from the EF-hands that were not mutated.
Because the N-lobe of CaM releases Ca2+ very rapidly, the kinetics of dissociation of Ca2+ from CaMWT was readily fit with a single-exponential function with a rate constant of 8.0 ± 0.3 s–1 [n = 5 (Figure 5A)], displaying the rate only from the C-lobe of CaM. An equivalent rate constant was obtained with CaM12, showing a koff value of 8.1 ± 0.2 s–1 [n = 4 (Figure 5B)]. The changes in the relative quin-2 fluorescence as a result of the release of Ca2+ from CaMWT and CaM12 were also similar (0.064 ± 0.016 and 0.061 ± 0.005, respectively). Therefore, the mutation in the N-lobe did not compromise the ability of the C-lobe to interact with Ca2+. Because the C-lobe of CaM contains two EF-hands that bind a Ca2+ ion each, the change in the amplitude of ∼0.06 was assumed to correspond to two Ca2+ ions released from CaM.
Figure 5.
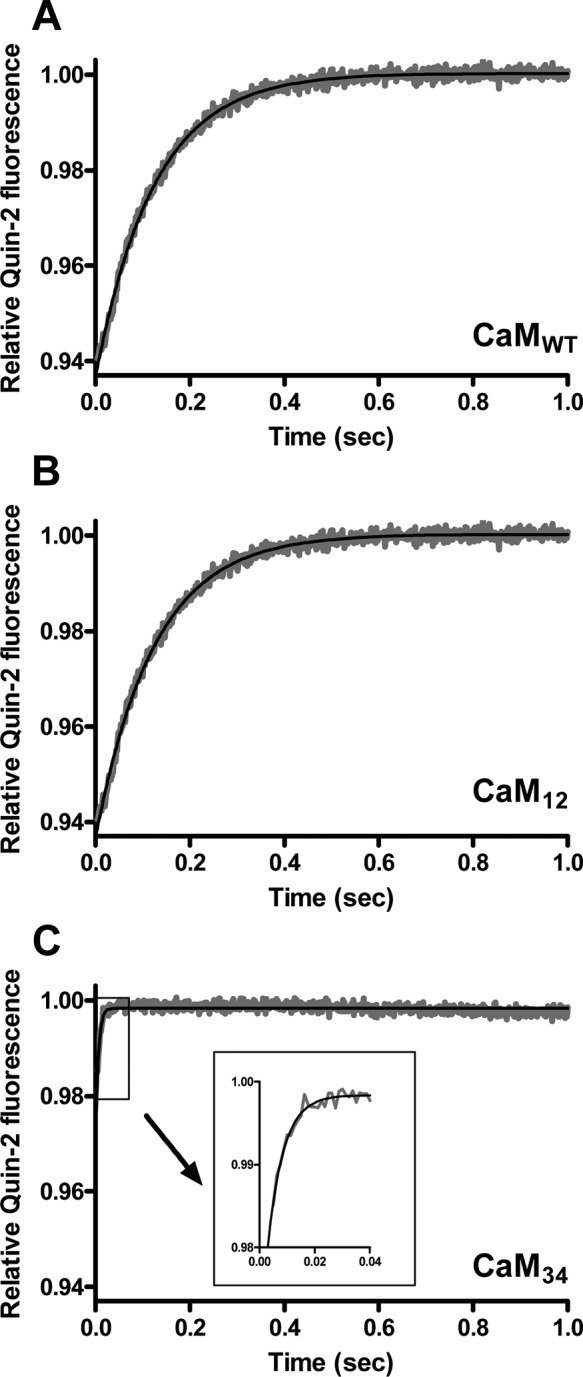
Ca2+ dissociation kinetics of CaM in the absence of peptide. Dissociation of Ca2+ from CaMWT (A), CaM12 (B), and CaM34 (C) was measured by monitoring changes in quin-2 fluorescence. Quin-2 (150 μM) was rapidly mixed with 20 μM Ca2+ and 2 μM CaM. The inset of panel C shows the close-up trace of dissociation of Ca2+ from CaM34 from 0 to 0.04 s. Each trace that represents an average of five to eight runs was fit using a single-exponential function. Data are representative of at least four experiments.
Because of its high off rate, Ca2+ released from the N-lobe of CaM and free Ca2+ are chelated by quin-2 within the dead time of the spectrofluorimeter, and thus, the fluorescence change cannot be observed unless the temperature is lowered to ∼10 °C.35,41,42 The C-lobe mutant of CaM, however, caused an increase in quin-2 fluorescence at 22 °C. Ca2+ dissociated from the N-lobe of CaM34 with a koff value of 195 ± 30 s–1 [n = 8 (Figure 5C)], in good agreement with an earlier study.43 The relative amplitude was only 0.030 ± 0.009, implying that one Ca2+ ion was released with the rate constant observed and that the other Ca2+ release event was too fast to be measured. Because the mutation within the EF-hands in the C-lobe affected the Ca2+ dissociation property of the N-lobe, we interpreted the data using CaM34 to examine the interaction between Ca2+ and the N-lobe of CaM with caution. Table 1 summarizes the rate constants and the change in the amplitude of dissociation of Ca2+ from CaMWT, CaM12, and CaM34.
Table 1. Ca2+ Dissociation Kinetics of CaMa.
| koff1 (s–1) | amplitude 1 | koff2 (s–1) | amplitude 2 | |
|---|---|---|---|---|
| CaM34 | >500 | (1) | 195 ± 29.7 | 0.030 ± 0.009 (1) |
| with AC1-C1b | >500 | (1) | 5.55 ± 0.63 | 0.032 ± 0.007 (1) |
| with AC8-Nt | >500 | (1) | 66.9 ± 10.4 | 0.027 ± 0.004 (1) |
| with AC8-C2b | 3.46 ± 0.61 | 0.049 ± 0.006 (2) | ||
| CaM12 | 8.12 ± 0.23 | 0.061 ± 0.003 (2) | ||
| with AC1-C1b | 5.17 ± 1.04 | 0.022 ± 0.010 (1) | 0.84 ± 0.27 | 0.012 ± 0.003 (0.5) |
| with AC8-Nt | 4.18 ± 1.04 | 0.017 ± 0.002 (0.5) | 0.29 ± 0.06 | 0.022 ± 0.06 (1) |
| with AC8-C2b | 8.45 ± 1.15 | 0.043 ± 0.007 (1.5) | 3.84 ± 1.27 | 0.021 ± 0.001 (0.5) |
| CaMWT | 7.95 ± 0.29 | 0.064 ± 0.016 (2) | ||
| with AC1-C1b | 7.98 ± 0.55 | 0.053 ± 0.001 (2) | 1.16 ± 0.29 | 0.024 ± 0.006 (1) |
| with AC8-Nt | 4.93 ± 0.86 | 0.029 ± 0.008 (1) | 0.49 ± 0.20 | 0.027 ± 0.007 (1) |
| with AC8-C2b | 8.38 ± 1.08 | 0.054 ± 0.006 (2) | 1.59 ± 0.58 | 0.053 ± 0.010 (2) |
Ca2+ dissociation kinetics were determined by measuring changes in quin-2 fluorescence at 22 °C and fit with a single- or double-exponential function. The average rate constants for Ca2+ dissociation and the amplitude are presented as means ± SD of three to eight independent experiments. A change in the relative amplitude of 0.03 corresponds to a release of one Ca2+ ion, and the number of Ca2+ ions released is shown in parentheses. The rate constant for Ca2+ dissociation, which was too fast to measure, is shown as >500.
Effect of CaMBD Peptides on the Release of Ca2+ from the N-Lobe of CaM34
Separating the role of each lobe of CaMWT can be difficult, as each target peptide interacts with CaM differently. The use of CaM mutants can overcome this issue and clarify how each lobe is influenced by peptides. For that reason, the effect of peptides on CaM mutants was established before examining the Ca2+ dissociation kinetics of wild-type CaM, to identify the rate that corresponds to each lobe’s action. CaM34 was first used to assess how the CaMBD peptides derived from AC1 and AC8 influence the ability of the N-lobe of CaM to interact with Ca2+. CaM34 can only bind Ca2+ at the N-lobe because of the mutations within EF3 and EF4 in the C-lobe and hence should provide a means of measuring the rate of dissociation of Ca2+ from the N-lobe, exclusive of the influence of Ca2+ binding events that take place at the C-lobe. The GST pull-down assay showed a strong interaction between CaM34 and GST-8C2b, but not GST-8Nt or GST-1C1b (panels D, C, and B of Figure 4, respectively). Therefore, an effect of AC8-C2b on the release of Ca2+ from CaM34 was expected, and accordingly, the impact that the C-terminal CaMBD of AC8 has on dissociation of Ca2+ from the N-lobe of CaM could be determined.
CaM34 releases one Ca2+ ion at a rate that is too fast to be measured under this condition (koff > 500 s–1) and another one with a koff value of ∼200 s–1, when mixed with quin-2 (Table 1). Thus, Ca2+ dissociates from CaM34 very rapidly; even the slower Ca2+ dissociation event is completed within the first 50 ms. Surprisingly, the rapid release of Ca2+ from the N-lobe of CaM34 was slowed not only by AC8-C2b but also by AC1-C1b and AC8-Nt (Figure 6A). All the data fit well to a single-exponential function (Figure 2A of the Supporting Information), which gave rate constants of 5.6 ± 0.6, 67 ± 10, and 3.5 ± 0.6 s–1 in the presence of AC1-C1b, AC8-Nt, and AC8-C2b, respectively [n = 3 (Figure 6A and Table 1)]. The relative amplitudes of dissociation of Ca2+ from the CaM34·AC1-C1b and CaM34·AC8-Nt complexes were 0.032 ± 0.007 and 0.027 ± 0.004, respectively [n = 3 (Figure 6A and Table 1)], demonstrating the involvement of only one EF-hand. Although the presence of AC8-Nt slowed the release of Ca2+ from one EF-hand of CaM34, the koff value of ∼70 s–1 indicates a very weak, possibly transient interaction that could not be detected by the GST pull-down assay.
Figure 6.
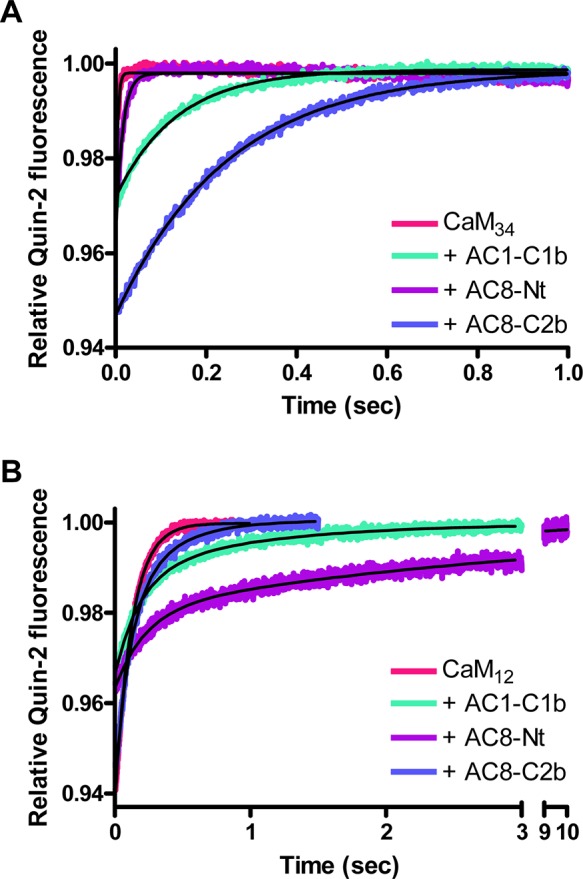
Effect of peptides on the Ca2+ dissociation kinetics of CaM mutants. Dissociation of Ca2+ from CaM34 (A) or CaM12 (B) in the absence or presence of peptide was measured by monitoring changes in quin-2 fluorescence. Quin-2 (150 μM) was rapidly mixed with 20 μM Ca2+ and 2 μM CaM in the absence or presence of 2 μM peptide. Each trace, which represents an average of five to eight runs, was fit using a single-exponential (A) or double-exponential (B) function. Representative experiments of three are shown.
In contrast to AC8-Nt, AC1-C1b and AC8-C2b reduced the speed of dissociation of Ca2+ from CaM34 considerably. A weak interaction caused by the involvement of only one EF-hand may explain why the GST pull-down assay showed no interaction between CaM34 and GST-1C1b (Figure 4B). Unlike AC1-C1b, AC8-C2b influenced both EF-hands in the N-lobe of CaM34. The change in the relative quin-2 fluorescence was 0.049 ± 0.006 in the presence of AC8-C2b [n = 3 (Figure 6A and Table 1)], illustrating that Ca2+ ions were released from both EF-hands in the N-lobe slowly. The cooperativity of the EF-hand pair was also restored, as the data could be fit only using a single-exponential function. Therefore, the C-terminal CaMBD of AC8 has a dramatic effect on the rate constants for dissociation of Ca2+ from the N-lobe of CaM.
Effect of CaMBD Peptides on the Release of Ca2+ from the C-Lobe of CaM12
Only GST-8Nt bound CaM12 (Figure 4C), and thus, AC8-Nt is expected to consolidate the ability of CaM12 to grasp Ca2+ in the presence of the Ca2+ chelator. CaM12 has a functional C-lobe; its Ca2+ dissociation kinetics can be determined accurately by monitoring quin-2 fluorescence. The release of Ca2+ from CaM12 alone gave a rate constant of ∼8 s–1, because of a slower off rate from the C-lobe, compared to CaM34, which contains a functional N-lobe. In the presence of peptides, the data were best fit with a double exponential (Figure 2C of the Supporting Information), as a single-exponential function failed to fit early time points (Figure 2B of the Supporting Information). The Ca2+ dissociation kinetics of each CaM12·peptide complex had two rates. However, for CaM12 in complex with AC8-C2b, the two kinetic phases were similar; the major fast phase had a koff1 value of 8.5 ± 1.2 s–1, with an amplitude of 0.043 ± 0.007, while the koff2 value was 3.8 ± 1.3 s–1, with an amplitude of 0.021 ± 0.001 [n = 3 (Figure 6B and Table 1)]. This result indicates that ∼1.5 mol of Ca2+ ions dissociates from the C-lobe with a rate almost identical to that observed in the absence of a peptide. In addition, the koff2 value, which showed ∼0.5 mol of Ca2+ released, is not very different from the koff1 value. The single-exponential fit, although not as good as the double-exponential fit, gave a rate constant of 7.0 ± 1.3 s–1 with an amplitude of 0.054 ± 0.004 (n = 3). Thus, the effect of AC8-C2b on Ca2+ binding of the C-lobe, if any, is minimal.
AC1-C1b and AC8-Nt had more influence on the release of Ca2+ from the C-lobe of CaM12 (Figure 6B). The curves were clearly biphasic, and data were fit to a double-exponential function. Rates of dissociation of Ca2+ from the CaM12·AC1-C1b complex were 5.2 ± 1.0 and 0.8 ± 0.3 s–1, corresponding to the release of Ca2+ from each EF-hand in the C-lobe [n = 3 (Figure 6B and Table 1)]. The marked reduction of the Ca2+ dissociation rate constants suggests an interaction between the C-lobe of CaM and the CaMBD of AC1, even though CaM12 was not pulled down by GST-1C1b (Figure 4B). However, as expected from the GST pull-down assay, the interaction of CaM12 with the N-terminal CaMBD of AC8 appears to be even stronger. Ca2+ dissociated with a rate constant of 4.2 ± 1.0 s–1 from one EF-hand and 0.3 ± 0.1 s–1 from the other EF-hand in the C-lobe [n = 3 (Figure 6B and Table 1)].
Effect of CaMBD Peptides on the Release of Ca2+ from CaMWT
Stopped-flow measurements with CaM mutants hitherto showed each lobe of CaM interacts differently with each peptide. Dissociation of Ca2+ from the N-lobe of CaM is slowed by AC1-C1b and even more by AC8-C2b. On the other hand, AC1-C1b and AC8-Nt, in particular, reduced the rate of release of Ca2+ from the C-lobe of CaM. To establish whether the interaction of each lobe observed was genuine or a consequence of the mutation in the other lobe, the Ca2+ dissociation kinetics was examined using CaMWT. In the absence of peptide, CaMWT releases Ca2+ with a rate constant of ∼8 s–1. In the presence of a peptide, a single-exponential function did not fit the data (Figure 2D of the Supporting Information), and subsequently, a double-exponential function was used (Figure 2E of the Supporting Information).
The Ca2+ dissociation kinetics of the CaMWT·AC1-C1b complex exhibited 2 mol of Ca2+ dissociated with a koff of 8.0 ± 0.6 s–1 and 1 mol dissociated with a koff of 1.2 ± 0.3 s–1 [n = 3 (Figure 7A and Table 1)]. The total change in the relative quin-2 fluorescence corresponds to 3 mol of Ca2+ ions released, and thus, 1 mol of Ca2+ was assumed to be released at >500 s–1 from one EF-hand in the N-lobe, which was too fast to be measured. The slow koff2 was assigned to the rate of dissociation of Ca2+ from one of the EF-hands in the C-lobe, based on the koff2 of the CaM12·AC1-C1b complex. Consequently, the koff1 represents the rate constant for the release of Ca2+ from one EF-hand in each lobe of CaM. In summary, CaMWT in complex with AC1-C1b exhibited Ca2+ dissociation rate constants of >500 and ∼8 s–1 for the N-lobe and ∼8 and ∼1 s–1 for the C-lobe.
Figure 7.
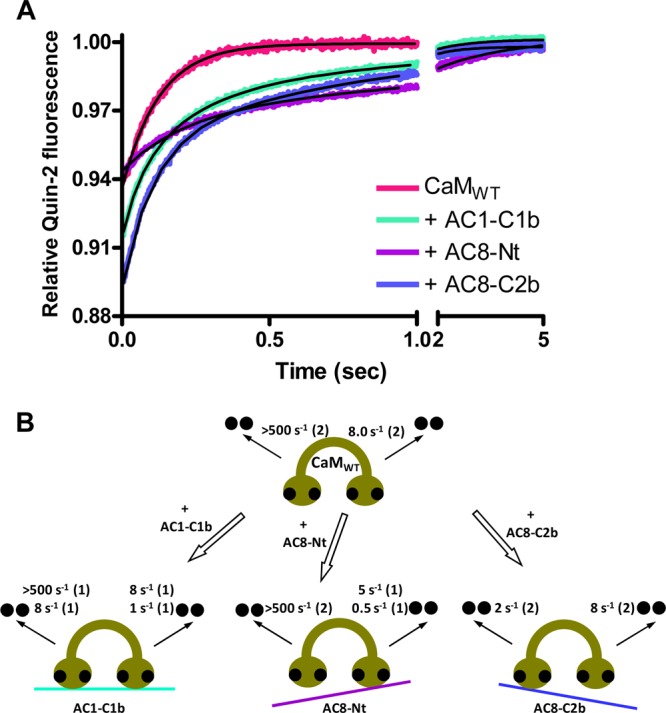
Effect of peptides on Ca2+ dissociation kinetics of CaMWT. (A) Dissociation of Ca2+ from CaMWT in the absence or presence of a peptide was measured by monitoring changes in quin-2 fluorescence. Quin-2 (150 μM) was rapidly mixed with 20 μM Ca2+ and 2 μM CaMWT in the absence or presence 2 μM peptide. Each trace that represents an average of five to eight runs was fit using a double-exponential function. A representative experiment of three is shown. (B) Summary showing the effect of AC1-C1b, AC8-Nt, and AC8-C2b on the rate constants for dissociation of Ca2+ from CaMWT. Ca2+ dissociates from the N- and C-lobes of peptide-free CaMWT with rate constants of >500 and 8 s–1, respectively, at 22 °C. When CaMWT binds to AC1-C1b, the rate of dissociation of Ca2+ from one EF-hand of the N-lobe is decreased to 8 s–1, and that for one EF-hand of the C-lobe is reduced to 1 s–1, without affecting the other EF-hand in each lobe. AC8-Nt reduced the rate of release of Ca2+ from the C-lobe of CaM only to 5 and 0.5 s–1. AC8-C2b influenced dissociation of Ca2+ only from the N-lobe of CaM at a rate of 2 s–1.
The CaMWT·AC8-Nt complex displayed Ca2+ dissociation rate constants of 4.9 ± 0.9 and 0.5 ± 0.2 s–1 with amplitudes of 0.029 ± 0.008 and 0.027 ± 0.007, respectively [n = 3 (Figure 7A and Table 1)], corresponding to the release of two Ca2+ ions. The rate constants measured with CaMWT were both concluded to correspond to the release of Ca2+ from the C-lobe of CaM, because they were very similar to the koff1 and koff2 of the CaM12·AC8-Nt complex [∼4 and ∼0.3 s–1, respectively (Table 1)]. Additionally, AC8-Nt did not affect dissociation of Ca2+ from the N-lobe of CaM34 to a great extent (Figure 6A), and hence, Ca2+ was presumed to be released from both EF-hands in the N-lobe with a koff of >500 s–1.
In contrast with AC8-Nt, AC8-C2b influenced the ability of the N-lobe of CaM34 (Figure 6A), but not the C-lobe of CaM12 (Figure 6B), to interact with Ca2+. The release of Ca2+ from the CaMWT·AC8-C2b complex was biphasic with a koff1 value of 8.4 ± 1.1 s–1 and a koff2 value of 1.6 ± 0.6 s–1 [n = 3 (Figure 7A and Table 1)]. Unlike CaMWT in complex with AC1-C1b or AC8-Nt, in which the stopped-flow measurement detected 3 or 2 mol of Ca2+ released, respectively, the relative amplitude was increased to ∼0.12 in the complex with AC8-C2b (Figure 7A), demonstrating that all four Ca2+ ions were released slowly, which can be measured by the spectrofluorimeter under this condition. Each rate constant was assumed to correspond to each lobe based on the Ca2+ dissociation kinetics of CaM34 and CaM12, and we concluded that the N-lobe of CaM released Ca2+ with the slower koff2 rate by interacting with AC8-C2b, while the C-lobe released Ca2+ with the koff1 rate, which was not affected because of the deficiency in associating with the peptide.
Discussion
Lobe independence and linker flexibility make CaM an exceptional Ca2+ sensor protein that interacts with a wide range of proteins.44 The use of Ca2+-binding deficient CaM mutants has revealed the regulation of a number of proteins by partially liganded CaM. For example, the C-lobe of CaM plays a key role in activating Bacillus anthracis exotoxin45 and Ca2+-CaM-dependent protein kinase II (CaMKII46), although CaMKII interacts with the N-lobe first before interacting with the C-lobe.47,48 On the other hand, Ca2+-independent tethering of small-conductance Ca2+-activated potassium (SK) channels requires the C-lobe of CaM, while Ca2+-dependent channel gating requires only the N-lobe of CaM.8 The Ca2+-mediated activation mechanism of SK channels is considered to be distinct from classical mechanisms because of the involvement of two subunits forming a dimer,49 yet the action of CaM in regulating AC8 may be more similar to that of SK channels than AC1, because AC8 tethers the C-lobe of CaM and is activated by the N-lobe of CaM.2 This study confirms that the lobe-specific regulation of CaM2 results from lobe-specific interactions; the stopped-flow data show interactions of CaM liganded at both lobes with AC1-C1b, the C-lobe occupied CaM with AC8-Nt, and the N-lobe occupied CaM with AC8-C2b, as summarized in Figure 7B. As demonstrated by connexin32,43 examining the release of Ca2+ from CaM34 (Figure 3A of the Supporting Information) and CaM12 (Figure 3B of the Supporting Information) allowed the effect of the peptides on each lobe to be isolated, and this simplified the analysis of the Ca2+ dissociation kinetics of the CaMWT·peptide complexes.
AC1-C1b influenced the ability of both lobes of CaM to interact with Ca2+. The interaction with AC1-C1b requires binding of Ca2+ to one EF-hand in each lobe of CaM, which can explain the complex of AC1-C1b with 2 mol of Ca2+-bound CaM observed in the ESI-MS experiment but the inadequate binding of GST-1C1b with the CaM mutants. The Ca2+ dissociation kinetics of the CaMWT·AC1-C1b complex was similar to the “3 + 1 kinetics” described for the CaMWT·skeletal muscle myosin light chain kinase complex, in which one Ca2+ ion is released very fast (rate constant of >500 s–1) in a manner independent of the other three Ca2+ ions.50 AC8-Nt slowed dissociation of Ca2+ from the C-lobe without affecting the N-lobe of CaM (Figure 7B). The result suggests that the interaction with the N-terminal CaMBD of AC8 requires at least two Ca2+ ions bound at the C-lobe of CaM. CaM often preassociates with proteins in a manner independent of Ca2+ concentrations, as seen with phosphorylase kinase,51 SK channels,8 and L-type, P/Q-type, and R-type Ca2+ channels.52 The N-terminus of AC8, however, contains an amphipathic 1-8-14 motif, in which CaM binding is dependent on Ca2+ as shown by GST pull-down assays (Figure 2) and offline nano-ESI-MS (Figure 3E,F), and the association with the N-lobe mutant of CaM also requires Ca2+ (Figure 4C). As suggested for a phosphorylated form of CaMKII,53 AC8 may be capable of associating with partially occupied CaM at a resting [Ca2+]i, which supports the inhibition of AC8 activity by CaM12.2,14 AC8-C2b exerts a dramatic effect on the ability of the N-lobe of CaM to interact with Ca2+. On the basis of the GST pull-down assay and stopped-flow measurements using the CaM mutants, the C-terminal CaMBD of AC8 was concluded to have little influence on the C-lobe of CaM (Figure 7B). However, the possibility that the lobe assignment was incorrect and that AC8-C2b caused slower release of Ca2+ from both lobes cannot be ruled out, because the slower Ca2+ dissociation rates are often considered to be the rates of the C-lobe.34,41 Even so, the reduction of the speed would be >60-fold by the N-lobe and 5-fold by the C-lobe, and thus, AC8-C2b preferentially increases the affinity of the N-lobe for Ca2+.
Our ESI-MS measurements illustrated that fully liganded and partially liganded CaM both have a higher affinity for AC1-C1b and AC8-Nt than for AC8-C2b (Figure 3). This could explain the residual Ca2+ stimulation observed in wild-type AC1 and AC8, but not the C1b domain mutant of AC1 or the N-terminal truncation mutant of AC8.2 The inhibition of their activities by M13 (Figure 1) suggests that the ability of AC1 and AC8 to compete for CaM with other Ca2+-CaM target proteins is equivalent in vitro, regardless of the available CaM concentrations. In the regions of the brain where AC1 and AC8 are distributed, a number of Ca2+-CaM target proteins also reside. It appears that AC1, which has a higher affinity for Ca2+ and partially liganded CaM, has the advantage of being maximally stimulated by the same concentrations of Ca2+-CaM. This may be the case in vitro; however, AC8 benefits from having the additional CaMBD that can tether C-lobe-occupied CaM in resting [Ca2+]i in vivo. Because of the very fast on rate of the N-lobe of CaM for Ca2+,54 CaM is fully liganded and AC8 is activated quickly by a Ca2+ transient. AC1 may need to have a high affinity for Ca2+ to compensate for the requirement of CaM with Ca2+ binding to both lobes, as this makes AC1 less able to compete with other high-affinity CaM-binding proteins, which sequester, tether, or compartmentalize the available pool of CaM.55
Unlike that of AC8, little is known about the activation mechanism of AC1. Because the CaMBD of AC1 is close to the catalytic site, AC1 is presumed to be activated by the relief of autoinhibition, like AC8 and many protein kinases. Ca2+ saturation of one lobe of CaM was not sufficient to interact with or regulate AC1, and thus, we speculated that all four EF-hands of CaM need to bind Ca2+ to activate AC1. However, a more comprehensive study of stopped-flow kinetics revealed that the binding of CaM to AC1 requires one Ca2+ ion in each lobe (Figure 7B). This unusual interaction supports an earlier study of AC1 using CaM mutants with a single-point mutation.56 Mutations at EF2 and EF4 severely compromised the ability of CaM to activate AC1,56 demonstrating that these two EF-hands require Ca2+ binding. The EF1 mutant had little effect,56 which is consistent with our stopped-flow data that show that one EF-hand in the N-lobe was not affected by the CaMBD peptide of AC1 (Figure 3A of the Supporting Information). Mutation at EF3 did not have a effect as dramatic as that at EF2 or EF4; nevertheless, this CaM mutant decreased the apparent affinity of CaM for AC1 by 5-fold.56 Stopped-flow kinetic data showed that the peptide-bound EF-hand in the N-lobe and the peptide-free EF-hand in the C-lobe of CaM released Ca2+ simultaneously (Figure 7B). A previous study showed that EF2 and EF3 make contact with a helix within the backbone of CaM, which could contribute to the cooperative binding of Ca2+ between the two lobes.5 Hence, EF2 and EF3 may communicate when interacting with AC1. The 5-fold decrease in the affinity by a mutation at EF3 may result from a disrupted communication; the binding of EF2 to CaMBD of AC1 may involve EF3.
AC1 displayed a slower activation by Ca2+ compared to that of AC8.2 We interpret these slower kinetics to reflect the slower interaction with CaM because of the involvement of both lobes and/or a conformational change, such as an isomerization, that stabilizes the complex as demonstrated by CaMKII.57,58 A recent study showed that the Ca2+ occupancy at EF1, EF3, and EF4 led to the full activation of CaMKII, while EF3 was the key for stabilizing ATP binding.59 The activation mechanism of CaMKII is very complex, and possibly unique. However, the CaM activation mechanism of AC1 may not be too dissimilar, as AC1 needs three EF-hands occupied with Ca2+. Because the CaMBD of AC1 is located very close to its catalytic core, ATP may also be involved in the stabilization of the complex, although more kinetic studies are required to determine this.
In response to Ca2+ oscillations, AC1 produced a steady cAMP increase, unlike AC8, which produced cAMP oscillations.2 We are inclined to speculate that this activation behavior might reflect the tight interaction between CaM and CaMBD of AC1 and/or the slower rate of Ca2+ association and dissociation, which could not respond to high-frequency Ca2+ transients. Of course, we must acknowledge that the time scales of these two processes, Ca2+-CaM binding and AC activation (not to mention the time scale of the two measurements, stopped-flow vs AC activity), render this speculation fanciful. Nevertheless, the differences in the ability of AC1 and AC8 to respond or not to Ca2+ transients must bear some relationship to the Ca2+-CaM binding events. Two isoforms of plasma membrane Ca2+-ATPase (PMCA), 4a and 4b, also exhibit different activation kinetics; PMCA4a is more efficient in handling transient Ca2+ spikes than PMCA4b.60 This is because PMCA4b, which involves the isomerization step,61 is activated and inactivated more slowly by addition and removal of CaM, respectively, than PMCA4a,62 which does not involve the isomerization.63 AC1 and AC8 may have activation kinetics similar to those of PMCA4b and PMCA4a, respectively. We propose that AC1 activation is initiated by binding of Ca2+ to EF2–EF4, and thus, the speed of activation is slow. These Ca2+ occupancies led to a conformational change so that neither CaM nor Ca2+ dissociates from the complex easily. The relief of autoinhibition then takes place to produce a nonoscillatory cAMP increase. Because of the strong interaction, [Ca2+]i needs to decrease considerably for CaM to dissociate to allow the inactivation of AC1.
AC8 possesses two CaMBDs, one at each end of the molecule.12 The findings in this study support an earlier proposal in which the N-terminal CaMBD of AC8 preassociates with C-lobe-liganded CaM, which can be passed onto, or shared with, the C-terminal CaMBD upon binding Ca2+ at the N-lobe to activate AC8 by removing the autoinhibitory domain.14 A recent study demonstrated that the two CaMBDs of AC8 could interact but only in the absence of Ca2+-CaM, and this interaction was specifically inhibited by Ca2+-saturated CaM, but not Ca2+-free CaM.13 A series of mutagenesis studies revealed that the stimulation by Ca2+-CaM of C-terminal CaMBD mutants of AC8 was only diminished when the N-terminal CaMBD was truncated or mutated so that it could no longer bind CaM.13 The binding at the N-terminus is apparently so tight that even though the independent ability of the C-terminus to bind CaM can be severely compromised, only when CaM is not available at the N-terminus can the consequences of C-terminal mutations be revealed.13 On this basis, it is proposed that the two CaMBDs associate at rest, with the C-lobe-saturated CaM binding to the N-terminal CaMBD. With increases in [Ca2+]i, the N-lobe of CaM binds Ca2+ and subsequently interacts with the C-terminal CaMBD of AC8, accompanied by a release of the N-terminal CaMBD from the complex.13 Because of the weak interaction between the two CaMBDs in the presence of Ca2+-saturated CaM, the translocation of CaM to the C-terminal CaMBD and the dissociation of the N-terminal CaMBD occur very rapidly. We assumed that this translocation takes place because of the higher affinity of the C-terminal CaMBD for CaM compared to that of the N-terminal CaMBD. The ESI-MS, however, clearly showed that the N-terminal CaMBD has a higher affinity for fully liganded CaM than the C-terminal CaMBD (Figure 3F,H). Therefore, a conformational change or dissociation of Ca2+ from the C-lobe of CaM needs to occur to decrease the affinity of the N-terminal CaMBD for CaM, so that CaM is released from the N- to C-terminal CaMBD. Recently, Black and Persechini showed that precisely where the N-lobe of CaM associates with IQ motifs and IQlm depends on the Ca2+ occupancy within the C-lobe of CaM.64 Although the C-lobe of CaM has a high affinity for Ca2+, Ca2+ may dissociate from the C-lobe transiently during a Ca2+ oscillation, when CaM associates with the C-terminal CaMBD of AC8 only. This may cause the switching of the interaction site of the N-lobe of CaM from upstream to downstream of the IQlm in the C2b domain of AC8, resulting in transient inactivation of AC8. This assumption is appealing as a reason for Ca2+-induced cAMP oscillations observed in AC83 notwithstanding the reservations raised above about the time scales of binding versus activation.
Although further studies such as equilibrium binding will help to establish the detailed CaM activation mechanisms of AC1 and AC8, this study reveals the essential EF-hands of CaM that interact with and regulate these two enzymes. This coupled with the relative affinity of their CaMBDs for CaM suggested how these two enzymes decode Ca2+ signals into cAMP signals. Note, however, that the properties of the CaMBDs inferred from these studies could be influenced within the context of the tertiary environment of the full-length AC molecules from which they are derived. Like many CaM-target proteins, AC1 and AC8 are involved in synaptic plasticity. Until recently, they were considered to have similar physiological roles, because of their high levels of expression in the brain and overlapping functions, because long-lasting long-term potentiation and fear-associated memory formation were impaired in double-knockout but not single-knockout mice of AC1 or AC8,65 implying that one of the ACs can compensate for the other. However, discrete neuronal functions are now becoming apparent for the two isoforms.66 The distinct activation mechanisms of AC1 and AC8 by CaM revealed here, which could decode Ca2+ signaling into different cAMP dynamics, would be expected to result in or contribute to different physiological possibilities for these enzymes. The different properties of AC1 and AC8 revealed here emanating from their differing dependencies on the two lobes of CaM illustrate the economical use that nature makes of simple motifs to yield distinct arrays of physiological outcomes.
Acknowledgments
We are grateful to Drs. Rachel Simpson and David MacDougall for preliminary data and Dr. Christian Ihling for help with nano-ESI-MS measurements. We also thank Drs. Debbie Willoughby, Katy Everett, and David MacDougall for helpful comments on the manuscript.
Glossary
Abbreviations
- AC
adenylyl cyclase
- CaM
calmodulin
- C-lobe and N-lobe
C-terminal and N-terminal lobes of CaM, respectively
- CaM1234
combined N- and C-lobe mutant of calmodulin
- CaM12
N-lobe mutant of calmodulin
- CaM34
C-lobe mutant of calmodulin
- CaMWT
wild-type calmodulin
- CaMBD
calmodulin binding domain
- CaMKII
Ca2+-CaM-dependent protein kinase II
- ESI
electrospray ionization
- GST
glutathione S-transferase
- IQlm
IQ-like motif
- M13
M13 skeletal muscle myosin light chain kinase peptide
- MS
mass spectrometry
- PBS
phosphate-buffered saline
- PMCA
plasma membrane Ca2+-ATPase
- SK
small-conductance Ca2+-activated potassium.
Supporting Information Available
Sequences of the peptides and GST fragments (Table 1), GST expression from the GST pull-down assay (Figure 1), residuals from the fit of dissociation of Ca2+ from CaM (Figure 2), and summary showing the effect of AC1-C1b, AC8-Nt, and AC8-C2b on the rate constants for dissociation of Ca2+ from CaM mutants. (Figure 3). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This paper published ASAP on September 21, 2012 with an error in the caption of Figure 5. The correct version was reposted on September 25, 2012.
This work was supported by Wellcome Trust Program Grant RG31760.
Footnotes
Transmembrane ACs consist of a cytosolic N-terminus followed by a six-transmembrane-spanning cassette (Tm1) and a large intracellular domain (C1a and -b). This arrangement is repeated in the second portion of the molecule, with a second six-transmembrane-spanning cassette (Tm2) followed by a large C-terminus (C2a and -b). The C1a and C2a regions are the most conserved domains between ACs and combine to form the catalytic core.56 The other regions of AC are less conserved between isoforms and are involved in subtype-specific regulation.
Supplementary Material
References
- Halls M. L.; Cooper D. M. F. (2011) Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harbor Perspect. Biol. 3, a004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masada N.; Ciruela A.; Macdougall D. A.; Cooper D. M. F. (2009) Distinct mechanisms of regulation by Ca2+/calmodulin of type 1 and 8 adenylyl cyclases support their different physiological roles. J. Biol. Chem. 284, 4451–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D.; Cooper D. M. F. (2006) Ca2+ stimulation of adenylyl cyclase generates dynamic oscillations in cyclic AMP. J. Cell Sci. 119, 828–836. [DOI] [PubMed] [Google Scholar]
- Yap K. L.; Kim J.; Truong K.; Sherman M.; Yuan T.; Ikura M. (2000) Calmodulin target database. J. Struct. Funct. Genomics 1, 8–14. [DOI] [PubMed] [Google Scholar]
- Babu Y. S.; Sack J. S.; Greenhough T. J.; Bugg C. E.; Means A. R.; Cook W. J. (1985) Three-dimensional structure of calmodulin. Nature 315, 37–40. [DOI] [PubMed] [Google Scholar]
- Geiser J. R.; van Tuinen D.; Brockerhoff S. E.; Neff M. M.; Davis T. N. (1991) Can calmodulin function without binding calcium?. Cell 65, 949–959. [DOI] [PubMed] [Google Scholar]
- Xia X. M.; Fakler B.; Rivard A.; Wayman G.; Johnson-Pais T.; Keen J. E.; Ishii T.; Hirschberg B.; Bond C. T.; Lutsenko S.; Maylie J.; Adelman J. P. (1998) Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507. [DOI] [PubMed] [Google Scholar]
- Keen J. E.; Khawaled R.; Farrens D. L.; Neelands T.; Rivard A.; Bond C. T.; Janowsky A.; Fakler B.; Adelman J. P.; Maylie J. (1999) Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. J. Neurosci. 19, 8830–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson B. Z.; DeMaria C. D.; Adelman J. P.; Yue D. T. (1999) Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22, 549–558. [DOI] [PubMed] [Google Scholar]
- Levin L. R.; Reed R. R. (1995) Identification of functional domains of adenylyl cyclase using in vivo chimeras. J. Biol. Chem. 270, 7573–7579. [DOI] [PubMed] [Google Scholar]
- Vorherr T.; Knopfel L.; Hofmann F.; Mollner S.; Pfeuffer T.; Carafoli E. (1993) The calmodulin binding domain of nitric oxide synthase and adenylyl cyclase. Biochemistry 32, 6081–6088. [DOI] [PubMed] [Google Scholar]
- Gu C.; Cooper D. M. F. (1999) Calmodulin-binding sites on adenylyl cyclase type VII. J. Biol. Chem. 274, 8012–8021. [DOI] [PubMed] [Google Scholar]
- Macdougall D. A.; Wachten S.; Ciruela A.; Sinz A.; Cooper D. M. F. (2009) Separate elements within a single IQ-like motif in adenylyl cyclase type 8 impart Ca2+/calmodulin binding and autoinhibition. J. Biol. Chem. 284, 15573–15588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson R. E.; Ciruela A.; Cooper D. M. F. (2006) The role of calmodulin recruitment in Ca2+-stimulation of adenylyl cyclase type 8. J. Biol. Chem. 281, 17379–17389. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Wong S. T.; Storm D. R. (1993) Modification of the calcium and calmodulin sensitivity of the type I adenylyl cyclase by mutagenesis of its calmodulin binding domain. J. Biol. Chem. 268, 23766–23768. [PubMed] [Google Scholar]
- Boyajian C. L.; Garritsen A.; Cooper D. M. F. (1991) Bradykinin stimulates Ca2+ mobilization in NCB-20 cells leading to direct inhibition of adenylylcyclase. A novel mechanism for inhibition of cAMP production. J. Biol. Chem. 266, 4995–5003. [PubMed] [Google Scholar]
- Salomon Y.; Londos C.; Rodbell M. (1974) A highly sensitive adenylate cyclase assay. Anal. Biochem. 58, 541–548. [DOI] [PubMed] [Google Scholar]
- Kasri N. N.; Török K.; Galione A.; Garnham C.; Callewaert G.; Missiaen L.; Parys J. B.; De Smedt H. (2006) Endogenously bound calmodulin is essential for the function of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 281, 8332–8338. [DOI] [PubMed] [Google Scholar]
- Findlay W. A.; Martin S. R.; Beckingham K.; Bayley P. M. (1995) Recovery of native structure by calcium binding site mutants of calmodulin upon binding of sk-MLCK target peptides. Biochemistry 34, 2087–2094. [DOI] [PubMed] [Google Scholar]
- Willoughby D.; Masada N.; Wachten S.; Pagano M.; Halls M. L.; Everett K. L.; Ciruela A.; Cooper D. M. F. (2010) AKAP79/150 interacts with AC8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J. Biol. Chem. 285, 20328–20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemirovskiy O. L.; Ramanathan R.; Gross M. L. (1997) Investigation of calcium-induced, noncovalent association of calmodulin with melittin by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 8, 809–812. [Google Scholar]
- Hill T. J.; Lafitte D.; Wallace J. I.; Cooper H. J.; Tsvetkov P. O.; Derrick P. J. (2000) Calmodulin-peptide interactions: Apocalmodulin binding to the myosin light chain kinase target-site. Biochemistry 39, 7284–7290. [DOI] [PubMed] [Google Scholar]
- Sinz A.; Kalkhof S.; Ihling C. (2005) Mapping protein interfaces by a trifunctional cross-linker combined with MALDI-TOF and ESI-FTICR mass spectrometry. J. Am. Soc. Mass Spectrom. 16, 1921–1931. [DOI] [PubMed] [Google Scholar]
- Pan J.; Konermann L. (2010) Calcium-induced structural transitions of the calmodulin-melittin system studied by electrospray mass spectrometry: Conformational subpopulations and metal-unsaturated intermediates. Biochemistry 49, 3477–3486. [DOI] [PubMed] [Google Scholar]
- Hu P.; Ye Q. Z.; Loo J. A. (1994) Calcium stoichiometry determination for calcium binding proteins by electrospray ionization mass spectrometry. Anal. Chem. 66, 4190–4194. [DOI] [PubMed] [Google Scholar]
- Zhu M. M.; Rempel D. L.; Du Z.; Gross M. L. (2003) Quantification of protein-ligand interactions by mass spectrometry, titration, and H/D exchange: PLIMSTEX. J. Am. Chem. Soc. 125, 5252–5253. [DOI] [PubMed] [Google Scholar]
- Nousiainen M.; Derrick P. J.; Lafitte D.; Vainiotalo P. (2003) Relative affinity constants by electrospray ionization and Fourier transform ion cyclotron resonance mass spectrometry: Calmodulin binding to peptide analogs of myosin light chain kinase. Biophys. J. 85, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milos M.; Comte M.; Schaer J. J.; Cox J. A. (1989) Evidence for four capital and six auxiliary cation-binding sites on calmodulin: Divalent cation interactions monitored by direct binding and microcalorimetry. J. Inorg. Biochem. 36, 11–25. [DOI] [PubMed] [Google Scholar]
- Shirran S. L.; Barran P. E. (2009) The use of ESI-MS to probe the binding of divalent cations to calmodulin. J. Am. Soc. Mass Spectrom. 20, 1159–1171. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Yu X.; Cui M.; Liu Z.; Song F.; Liu S. (2009) Investigation of calmodulin-peptide interactions using matrix-assisted laser desorption/ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 20, 576–583. [DOI] [PubMed] [Google Scholar]
- Maulet Y.; Cox J. A. (1983) Structural changes in melittin and calmodulin upon complex formation and their modulation by calcium. Biochemistry 22, 5680–5686. [DOI] [PubMed] [Google Scholar]
- Olwin B. B.; Storm D. R. (1985) Calcium binding to complexes of calmodulin and calmodulin binding proteins. Biochemistry 24, 8081–8086. [DOI] [PubMed] [Google Scholar]
- Yazawa M.; Vorherr T.; James P.; Carafoli E.; Yagi K. (1992) Binding of calcium by calmodulin: Influence of the calmodulin binding domain of the plasma membrane calcium pump. Biochemistry 31, 3171–3176. [DOI] [PubMed] [Google Scholar]
- Peersen O. B.; Madsen T. S.; Falke J. J. (1997) Intermolecular tuning of calmodulin by target peptides and proteins: Differential effects on Ca2+ binding and implications for kinase activation. Protein Sci. 6, 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley P.; Ahlstrom P.; Martin S. R.; Forsen S. (1984) The kinetics of calcium binding to calmodulin: Quin 2 and ANS stopped-flow fluorescence studies. Biochem. Biophys. Res. Commun. 120, 185–191. [DOI] [PubMed] [Google Scholar]
- Martin S. R.; Andersson Teleman A.; Bayley P. M.; Drakenberg T.; Forsen S. (1985) Kinetics of calcium dissociation from calmodulin and its tryptic fragments. A stopped-flow fluorescence study using Quin 2 reveals a two-domain structure. Eur. J. Biochem. 151, 543–550. [DOI] [PubMed] [Google Scholar]
- Suko J.; Pidlich J.; Bertel O. (1985) Calcium release from intact calmodulin and calmodulin fragment 78–148 measured by stopped-flow fluorescence with 2-p-toluidinylnaphthalene sulfonate. Effect of calmodulin fragments on cardiac sarcoplasmic reticulum. Eur. J. Biochem. 153, 451–457. [DOI] [PubMed] [Google Scholar]
- Tsien R. Y.; Pozzan T.; Rink T. J. (1982) Calcium homeostasis in intact lymphocytes: Cytoplasmic free calcium monitored with a new, intracellularly trapped fluorescent indicator. J. Cell Biol. 94, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast U.; Labhardt A. M.; Doyle V. M. (1984) Stopped-flow kinetics of the interaction of the fluorescent calcium indicator Quin 2 with calcium ions. Biochem. Biophys. Res. Commun. 123, 604–611. [DOI] [PubMed] [Google Scholar]
- Haiech J.; Kihoffer M. C.; Lukas T. J.; Craig T. A.; Roberts D. M.; Watterson D. M. (1991) Restoration of the calcium binding activity of mutant calmodulins toward normal by the presence of a calmodulin binding structure. J. Biol. Chem. 266, 3427–3431. [PubMed] [Google Scholar]
- Johnson J. D.; Snyder C.; Walsh M.; Flynn M. (1996) Effects of myosin light chain kinase and peptides on Ca2+ exchange with the N- and C-terminal Ca2+ binding sites of calmodulin. J. Biol. Chem. 271, 761–767. [DOI] [PubMed] [Google Scholar]
- Gaertner T. R.; Putkey J. A.; Waxham M. N. (2004) RC3/Neurogranin and Ca2+/calmodulin-dependent protein kinase II produce opposing effects on the affinity of calmodulin for calcium. J. Biol. Chem. 279, 39374–39382. [DOI] [PubMed] [Google Scholar]
- Dodd R.; Peracchia C.; Stolady D.; Török K. (2008) Calmodulin association with connexin32-derived peptides suggests trans-domain interaction in chemical gating of gap junction channels. J. Biol. Chem. 283, 26911–26920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamniuk A. P.; Vogel H. J. (2004) Calmodulin’s flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol. Biotechnol. 27, 33–57. [DOI] [PubMed] [Google Scholar]
- Drum C. L.; Yan S. Z.; Sarac R.; Mabuchi Y.; Beckingham K.; Bohm A.; Grabarek Z.; Tang W. J. (2000) An extended conformation of calmodulin induces interactions between the structural domains of adenylyl cyclase from Bacillus anthracis to promote catalysis. J. Biol. Chem. 275, 36334–36340. [DOI] [PubMed] [Google Scholar]
- Shifman J. M.; Choi M. H.; Mihalas S.; Mayo S. L.; Kennedy M. B. (2006) Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by calmodulin with two bound calciums. Proc. Natl. Acad. Sci. U.S.A. 103, 13968–13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forest A.; Swulius M. T.; Tse J. K.; Bradshaw J. M.; Gaertner T.; Waxham M. N. (2008) Role of the N- and C-lobes of calmodulin in the activation of Ca2+/calmodulin-dependent protein kinase II. Biochemistry 47, 10587–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M. J.; Putkey J. A.; Waxham M. N.; Kubota Y. (2009) Dissecting cooperative calmodulin binding to CaM kinase II: A detailed stochastic model. J. Comput. Neurosci. 27, 621–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M. A.; Rivard A. F.; Bachinger H. P.; Adelman J. P. (2001) Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 410, 1120–1124. [DOI] [PubMed] [Google Scholar]
- Persechini A.; White H. D.; Gansz K. J. (1996) Different mechanisms for Ca2+ dissociation from complexes of calmodulin with nitric oxide synthase or myosin light chain kinase. J. Biol. Chem. 271, 62–67. [DOI] [PubMed] [Google Scholar]
- Cook A. G.; Johnson L. N.; McDonnell J. M. (2005) Structural characterization of Ca2+/CaM in complex with the phosphorylase kinase PhK5 peptide. FEBS J. 272, 1511–1522. [DOI] [PubMed] [Google Scholar]
- Erickson M. G.; Alseikhan B. A.; Peterson B. Z.; Yue D. T. (2001) Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron 31, 973–985. [DOI] [PubMed] [Google Scholar]
- Tzortzopoulos A.; Best S. L.; Kalamida D.; Török K. (2004) Ca2+/calmodulin-dependent activation and inactivation mechanisms of αCaMKII and phospho-Thr286-αCaMKI. Biochemistry 43, 6270–6280. [DOI] [PubMed] [Google Scholar]
- Faas G. C.; Raghavachari S.; Lisman J. E.; Mody I. (2011) Calmodulin as a direct detector of Ca2+ signals. Nat. Neurosci. 14, 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran Q. K.; Black D. J.; Persechini A. (2003) Intracellular coupling via limiting calmodulin. J. Biol. Chem. 278, 24247–24250. [DOI] [PubMed] [Google Scholar]
- Gao Z. H.; Krebs J.; VanBerkum M. F.; Tang W. J.; Maune J. F.; Means A. R.; Stull J. T.; Beckingham K. (1993) Activation of four enzymes by two series of calmodulin mutants with point mutations in individual Ca2+ binding sites. J. Biol. Chem. 268, 20096–20104. [PubMed] [Google Scholar]
- Török K.; Tzortzopoulos A.; Grabarek Z.; Best S. L.; Thorogate R. (2001) Dual effect of ATP in the activation mechanism of brain Ca2+/calmodulin-dependent protein kinase II by Ca2+/calmodulin. Biochemistry 40, 14878–14890. [DOI] [PubMed] [Google Scholar]
- Tzortzopoulos A.; Török K. (2004) Mechanism of the T286A-mutant αCaMKII interactions with Ca2+/calmodulin and ATP. Biochemistry 43, 6404–6414. [DOI] [PubMed] [Google Scholar]
- Jama A. M.; Gabriel J.; Al-Nagar A. J.; Martin S.; Baig S. Z.; Soleymani H.; Chowdhury Z.; Beesley P.; Török K. (2011) Lobe-specific functions of Ca2+·calmodulin in αCa2+·calmodulin-dependent protein kinase II activation. J. Biol. Chem. 286, 12308–12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M.; Coletto L.; Pierobon N.; Kraev N.; Guerini D.; Carafoli E. (2003) A comparative functional analysis of plasma membrane Ca2+ pump isoforms in intact cells. J. Biol. Chem. 278, 24500–24508. [DOI] [PubMed] [Google Scholar]
- Penheiter A. R.; Bajzer Z.; Filoteo A. G.; Thorogate R.; Török K.; Caride A. J. (2003) A model for the activation of plasma membrane calcium pump isoform 4b by calmodulin. Biochemistry 42, 12115–12124. [DOI] [PubMed] [Google Scholar]
- Caride A. J.; Elwess N. L.; Verma A. K.; Filoteo A. G.; Enyedi A.; Bajzer Z.; Penniston J. T. (1999) The rate of activation by calmodulin of isoform 4 of the plasma membrane Ca2+ pump is slow and is changed by alternative splicing. J. Biol. Chem. 274, 35227–35232. [DOI] [PubMed] [Google Scholar]
- Caride A. J.; Filoteo A. G.; Penniston J. T.; Strehler E. E. (2007) The plasma membrane Ca2+ pump isoform 4a differs from isoform 4b in the mechanism of calmodulin binding and activation kinetics: Implications for Ca2+ signaling. J. Biol. Chem. 282, 25640–25648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D. J.; Persechini A. (2011) In calmodulin-IQ domain complexes, the Ca2+-free and Ca2+-bound forms of the calmodulin C-lobe direct the N-lobe to different binding sites. Biochemistry 50, 10061–10068. [DOI] [PubMed] [Google Scholar]
- Wong S. T.; Athos J.; Figueroa X. A.; Pineda V. V.; Schaefer M. L.; Chavkin C. C.; Muglia L. J.; Storm D. R. (1999) Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23, 787–798. [DOI] [PubMed] [Google Scholar]
- Sadana R.; Dessauer C. W. (2009) Physiological roles for G protein-regulated adenylyl cyclase isoforms: Insights from knockout and overexpression studies. Neurosignals 17, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.