

Abstract
MtATP-phosphoribosyltransferase catalyzes the first and committed step in l-histidine biosynthesis in Mycobacterium tuberculosis and is therefore subjected to allosteric feedback regulation. Because of its essentiality, this enzyme is being studied as a potential target for novel anti-infectives. To understand the basis for its regulation, we characterized the allosteric inhibition using gel filtration, steady-state and pre-steady-state kinetics, and the pH dependence of inhibition and binding. Gel filtration experiments indicate that MtATP-phosphoribosyltransferase is a hexamer in solution, in the presence or absence of l-histidine. Steady-state kinetic studies demonstrate that l-histidine inhibition is uncompetitive versus ATP and noncompetitive versus PRPP. At pH values close to neutrality, a Kii value of 4 μM was obtained for l-histidine. Pre-steady-state kinetic experiments indicate that chemistry is not rate-limiting for the overall reaction and that l-histidine inhibition is caused by trapping the enzyme in an inactive conformation. The pH dependence of binding, obtained by nuclear magnetic resonance, indicates that l-histidine binds better as the neutral α-amino group. The pH dependence of inhibition (Kii), on the contrary, indicates that l-histidine better inhibits MtATP-phosphoribosytransferase with a neutral imidazole and an ionized α-amino group. These results are combined into a model that accounts for the allosteric inhibition of MtATP-phosphoribosyltransferase.
Feedback allosteric inhibition of metabolic enzymes is one of the most efficient mechanisms maintained through evolution for control of flux through biochemical pathways.1−3 Efficiency comes from a direct, specific, and nearly instantaneous effect, which permits dynamic control of the flux through biochemical pathways. In addition, feedback inhibition is independent of transcription, translation, and intricate signal transduction cascades.
Natural feedback inhibitors can also serve as templates for chemically diverse, allosteric small molecule inhibitors, which are currently intensely sought as probes for chemical biology and chemical genetics, as regulators for synthetic biology, and as therapeutics for several diseases. In addition to the qualities described above, allosteric inhibitors might display other appealing features as small molecule therapeutics. For instance, allosteric inhibitors have the potential to be noncompetitive or uncompetitive versus a particular substrate4 and time-dependent5,6 and to target sites in the enzyme other than the active site. For these reasons, allosteric inhibitors that target the feedback site might become compounds of choice when the active site of an enzyme is deemed “not targetable” or of poor “ligandability”.7,8 A few examples of important enzymes that have been successfully targeted at an allosteric site are pyruvate kinase (as a potential antihyperglycemic therapy),9 mitotic kinesin (involved in centrosome separation and formation of the bipolar mitotic spindle),10Staphylococcus aureusd-alanine:d-alanine ligase (involved in peptidoglycan biosynthesis),11 the Cdc34 ubiquitin-conjugatin enzyme (active against certain prostate and colorectal cancer cell lines),12 and the Haemophilus influenzae HslUV protease (a bacterial version of the proteosome).13
A remarkable example of an allosterically regulated enzyme is ATP phosphoribosyltransferase (ATP-PRT), encoded by the hisG gene in Mycobacterium tuberculosis. MtATP-PRT catalyzes the first committed step in l-histidine biosynthesis (Scheme 1) and is therefore inhibited by the end product of the pathway, l-histidine.14,15 In M. tuberculosis, the hisG gene is predicted to share an operon with the next enzyme on the pathway, encoded by the hisE gene. MtATP-PRT has been proposed as a potential target for the development of novel anti-TB agents.16,17 Target validation, i.e., the essentiality of the reaction and the pathway for M. tuberculosis, has been confirmed by high-resolution transposon mutagenesis18 and clean genetic knockouts of other enzymes in the pathway.19 In addition, this pathway is absent in humans, which suggests no a priori toxicity associated with potential inhibitors.
Scheme 1.

ATP-PRTs (EC 2.4.2.17) catalyze the reversible, Mg2+-dependent formation of 1-(5-phospho-α-d-ribosyl)-ATP (PR-ATP) and inorganic pyrophosphate (PPi) from ATP and 5-phospho-α-d-ribosyl pyrophosphate (PRPP) (Scheme 2). Some ATP-PRTs are also activated by K+.20 This reaction was shown to proceed directly, without the generation of a covalent intermediate,21 and to follow a sequential ordered kinetic mechanism with ATP as the first substrate to bind and PR-ATP the last product to dissociate.22 Interestingly, ATP-PRT and anthranilate-PRT are the only phosphoribosyltransferases (PRTs) described to this day that are not involved in nucleoside or NAD+/NADP+ metabolism. Common ribosyl acceptors used by other PRTs are orotate, nicotinamide/nicotinate, adenine, and hypoxanthine/guanine.
Scheme 2.

With regard to tertiary and quaternary structure, there are two types of ATP-PRTs. They are the more widespread homohexameric form, encountered in bacteria, fungi, and plants, and the hetero-octameric form, restricted to some bacteria. MtATP-PRT belongs to the homohexameric class,16 which is also found in Salmonella enterica and Escherichia coli.23,24 Crystallographic data suggested that MtATP-PRT is an active dimer that associates into an inactive hexamer, upon l-histidine binding.16 In addition, the l-histidine-bound hexameric form displays a major conformational change, a twist of domain III relative to domains I and II. Crystallographic studies also revealed that l-histidine binds at a site ∼30 Å from the active site, confirming that inhibition is allosteric in nature. Finally, it was shown that l-histidine is held at the allosteric site by interactions with the carboxyl group from Asp218, the hydroxyl from Thr238, and the backbone amide oxygen from Ala273.16
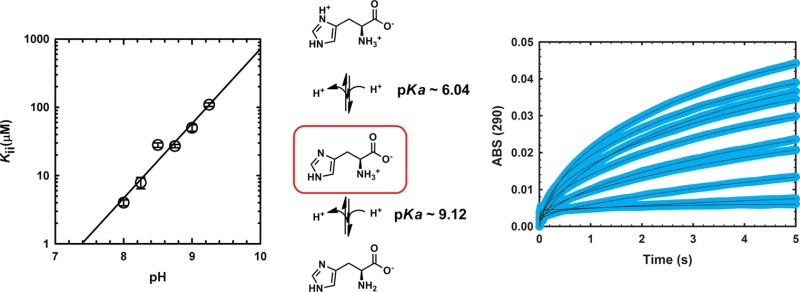
Because l-histidine’s imidazole group displays a pKa near neutrality (∼6.0), it is of interest to define which ionic form binds to and inhibits MtATP-PRT. This question has been partially answered for the homologous enzymes from S. enterica (SeATP-PRT) and Corynebacterium glutamicum (CgATP-PRT).23,25 With SeATP-PRT, l-histidine was shown to inhibit better at higher pH values, indicating that the imidazole ring is deprotonated. In contrast, with CgATP-PRT, lower pH values increased potency, suggesting that a protonated imidazole form inhibits better. Unfortunately, both studies used “IC50” or “% activity” as surrogates of affinity, instead of Ki or Kd values, making the interpretation of the results difficult. Currently, no information about the ionic state of l-histidine inhibition of MtATP-PRT is available.
In this work, we describe a detailed characterization of the mechanism of allosteric inhibition of MtATP-PRT by l-histidine. We use a combination of steady-state and pre-steady-state kinetics, pH studies, 1H NMR spectroscopy, and gel filtration experiments to characterize how l-histidine allosterically controls MtATP-PRT. Our results are combined with the structural data available to generate a new model that accounts for the inhibition.
Materials and Methods
Materials
Buffers were purchased from Fisher Scientific. Unless otherwise stated, all other chemicals were purchased from Sigma-Aldrich. Baker’s yeast inorganic pyrophosphatase was purchased from Sigma-Aldrich. M. tuberculosis inorganic pyrophosphatase (Rv3628) was produced in the laboratory (L. P. Carvalho, unpublished results). Chromatographic columns were purchased from GE, and Ni-NTA resin was purchased from EMD. Complete EDTA-Free protease inhibitor was purchased from Roche. BL21(DE3)pLysS was from EMD.
General Methods and Equipment
The protein concentration was measured using the BCA assay from Pierce, using bovine serum albumin as a standard. Protein purification was performed using an AKTA purifier 10 (GE Healthcare). SDS–PAGE was performed on a PhastSystem (GE Healthcare). Spectrophotometry was conducted with a Shimadzu UV-2550 spectrophotometer equipped with dual-beam optics and a Peltier system for temperature control. Stopped-flow absorbance spectroscopy was conducted on an Applied Photophysics SX-20 stopped-flow spectrophotometer, equipped with a circulating water bath. Under these conditions, the dead time was estimated to be 3 ms. Experiments were conducted by mixing equal volumes of two solutions, one containing enzyme and the other the variable substrate with or without l-histidine. NMR experiments were conducted using a Varian Inova spectrometer at 14.4 T. All assays were conducted at 25 °C. All concentrations of MtATP-PRT reported are the final concentrations used, for the monomer.
Plasmid Preparation, Protein Expression, and Purification
The Rv2121c gene sequence from M. tuberculosis H37Rv was codon adapted to E. coli, and its nucleotide sequence was synthetically prepared and ligated into the pJ411 plasmid (DNA 2.0). DNA sequence was confirmed by sequencing. This construct contained a noncleavable N-terminal hexahistidine tag to facilitate purification. The N-terminal hexahistidine tag was shown not to affect the structure or activity of MtATP-PRT.16
During MtATP-PRT purification, all steps were performed at 4 °C. Frozen BL21(DE3)pLysS (pJ411::hisG) cells were thawed on ice, and lysed by sonication, in the presence of buffer A [20 mM triethanolamine (TEA) (pH 7.8), 300 mM NaCl, and 50 mM imidazole] containing lysozyme and Complete EDTA-free protease inhibitor cocktail. Soluble protein was obtained by centrifugation at 25000g for 30 min. The soluble fraction was loaded into a 50 mL Ni-NTA column and the protein separated by a gradient using buffer B [20 mM TEA (pH 7.8), 300 mM NaCl, and 500 mM imidazole]. Peak fractions were analyzed by SDS–PAGE. Fractions containing only MtATP-PRT were pooled together, concentrated, dialyzed in 20 mM TEA (pH 7.8), and stored in 50% glycerol at −20 °C or, alternatively, stored at −80 °C after being flash-frozen in liquid nitrogen. This protocol allowed the purification to homogeneity of 114 mg of MtATP-PRT from 15 g of wet cell pellet. Purified ATP-PRT was analyzed by electrospray ionization mass spectrometry (ESI-MS) to confirm protein identity (NIMR Mass Spectrometry Facility).
For pre-steady-state kinetic experiments, in which larger amounts of inorganic pyrophosphatase were required, we used recombinant inorganic pyrophosphatase from M. tuberculosis produced in our laboratory (L. P. Carvalho, unpublished results).
Gel Filtration
The solution oligomeric state of MtATP-PRT was determined using a Sephacryl S-200 column and molecular weight standards (Bio-Rad). Typically, 100 μL of MtATP-PRT at concentrations ranging from 0.1 to 10 mg/mL was injected. Protein was eluted isocratically for 1.5 column volumes, at a rate of 0.5 mL/min. The buffer was 20 mM Tris-HCl (pH 8) containing 100 mM NaCl, with or without 2 mM l-histidine.
Measurement of Enzymatic Activity
Initial velocities for the forward reaction of MtATP-PRT were measured by following the formation of PR-ATP (ε290 = 3600 M–1 cm–1),26 in the presence of inorganic pyrophosphatase. Pyrophosphatase is essential for this assay, as the equilibrium constant lies toward formation of ATP and PRPP. A typical reaction mixture contained 50 mM Tris-HCl (pH 8.5), 7 mM MgCl2, 200 mM KCl, 800 μM ATP, 400 μM PRPP, 3 milliunits of pyrophosphatase, and 150 nM MtATP-PRT. All activity assays were performed at 25 ± 0.2 °C.
1H NMR Spectroscopy
One-dimensional spectra of l-histidine as a function of pH were recorded using excitation sculpting27 for water suppression. STD spectra were recorded in an interleaved manner with a range of saturation times (0.2–2.0 s in 0.2 s intervals) with on-resonance presaturation of the enzyme at 0.8 ppm. A single off-resonance control spectrum was recorded with a saturation time of 2 s. Water suppression was achieved by excitation sculpting. Data processing, spectral subtractions, and quantitation were performed using VNMRJ 1.1D. The physical basis for saturation transfer difference NMR is described elsewhere.28,29
Inhibition Studies
To determine inhibition constants and inhibition patterns associated with l-histidine, MtATP-PRT activity was studied in the presence of variable concentrations of one substrate, fixed saturating concentrations of the other substrate and metals, and several fixed concentrations of l-histidine.
pH Studies
To determine the effect of pH on the inhibitory activity of l-histidine, MtATP-PRT activity was studied in the presence of variable concentrations of ATP, fixed saturating concentrations of PRPP and metals, and several fixed concentrations of l-histidine, at different pHs ranging from 6.5 to 9.5 (in HEPES, PIPES, TAPS, and CHES buffers). Because of the narrow pH range of MtATP-PRT, only the inhibition data from pH 8 to 9.25 were analyzed (TAPS from pH 8.0 to 8.75 and CHES from pH 9.0 to 9.25). Between pH 8 and 9.25, PRPP concentrations were tested and shown to be saturating.
Pre-Steady-State Kinetics
Photomultiplier voltage was zeroed with buffer. A cell with a 10 mm path length was used for all experiments. All experiments were performed at 25 °C in 50 mM Tris-HCl (pH 8.5) containing 150 mM KCl, 10 mM MgCl2, 1.5 mM PRPP, 60 μM pyrophosphatase, and 10 μM MtATP-PRT. Higher concentrations of substrates and metals were not used, because of precipitation. Typically, 10 progress curves with 1000 points each were averaged before data were fit. Rates and amplitudes were obtained by fitting the average to the burst equation (eq 7). Mixing was achieved by rapid combination of 60 μL from each syringe. The effect of increasing concentrations of ATP (from 0.025 to 4 mM) and l-histidine (from 5 to 80 μM) on the rate of the reaction was measured by monitoring the conversion of ATP to PR-ATP, by observing the increase in absorbance at 290 nm.
Data Analysis
Steady-state and NMR data were fit using the nonlinear, least-squares, curve-fitting programs of SigmaPlot for Windows, version 11.0. Pre-steady-state data were fit using KinTek Global Kinetic Explorer,30,31 version 3.0. Individual saturation curves were fit to eq 1
| 1 |
where V is the maximal velocity, A is the substrate or metal concentration, and K is the Michaelis constant for the substrate (Km) or constant for the metal (Kact). Individual saturation curves showing substrate inhibition pattern were fit to eq 2
| 2 |
where Kiapp is the apparent inhibition constant for A. Inhibition data, recorded under saturating concentrations of ATP, PRPP, Mg2+, and K+ and variable concentration of l-histidine, were fit to eq 3
| 3 |
where v0 is the uninhibited velocity, I is the l-histidine concentration, IC50 is the concentration of l-histidine necessary to give 50% inhibition, and nH is the Hill number. Inhibition data showing linear, noncompetitive, or uncompetitive patterns in double-reciprocal plots were fit to eqs 4 and 5
| 4 |
| 5 |
where Kis and Kii are the slope and intercept inhibition constants, respectively. NMR chemical shift data showing a single titration event were fit to a variation of the Henderson–Hasselbalch equation32
| 6 |
where δ is the instantaneous chemical shift in parts per million, δacid is the chemical shift of the protonated form, δbase is the chemical shift of the deprotonated form, pH is the antilogarithm of the concentration of protons, and pKa is the acid dissociation constant for the group being titrated. Pre-steady-state burst kinetic data were fit to eq 7
| 7 |
where Yt is the observed signal at time t, A is the amplitude of the transient, kobs is the observed burst rate for the transient, v is the linear rate, and C is the offset of the linear phase. The dependence of the burst amplitude on ATP concentration under conditions where substrate and enzyme concentration were comparable is described by eq 8(33)
| 8 |
where Amax is the amplitude maximum, E is the concentration of the enzyme, S is the total concentration of ATP, and Kdapp is the apparent dissociation constant for ATP. Similarly, the dependence of the kobs on ATP concentration under conditions where substrate and enzyme concentrations are comparable was fit to eq 9
| 9 |
where kburst is the maximal burst rate. The dependence of the burst amplitude on l-histidine concentration was fit to
| 10 |
where A0 is the maximal amplitude (in the absence of l-histidine) and [I] is the concentration of l-histidine. The dependence of the kobs on the concentration of l-histidine was described by a one-step binding mechanism, according to eq 11
| 11 |
where kon is the second-order association rate constant and koff is the unimolecular dissociation rate constant.
Results
MtATP-PRT Purification
The hisG (rv2121) gene sequence from M. tuberculosis H37Rv was codon-adapted to E. coli and then chemically synthesized and ligated into pJ411, and the nucleotide sequence was confirmed (DNA 2.0); 7.6 mg of MtATP-PRT were purified per gram of BL21(DE3)pLysS (pJ411::hisG), using a single Ni-NTA chromatographic step. Purified MtATP-PRT was subjected to ESI-MS for identification. The purified protein exhibited a molecular mass of 31,515.1 Da, which is within experimental error of the calculated molecular mass of 31515.6 Da. This result confirms the identity of the enzyme used in the following experiments.
Oligomeric State of MtATP-PRT
We next determined the apparent solution molecular mass of MtATP-PRT using gel filtration, on a Sephadex S200 column. Figure 1 depicts representative chromatograms obtained, showing that the mycobacterial enzyme behaves as a single species in solution, with an apparent molecular mass of ∼180 kDa. This molecular mass is consistent with a hexameric form. The repetition of this experiment at lower concentrations of protein yields the same result, indicating that MtATP-PRT does not display a concentration-dependent change in the oligomeric state at protein concentrations ranging from 0.1 to 10 mg/mL. Repetition of this experiment in the presence of 2 mM l-histidine (Figure 1) showed the same overall result. Repetition of the experiment in the presence of ATP (the first substrate to bind to ATP-PRTs) did not yield an observable change in the apparent molecular mass (data not shown). These results indicate that MtATP-PRT is a hexamer in solution and this oligomeric state is not influenced by the allosteric inhibitor or by ATP, the first substrate.
Figure 1.

MtATP-PRT is hexameric in solution in the presence and absence of l-histidine. Gel filtration experiment illustrating the lack of change in the oligomeric state of MtATP-PRT. Solid and dashed lines are representative chromatographic traces of MtATP-PRT (100 μg) analyzed with a Sephacryl S200 in the absence and presence of 2 mM l-histidine, respectively. These experiments were performed in 20 mM Tris-HCl (pH 8) containing 100 mM NaCl. Results are representative of four independent experiments.
Steady-State Kinetics and Inhibition Studies
MtATP-PRT displays Michaelis–Menten kinetics with both substrates (Figure 2). Time courses are only linear for a few minutes (data not shown), which is consistent with previous observations that the product, PR-ATP, is a potent inhibitor of ATP-PRTs.34 Interestingly, MtATP-PRT displays very low activity at neutral and acidic pH. Therefore, the lowest pH used in our kinetic studies was 8.0. No activity is observed in the presence of metal-chelating agent EDTA, indicating that Mg2+ is essential for activity and is rapidly dissociating from the enzyme (data not shown). Addition of the thiol reducing agent DTT did not alter the time course (data not shown), indicating that this enzyme preparation did not contain oxidized cysteine residues or disulfide bonds that could inhibit activity.
Figure 2.

Steady-state kinetics of MtATP-PRT and its inhibition by l-histidine. The top and middle panels show the saturation curves for ATP (●) and PRPP (○), respectively. These experiments were performed at 25 °C, in 50 mM Tris-HCl (pH 8.5) containing 7 mM MgCl2 and 200 mM KCl. When the ATP concentration was varied, the PRPP concentration was 0.8 mM. When the PRPP concentration was varied, the ATP concentration was 1.6 mM. The bottom panel shows the inhibition (at a single substrate concentration) of MtATP-PRT by l-histidine (●). This experiment was performed at 25 °C, in 50 mM Tris-HCl (pH 8.5) containing 7 mM MgCl2, 200 mM KCl, 1.6 mM ATP, and 0.8 mM PRPP. Points are data and lines best fit to eq 1 (top and middle panels) and eq 2 (bottom panel). Results are representative of two independent experiments.
Apparent Km values were obtained for ATP (263 ± 63 μM) in the presence of saturating concentrations of PRPP and MgCl2 and 200 mM KCl (Figure 2, top panel) and for PRPP (49 ± 6 μM) in the presence of saturating levels of ATP, MgCl2, and KCl (Figure 2, middle panel). As observed with SeATP-PRT,22 high concentrations of MgCl2 led to inhibition of MtATP-PRT. Apparent Kact and Ki values were obtained for MgCl2 (1.9 ± 0.6 and 23.3 ± 8.6 mM, respectively) in the presence of saturating concentrations of both substrates and 200 mM KCl (data not shown). The effect of K+ on the rate of MtATP-PRT was not saturable; therefore, we performed all the experiments at 200 mM KCl (data not shown). Steady-state data are summarized in Table 1.
Table 1. Steady-State and Pre-Steady-State Kinetic Parameters for MtATP-PRTa.
| parameter | best fit | equation |
|---|---|---|
| Vmax (s–1) | 0.31 ± 0.01 | 1 |
| Km,ATP (μM) | 263 ± 63 | 1 |
| Km,PRPP (μM) | 49 ± 6 | 1 |
| Kact,Mg2+ (μM) | 1900 ± 600 | 2 |
| Ki,Mg2+ (μM) | 23000 ± 900 | 2 |
| IC50,l-histidine (μM) | 33 ± 3 | 3 |
| Amax (μM) | 7.8 ± 0.3 | 8 |
| Kd,ATPapp (μM) | 47.2 ± 10.3 | 8 |
| Kburst (s–1) | 0.67 ± 0.02 | 9 |
| Kd,l-histidine (μM) | 27.9 ± 3.2 | 10 |
| kon,l-histidine(M–1 s–1) | 4885 ± 717 | 11 |
| koff,l-histidine (s–1) | 0.95 ± 0.03 | 11 |
Assays performed in 50 mM buffer, containing MgCl2 and KCl, at the specified values, and 25 °C. See Materials and Methods for the buffers and concentrations of substrates, metals, and l-histidine.
We then confirmed the inhibition of MtATP-PRT by l-histidine by determining its IC50. As can be seen in Figure 2 (bottom panel), our preparation of MtATP-PRT was readily inhibited by l-histidine. Under our experimental conditions, the IC50 obtained for l-histidine is 33.3 ± 3.5 μM and the Hill number is 1.5 (Table 1). To obtain inhibition constants (Ki), which are independent of the concentration of enzyme, substrates, and metal ions used,35 and to define the nature of inhibition, we performed single inhibition studies at a fixed saturating concentration of one substrate and fixed variable concentrations of the second substrate. Inhibition of l-histidine versus PRPP was linear and noncompetitive (Figure 3), with a set of lines intersecting on the left of the Y-axis in a double-reciprocal plot, with a Kii value of 23.5 ± 6.5 μM and a Kis value of 25.7 ± 12.8 μM. As the Kii and Kis values are identical within experimental error, l-histidine appears to have the same affinity for the enzyme in the presence and absence of PRPP. When the ATP concentration was varied, l-histidine inhibition was linear and uncompetitive (Figure 4), with a set of parallel lines in a double-reciprocal plot, with a Kii value of 27.9 ± 1.9 μM. This result indicates that binding of ATP is essential for inhibition by l-histidine. Inhibition data are summarized in Table 2.
Figure 3.

Inhibition of MtATP-PRT by l-histidine vs PRPP. (A) Double-reciprocal plot illustrating the linear noncompetitive inhibition pattern obtained when varying the concentration of l-histidine at fixed variable concentrations of PRPP and fixed saturating concentrations of ATP, Mg2+, and K+. Data were obtained with 50 mM Tris-HCl (pH 8.5) at 25 °C. Results are representative of two independent experiments. Points are data obtained with 0 (●), 15 (○), 30 (■), and 45 μM l-histidine (□), and error bars indicate the standard deviation. Lines are the best fit of the entire data set to eq 3. (B) Replot of the intercept of the data shown in panel A (top), showing the linear dependence on inhibitor concentration. Replot of the slope of the data shown in panel A (bottom), showing the linear dependence on inhibitor concentration. Points are data, and the line is the linear regression of the data.
Figure 4.

Inhibition of MtATP-PRT by l-histidine vs ATP. (A) Double-reciprocal plot illustrating the linear uncompetitive inhibition pattern obtained when varying the concentration of l-histidine at fixed variable concentrations of ATP and fixed saturating concentrations of PRPP, Mg2+, and K+. Data were obtained with 50 mM Tris-HCl (pH 8.5) at 25 °C. This result is representative of two independent experiments. Points are data obtained with 0 (●), 15 (○), 30 (■), and 45 μM l-histidine (□), and error bars indicate the standard deviation. Lines are the best fit of the entire data set to eq 4. (B) Replot of the intercept of the data shown in panel A, showing the linear dependence on inhibitor concentration. Points are data, and the line is the linear regression of the data.
Table 2. Steady-State Inhibition Constants for MtATP-PRT and l-Histidinea.
| pH | varied substrateb | inhibition patternc | Kii (μM) | Kis (μM) |
|---|---|---|---|---|
| 8.50 | PRPP | NC | 23.5 ± 6.5 | 25.7 ± 12.8 |
| 8.50 | ATP | UC | 27.9 ± 1.9 | – |
| 8.00 | ATP | UC | 4.0 ± 0.4 | – |
| 8.25 | ATP | UC | 7.9 ± 1.5 | – |
| 8.50 | ATP | UC | 28.3 ± 2.1 | – |
| 8.75 | ATP | UC | 26.8 ± 1.5 | – |
| 9.00 | ATP | UC | 50.4 ± 5.5 | – |
| 9.25 | ATP | UC | 109.4 ± 6.8 | – |
Assays performed in 50 mM buffer containing 7 mM MgCl2 and 200 mM KCl, at the specified pH values, and 25 °C. See Materials and Methods for the buffers for each pH.
The concentrations of cosubstrates were held fixed (0.8 mM PRPP and 1.6 mM ATP).
NC, noncompetitive; UC, uncompetitive.
Ionic States of l-Histidine and Interaction with MtATP-PRT
To define the ionic state of l-histidine bound to MtATP-PRT, we first employed equilibrium fluorescence measurements. Unfortunately, binding of l-histidine only modestly alters the intrinsic tryptophan fluorescence spectrum of MtATP-PRT (data not shown), precluding quantitative analysis. As an alternative to fluorescence, we employed 1H NMR to characterize this interaction. We reasoned that STD-NMRa could constitute a simple and efficient way of monitoring ligand binding at different pH values. Before the STD experiments, we recorded spectra of l-histidine as a function of pH to determine the ionization states under our experimental conditions and the pH-dependent changes in the chemical shifts of the four sets of protons from l-histidine (Figure 5A). The chemical shift of these protons is influenced by pH as shown in Figure 5B. Taken together, these results indicate that under these experimental conditions protons Cδ2-H, Cε1-H, and Cα-H are coupled to and therefore report on the ionization of the imidazole ring. On the other hand, protons Cβ-H2 inform on the ionization of the α-amino group. Data shown in Figure 5C and Table 3 indicate that l-histidine bound to MtATP-PRT is subjected to saturation transfer, and as expected, this process is influenced by pH. Specifically, saturation transfer is neglible at lower pH values (<7.5) and maximal at high pH values (∼9), indicating that a neutral imidazole and deprotonated α-amino group favor binding. The aliphatic protons do not appear to pick up saturation (Table 3), indicating a looser contact with the protein, compared with that of the imidazole protons.
Figure 5.

Ionic states and STD-NMR spectroscopy studies of l-histidine binding to MtATP-PRT. (A) 1H NMR spectrum of l-histidine at 10 mM, in 50 mM KPi (pH 8). The asterisk indicates the water signal. (B) Chemical shift dependence of pH for l-histidine. The top panel shows data for ring protons, Cδ2-H (○) and Cε1-H (●). The bottom panel shows data for aliphatic protons, Cα-H (□) and Cβ-H (■). Experiments were performed in 50 mM sodium phosphate buffer at the indicated pH values, in the presence 0.4 mM l-histidine. Symbols are data, and solid lines are the fits to eq 5. (C) pH dependence of STD-NMR of l-histidine with 30 μM MtATP-PRT. Aromatic resonances (Cδ2 and Cε1) are shown.
Table 3. Saturation Transfer Data for l-Histidine and MtATP-PRTa.
| % saturation
transfer to l-histidine protons |
||||
|---|---|---|---|---|
| pH | Cα-H | Cβ-H2 | Cδ2-H | Cε1-H |
| 5.5 | 0 | 0 | 0 | 0 |
| 6.0 | 0 | 0 | 0 | 0 |
| 6.5 | 0 | 0 | 0 | 0 |
| 7.5 | 0 | 0 | 0 | 0 |
| 8.0 | 0 | 0 | 36.9 | 40.4 |
| 8.5 | 0 | 0 | 81.6 | 88.2 |
| 9.0 | 0 | 0 | 100 | 100 |
Assays performed in 50 mM sodium phosphate buffer at the specified pH values at 25 °C, containing 0.6 mM l-histidine and 30 μM HisG.
pH–Rate Studies
To investigate the effect of pH on the allosteric inhibition, we determined the Kii values for l-histidine, varying the ATP concentration at a fixed saturating concentration of PRPP, at various pH values. As one can see in Figure 6 and Table 2, l-histidine inhibition is dependent on pH, but surprisingly, the pH dependence of inhibition is the opposite of what was observed for binding (inhibition is weaker at higher pH values). In the pH range of 8–9.25, the group being titrated is the α-amino group of l-histidine, and therefore, we are most likely observing a direct effect of its protonation state on inhibition. Unfortunately, because of the low activity of MtATP-PRT at neutral and acidic pH values, we could not determine any Kii values below pH 8.0. Also, no pKa values could be derived from the analysis, even though a slope of 1 was obtained. This result suggests that deprotonation of the α-amino group decreases inhibition potency.
Figure 6.

pH dependence of MtATP-PRT inhibition by l-histidine. Kii values obtained by varying the l-histidine concentration at various fixed concentrations of ATP and fixed saturating concentrations of PRPP, MgCl2, and KCl. All experiments were performed at 25 °C. Symbols are values obtained at the specified pH values by fitting data to eq 4; error bars represent the standard error obtained from the fit to eq 4, and the solid line is the linear regression of the data.
Pre-Steady-State Inhibition Studies
To investigate which elementary steps of the catalytic cycle are directly affected by l-histidine, we studied inhibition of MtATP-PRT using pre-steady-state kinetics (Table 1). First, we characterized the kinetics in the absence of inhibitor, to determine whether chemistry is rate-limiting. As one can see in Figure 7, under multiple-turnover conditions the progress curve of MtATP-PRT displays burst kinetics. It is described by a single-exponential increase followed by a steady-state phase (eq 7). Burst kinetics is diagnostic of chemistry being faster than subsequent steps.36,37 In this particular case, product (PR-ATP) formation is directly observed, and therefore, a buildup of product indicates slower steps after the chemical step. Burst kinetics was studied by varying the concentration of ATP. As one can see in Figure 8, there is a quadratic dependence of the burst amplitude on ATP concentration under these experimental conditions (Figure 8B, top panel) with an amplitude maximum of 7.8 ± 0.3 μM (Table 3). This value is very close to the actual concentration of active sites used (10 μM), suggesting that most MtATP-PRT is active during catalysis.b The observed dependence of kobs on ATP concentration could also be fit by a quadratic equation (Figure 8B, bottom panel), giving a maximal kburst of 0.67 ± 0.02 s–1 (Table 1). It is worth mentioning that, under these conditions, the Y intercept for the plot of burst dependence on ATP concentration is zero, indicating very slow ATP dissociation.
Figure 7.

Stopped-flow absorbance time course of the MtATP-PRT reaction. Representative trace of the MtATP-PRT progress curve (10 μM enzyme), under multiple-turnover conditions, obtained at saturating concentrations of ATP and PRPP, in 50 mM Tris-HCl (pH 8.5) at 25 °C. The gray line shows data, and the black line represents the best fit to the burst equation (eq 7). The inset shows the residuals from the fit to the burst equation.
Figure 8.
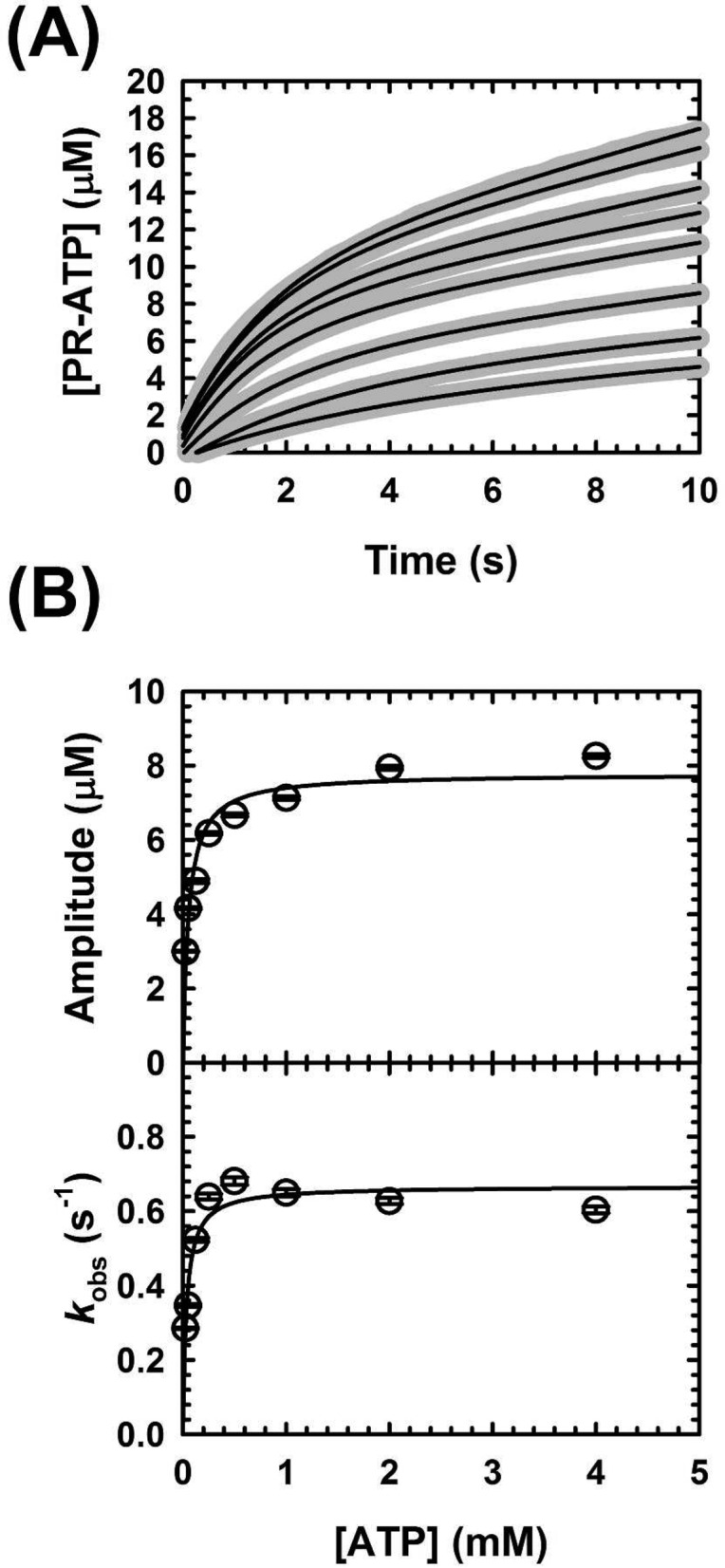
Pre-steady-state kinetics of MtATP-PRT with a varying ATP concentration. (A) Representative traces of the MtATP-PRT reaction under multiple-turnover conditions obtained at different concentrations of ATP and fixed saturating concentrations of PRPP and metals. Gray thick lines represent data and black lines the best fit to eq 6. Data are averages of 10 traces. (B) Plot of the burst amplitude vs ATP concentration (top) and plot of kobs vs ATP concentration (bottom). Symbols represent data and solid lines the best fit to eqs 8 and 9 for burst amplitude and burst rate dependencies, respectively. Reactions were conducted in 50 mM Tris-HCl (pH 8.5), 150 mM KCl, 10 mM MgCl2, 1.5 mM PRPP, and 10 μM MtATP-PRT, at 25 °C. Results are representative of two independent experiments.
We next probed the effect of l-histidine, at a fixed saturating ATP concentration. Two different effects were observed under multiple-turnover conditions. A marked effect on burst amplitude is apparent with increasing concentrations of l-histidine (Figure 9A,B). A decrease in the burst amplitude indicates that l-histidine affects MtATP-PRT largely by trapping it in an inactive complex. Slower product release or chemical steps would be characterized by a decrease in the steady-state rate or the burst rate, respectively. A Kd value of 27.9 ± 3.2 μM was obtained by fitting the data to eq 10, which is in good agreement with the Ki value obtained by steady-state kinetics. In addition, a linear decrease in kobs is observed (Figure 9C,D).
Figure 9.
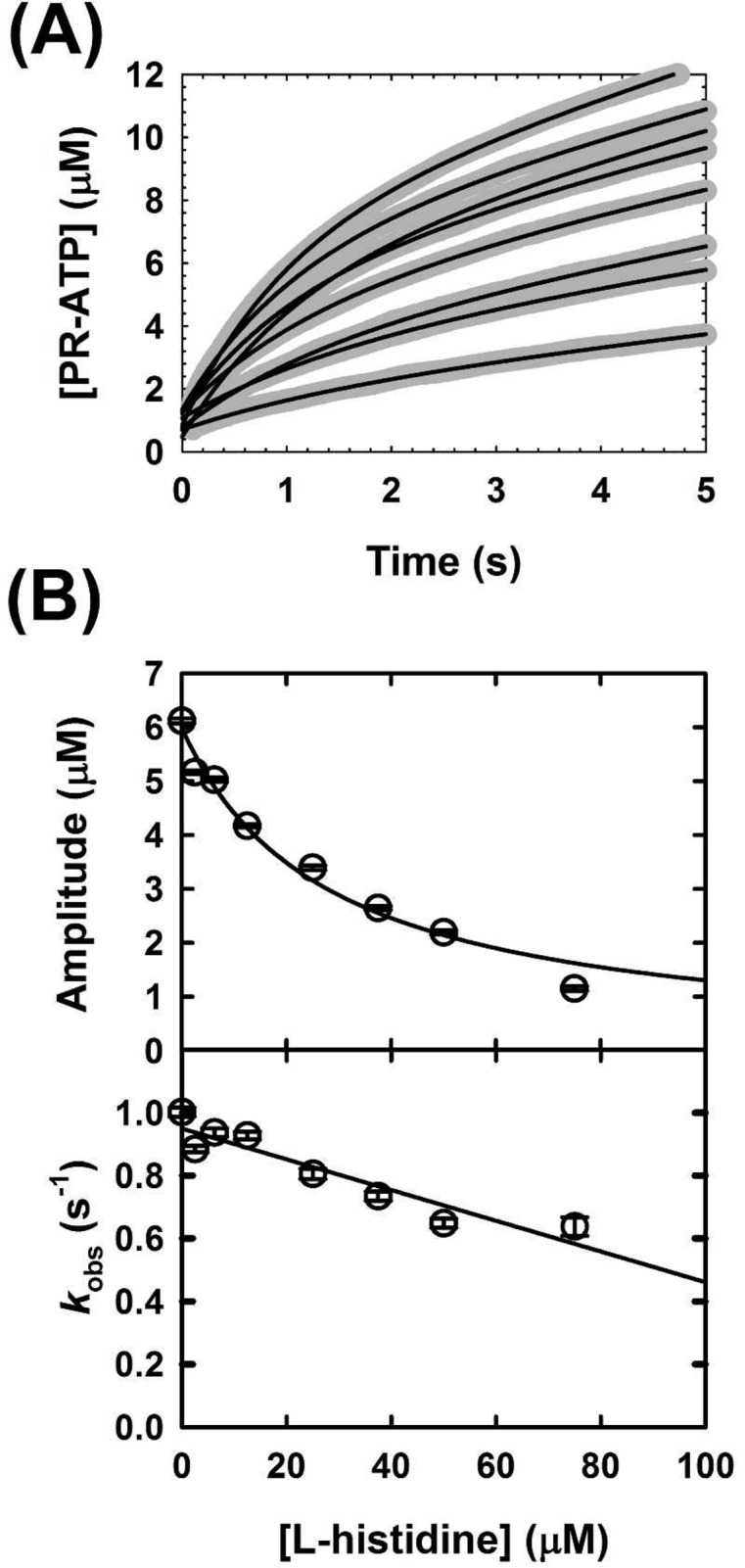
Pre-steady-state kinetics of MtATP-PRT inhibition by l-histidine. (A) Representative traces of the MtATP-PRT reaction under pre-steady-state conditions at various l-histidine concentrations, and fixed saturating concentrations of ATP, PRPP, and metals. Gray thick lines represent data and black lines the best fits to eq 6. Data are averages of 10 traces. (B) Plot of the burst amplitude vs l-histidine concentration (top) and plot of kobs vs l-histidine concentration (bottom). Symbols represent data and solid lines best fits to eqs 10 and 11 for burst amplitude and burst rate dependencies, respectively. Reactions were conducted in 50 mM Tris-HCl (pH 8.5), 150 mM KCl, 10 mM MgCl2, 1.5 mM PRPP, 4 mM ATP, and 10 μM MtATP-PRT, at 25 °C. Results are representative of two independent experiments.
Discussion
Oligomeric State of MtATP-PRT
Our gel filtration experiments indicate that MtATP-PRT is mainly hexameric in the absence of ligands or in the presence of l-histidine or ATP, at pH 8. Although this result appears to contrast with the results obtained by Cho and colleagues,16 a dimer–hexamer equilibrium, we believe this can be explained by the experimental conditions used. It is worth mentioning that no shift in the oligomeric state was observed for the hexameric homologous enzyme from S. enterica.20,23 Our interpretation of these results is that lower pH values can lead to dissociation of MtATP-PRT into dimers, as observed by Cho et al. However, there is no evidence that the dimer represents a kinetically competent form of MtATP-PRT, as activity decreases drastically as the pH drops below 8. Similar results have been recently reported for ATP-PRT from C. glutamicum. Although there is some dimer present during gel filtration (at pH 7.2), CgATP-PRT loses most of it activity below pH 8.25
If the dimer–hexamer equilibrium does not account for the allosteric inhibition, another mechanism has to be envisioned. It is noteworthy to mention that Cho and colleagues16 observed a large domain shift, on domain III, in the dimeric form compared with the hexameric form. Although this dimeric form is not present under our experimental conditions, the domain shift could still take place, generating an alternative hexamer, which could be inhibited. Regardless of whether this conformational change is the one responsible for l-histidine inhibition, we propose that MtATP-PRT is a functional hexamer and conformational changes have to be invoked to explain the allosteric inhibition.
Inhibition of MtATP-PRT by l-Histidine
Steady-state kinetic analysis indicates that l-histidine inhibition versus PRPP is linear and noncompetitive, where Kis = Kii. The equality of these two inhibition constants indicates that l-histidine has the same affinity for ATP-bound and ATP-PRPP-bound enzyme forms. l-Histidine inhibition versus ATP was linear and uncompetitive, indicating that l-histidine binds to MtATP-PRT only when ATP is present at the enzyme active site. These results are fully consistent with the allosteric inhibition proposed on the basis of the structure. In addition, uncompetitive inhibition versus ATP is in agreement with the l-histidine–AMP ternary complex structure determined by X-ray crystallography.16
Pre-steady-state kinetic analysis of enzyme inhibition is a powerful approach for determining which step or steps are affected by a given inhibitor, and it has been applied to study a number of systems, including DNA polymerase γ,38 HIV-1 reverse transcriptase,39d-3-phosphoglycerate dehydrogenase,40 and BACE.41 To gather further information about how l-histidine inhibits MtATP-PRT, we performed pre-steady-state kinetic studies, using stopped-flow absorbance, monitoring PR-ATP formation. l-Histidine inhibition under pre-steady-state conditions is complex, affecting multiple steps. The dominant effect observed is a decrease in the amplitude of the burst, rather than a decrease in the burst or steady-state rates. A decrease in burst amplitude indicates that l-histidine inhibits MtATP-PRT by trapping it on a form that is catalytic and inactive, and not by severely slowing chemistry or product release. On the basis of our results and the sequential ordered kinetic mechanismc previously proposed for ATP-PRTs, we can describe the inhibition using Scheme 3. Burton et al. elegantly demonstrated a similar mechanism for feedback allosteric inhibition of E. colid-3-phosphoglycerate dehydrogenase (PGDH) by l-serine.40 It is important to stress that MtATP-PRT and PGDH are structurally unrelated enzymes, which catalyze very different types of chemistry with distinct substrates. However, the conservation of the allosteric mechanism, e.g., sequestration of the enzyme–substrate–inhibitor complex into a nonproductive form, appears to be a successful case of convergent evolution.
Scheme 3.

On the basis of our steady-state data, the extent of l-histidine inhibition is maximal at saturating ATP concentrations (uncompetitive kinetic pattern) and is independent of the concentration of PRPP [noncompetitive kinetic pattern (Kii = Kis)]. Taken together, results from steady-state and pre-steady-state experiments indicate that the k–6/k6 and k–7/k7 ratios (Scheme 3) must be very similar, and that both E–ATP–histidine and E–ATP–PRPP–histidine complexes must be inactive forms of MtATP-PRT. The structural basis for this effect cannot yet be defined because of the lack of structural information regarding complexes of MtATP-PRT and the two substrates or products, with and without l-histidine.
Ionic States of l-Histidine Bound to ATP-PRT
The definition of the ionic state of l-histidine that best binds to and best inhibits MtATP-PRT is an essential step toward the molecular description of its inhibition, and it paves the way for any rational approach to target this site. We first analyzed binding, using STD-NMR. The pH dependence of the STD-NMR clearly demonstrates that l-histidine binds better to MtATP-PRT at higher pH values. More specifically, deprotonation of the α-amino group of l-histidine increases the extent of saturation transfer, which correlates with an increased level of binding. On the basis of the identity of the residues involved in l-histidine binding, Asp218, Thr238, and Ala273, we tentatively assign this effect to titration of the α-amino group of l-histidine (pKa ∼ 9). Although this result most likely has no physiological importance, as the cytoplasm will not reach pH 9, it hints about a potential mechanism for preparing l-histidine analogues that bind tightly to MtATP-PRT.
Our kinetic results demonstrate that l-histidine inhibition is stronger (lower Ki values) near pH 8. This is the opposite result obtained with binding and indicates that the form of l-histidine that best inhibits MtATP-PRT is different from the form of l-histidine that best binds it. Although IC50 measurements of l-histidine inhibition at different pH values have been performed with other ATP-PRTs, this is the first report of the pH dependence of Ki for l-histidine and ATP-PRT. Bearing in mind that IC50 values could be misleading as they do not take into account the concentration of the enzyme, substrates and their saturation, inhibition type, etc., we note that the inhibitory behavior seen with MtATP-PRT appears to follow the same trend observed with the homologous enzymes from S. enterica and C. glutamicum.23,25 Specifically, higher pH values decrease the inhibitory effect of l-histidine, and it appears that this correlates with the pKa of the α-amino group. We emphasize that other ionizable groups responsible for the transmission of the inhibitory signal from the allosteric site to the active site are certainly also important, but unfortunately, they were not observed because of the narrow pH range that could be employed to study catalysis and inhibition.
Finally, it is important to explain that this “reverse protonation” behavior is most likely not unique to l-histidine and MtATP-PRT. Usually, allosteric regulation of enzymes is studied by either analysis of binding or inhibition, and rarely is the pH dependence of either or both investigated. The assumption that the dissociation constant (Kd) and inhibition constant (Ki) for allosteric inhibitors are identical might be an oversimplification. This is likely to be the case when dealing with inhibitors and allosteric sites that contain several groups with ionization constants near neutrality. As more allosteric inhibitors are discovered and characterized, more examples such as this are likely to be found.
l-Histidine Biosynthesis in M. tuberculosis
Two conclusions regarding l-histidine levels can be drawn from our studies. Assuming that ATP is at saturating concentration in the cell,d we would have half-maximal inhibition with 4 μM l-histidine, at pH 8. This indicates that physiological concentrations of l-histidine must be varying around this value to be able to feedback control the pathway. Specifically, concentrations of ≤0.4 μM would make MtATP-PRT mostly active, and conversely, concentrations of l-histidine of ≥40 μM would inhibit MtATP-PRT and therefore shut down l-histidine biosynthesis in M. tuberculosis. Therefore, this range, between 0.4 and 40 μM, should correlate with the maximal variation expected for l-histidine in M. tuberculosis.
Only one other ATP-PRT has been characterized with regard to inhibition constants. Comparison of the value reported for Lactococcus lactis ATP-PRT indicates that l-histidine levels might fluctuate at least 10-fold across bacteria, as a Ki of 81 μM was determined, at pH 8.42 In S. enterica serovar Typhimurium, the intracellular concentration of l-histidine was shown to vary between 15 and 100 μM, depending on the growth conditions.15 On the basis of these values, the concentration of l-histidine in M. tuberculosis might be in the same range as in Salmonella and Lactococcus.
Chemical Mechanism
A few observations were made regarding the chemical mechanism of MtATP-PRT. First, MtATP-PRT displays an unusual pH dependence, which has been observed with other homologous enzymes.25 MtATP-PRT is inactive at pH ≤7, but active at neutral and alkaline pH values up to 9.5. The basis for this alkaline pH dependence is unknown, but it could be caused by a highly basic active site, which has to bind six phosphates from the two substrates, or by a missing protein partner, which could alter this toward more acidic pH.
Second, multiple-turnover experiments showed a burst of product formation, indicating that a step after chemistry is partially rate-limiting for MtATP-PRT. Unfortunately, there are no studies of other ATP-PRTs under pre-steady-state conditions, and slow product release has been shown not to be a ubiquitous trait among other phosphoribosyltransferases. For example, quinolinate phosphoribosyltransferase has been shown not to display burst kinetics, indicating that chemistry is rate-limiting and relatively slow (kobs = kcat = 0.4 s–1).43 In contrast, nicotinate phosphoribosyltransferase displayed burst kinetics, indicating fast chemistry (kobs ≥ 500 s–1) and slow product release (kcat = 1.2 s–1).44
Summary
Results presented here indicate that active and l-histidine-inhibited MtATP-PRT are hexameric in solution. l-Histidine inhibition is noncompetitive versus PRPP and uncompetitive versus ATP. Inhibition of MtATP-PRT by l-histidine is strongly influenced by pH, reaching higher potency closer to physiological pH. Binding of l-histidine displays the opposite pH dependence observed for inhibition, indicating that deprotonation of the α-amino group increases the level of binding but decreases the level of inhibition. Therefore, l-histidine binds and inhibits MtATP-PRT with a neutral imidazole and ionized α-amino and α-carboxyl groups. Pre-steady-state kinetic studies revealed that a step after chemistry is partially rate-limiting and that l-histidine exerts its inhibition by decreasing the amount of catalytically active MtATP-PRT (Scheme 3).
Acknowledgments
We thank Dr. Steve Howell for ESI-MS analysis of MtATP-PRT and NIMR’s Large Scale Laboratory for E. coli growth. We thank Drs. Clarissa M. Czekster and Rafael G. da Silva for thoughtful discussions. All NMR spectra were recorded at the MRC Biomedical NMR Centre, NIMR, Mill Hill (funded by MRC Grant-in-Aid U117533887).
Glossary
Abbreviations
- ATP-PRT
ATP phosphoribosyltransferase
- PR-ATP
1-(5-phospho-α-d-ribosyl)-ATP
- PPi
inorganic pyrophosphate
- PRPP
5-phospho-α-d-ribosyl pyrophosphate
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TEA
triethanolamine
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- Tris
tris(hydroxymethyl)aminomethane
- ESI-MS
electrospray ionization mass spectrometry
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- NMR
nuclear magnetic resonance
- STD
saturation transfer difference
- PIPES
1,4-piperazinediethanesulfonic acid
- TAPS
N-[tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid
- CHES
2-(cyclohexylamino)ethanesulfonic acid.
The authors declare no competing financial interest.
Work in the L.P.S.d.C. laboratory is funded by the MRC (MC_UP_A253_1111). S.P. and J.P.P. were recipients of Erasmus Fellowships, from the European Commission.
Footnotes
Saturation transfer difference NMR (STD-NMR) is a 1H NMR method developed for monitoring the interaction of small molecules with proteins, without the resonance assignments of the protein being necessary.28,29 In this method, a first spectrum of protein with ligand is obtained, followed by a second spectrum where a radiofrequency has ablated all resonances from the protein and by spin diffusion of any bound ligand (saturation transfer). As the ligand is in equilibrium between the bound and free states, all signals are perturbed, not only the ones from the bound ligand. Once these spectra are obtained, they are subtracted, and the resulting difference spectrum reveals which resonances of the ligand are affected, indicating a direct contact with protein.
The amplitude of the burst observed under pre-steady-state multiple-turnover conditions is a function of three factors: (i) the rate of the chemical step relative to the rate of product release, (ii) the internal equilibrium constant for the chemical step, at the active site, and (iii) the fraction of active enzyme.37 As the goal of these studies was not to describe the complete kinetic mechanism of MtATP-PRT, we did not attempt to determine why the amplitude of the burst is lower than the concentration of MtATP-PRT.
Scheme 3 shows the sequential ordered kinetic mechanism, with binding of ATP followed by binding of PRPP. Product release is also shown as ordered, with PPi dissociating first followed by PR-ATP. This kinetic mechanism was observed with SeATP-PRT.19
The concentration of ATP in M. tuberculosis is unknown, but cells typically contain between 1 and 10 mM ATP. This range corresponds to roughly 5–50 times the Km for ATP.
References
- Pardee A. B.; Yates R. A. (1956) Control of pyrimidine biosynthesis in Escherichia coli by a feedback mechanism. J. Biol. Chem. 221, 757–770. [PubMed] [Google Scholar]
- Gerhart J. C.; Pardee A. B. (1962) The enzymology of control by feedback inhibition. J. Biol. Chem. 237, 891–896. [PubMed] [Google Scholar]
- Umbarger H. E. (1956) Evidence for a negative-feedback mechanism in the biosynthesis of isoleucine. Science 123, 848. [DOI] [PubMed] [Google Scholar]
- Cook P. F., and Cleland W. W. (2007) Enzyme Kinetics and Mechanism, Garland Science Publishing, New York. [Google Scholar]
- Bearer C. F.; Neet K. E. (1978) Threonine inhibition of the aspartokinase-homoserine dehydrogenase I of Escherichia coli. A slow transient and cooperativity of inhibition of the aspartokinase activity. Biochemistry 17, 3523–3530. [DOI] [PubMed] [Google Scholar]
- de Carvalho L. P.; Argyrou A.; Blanchard J. S. (2005) Slow-onset feedback inhibition: Inhibition of Mycobacterium tuberculosis α-isopropylmalate synthase by l-leucine. J. Am. Chem. Soc. 127, 10004–10005. [DOI] [PubMed] [Google Scholar]
- Edfeldt F. N.; Folmer R. H.; Breeze A. L. (2011) Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discovery Today 16, 284–287. [DOI] [PubMed] [Google Scholar]
- Surade S.; Blundell T. L. (2012) Structural biology and drug discovery of difficult targets: The limits of ligandability. Chem. Biol. 19, 42–50. [DOI] [PubMed] [Google Scholar]
- Fenton A. W. (2012) Identification of allosteric-activating drug leads for human liver pyruvate kinase. Methods Mol. Biol. 796, 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L.; Parrish C. A.; Nevins N.; McNulty D. E.; Chaudhari A. M.; Carson J. D.; Sudakin V.; Shaw A. N.; Lehr R.; Zhao H.; Sweitzer S.; Lad L.; Wood K. W.; Sakowicz R.; Annan R. S.; Huang P. S.; Jackson J. R.; Dhanak D.; Copeland R. A.; Auger K. R. (2007) ATP-competitive inhibitors of the mitotic kinesin KSP that function via an allosteric mechanism. Nat. Chem. Biol. 3, 722–726. [DOI] [PubMed] [Google Scholar]
- Liu S.; Chang J. S.; Herberg J. T.; Horng M. M.; Tomich P. K.; Lin A. H.; Marotti K. R. (2006) Allosteric inhibition of Staphylococcus aureusd-alanine:d-alanine ligase revealed by crystallographic studies. Proc. Natl. Acad. Sci. U.S.A. 103, 15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli D. F.; Tang X.; Pelletier B.; Orlicky S.; Xie W.; Plantevin V.; Neculai D.; Chou Y. C.; Ogunjimi A.; Al-Hakim A.; Varelas X.; Koszela J.; Wasney G. A.; Vedadi M.; Dhe-Paganon S.; Cox S.; Xu S.; Lopez-Girona A.; Mercurio F.; Wrana J.; Durocher D.; Meloche S.; Webb D. R.; Tyers M.; Sicheri F. (2011) An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 145, 1075–1087. [DOI] [PubMed] [Google Scholar]
- Sousa M. C.; Kessler B. M.; Overkleeft H. S.; McKay D. B. (2002) Crystal structure of HslUV complexed with a vinyl sulfone inhibitor: corroboration of a proposed mechanism of allosteric activation of HslV by HslU. J. Mol. Biol. 318, 779–785. [DOI] [PubMed] [Google Scholar]
- Ames B. N.; Martin R. G.; Garry B. J. (1961) The first step of histidine biosynthesis. J. Biol. Chem. 236, 2019–2026. [PubMed] [Google Scholar]
- Winkler M. E. (1987) Biosynthesis of Histidine. In Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology (Neidhardt F. C., Ed.) pp 395–411, American Society for Microbiology, Washington, DC. [Google Scholar]
- Cho Y.; Sharma V.; Sacchettini J. C. (2003) Crystal structure of ATP phosphoribosyltransferase from Mycobacterium tuberculosis. J. Biol. Chem. 278, 8333–8339. [DOI] [PubMed] [Google Scholar]
- Cho Y.; Ioerger T. R.; Sacchettini J. C. (2008) Discovery of novel nitrobenzothiazole inhibitors for Mycobacterium tuberculosis ATP phosphoribosyl transferase (HisG) through virtual screening. J. Med. Chem. 51, 5984–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; Dejesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish T. (2003) Starvation survival response of Mycobacterium tuberculosis. J. Bacteriol. 185, 6702–6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll M. J.; Appella E.; Martin R. G. (1967) Purification and composition studies of phosphoribosyladenosine triphosphate:pyrophosphate phosphoribosyltransferase, the first enzyme of histidine biosynthesis. J. Biol. Chem. 242, 1760–1767. [PubMed] [Google Scholar]
- Brashear W. T.; Parsons S. M. (1975) Evidence against a covalent intermediate in the adenosine triphosphate phosphoribosyltransferase reaction of histidine biosynthesis. J. Biol. Chem. 250, 6885–6890. [PubMed] [Google Scholar]
- Morton D. P.; Parsons S. M. (1976) Biosynthetic direction substrate kinetics and product inhibition studies on the first enzyme of histidine biosynthesis, adenosine triphosphate phosphoribosyltransferase. Arch. Biochem. Biophys. 175, 677–686. [DOI] [PubMed] [Google Scholar]
- Martin R. G. (1963) The First Enzyme in Histidine Biosynthesis: The Nature of Feedback Inhibition by Histidine. J. Biol. Chem. 238, 257–268. [Google Scholar]
- Lohkamp B.; Coggins J. R.; Lapthorn A. J. (2000) Purification, crystallization and preliminary X-ray crystallographic analysis of ATP-phosphoribosyltransferase from Escherichia coli. Acta Crystallogr. D56, 1488–1491. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Shang X.; Deng A.; Chai X.; Lai S.; Zhang G.; Wen T. (2012) Genetic and biochemical characterization of Corynebacterium glutamicum ATP phosphoribosyltransferase and its three mutants resistant to feedback inhibition by histidine. Biochimie 94, 829–838. [DOI] [PubMed] [Google Scholar]
- Ames B. N.; Hartman P. E.; Jacob F. (1963) Chromosomal alterations affecting the regulation of histidine biosynthetic enzymes in Salmonella. J. Mol. Biol. 7, 23–42. [DOI] [PubMed] [Google Scholar]
- Hwang T.-L.; Shaka A. J. (1995) Water Suppression That Works. Excitation Sculpting Using Arbitrar Waveforms and Pulsed Field Gradients. J. Magn. Reson. 112, 275–279. [Google Scholar]
- Mayer M.; Meyer B. (1999) Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem., Int. Ed. 38, 1784–1788. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. (2001) Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 123, 6108–6117. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. (2009) Global kinetic explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29. [DOI] [PubMed] [Google Scholar]
- Johnson K. A. (2009) Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods Enzymol. 467, 601–626. [DOI] [PubMed] [Google Scholar]
- Chen H. A.; Pfuhl M.; McAlister M. S.; Driscoll P. C. (2000) Determination of pKa values of carboxyl groups in the N-terminal domain of rat CD2: Anomalous pKa of a glutamate on the ligand-binding surface. Biochemistry 39, 6814–6824. [DOI] [PubMed] [Google Scholar]
- Schuckmann M. M.; Marchand B.; Hachiya A.; Kodama E. N.; Kirby K. A.; Singh K.; Sarafianos S. G. (2010) The N348I mutation at the connection subdomain of HIV-1 reverse transcriptase decreases binding to nevirapine. J. Biol. Chem. 285, 38700–38709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeman J. E.; Parsons S. M. (1976) Reverse direction substrate kinetics and inhibition studies on the first enzyme of histidine biosynthesis, adenosine triphosphate phosphoribosyltransferase. Arch. Biochem. Biophys. 175, 687–693. [DOI] [PubMed] [Google Scholar]
- Copeland R. A. (2005) Evaluation of enzyme inhibitors in drug discovery: A guide for medicinal chemists and pharmacologists, John Wiley & Sons, Inc., Hoboken, NJ. [PubMed] [Google Scholar]
- Fersht A. (1999) Structure and Mechanism in Protein Science, W. H. Freeman and Co., New York. [Google Scholar]
- Johnson K. A. (2003) Introduction to kinetic analysis of enzyme systems. In Kinetic Analysis of Macromolecules (Johnson K. A., Ed.) pp 10–12, Oxford University Press Inc., New York. [Google Scholar]
- Fowler J. D.; Brown J. A.; Johnson K. A.; Suo Z. (2008) Kinetic investigation of the inhibitory effect of gemcitabine on DNA polymerization catalyzed by human mitochondrial DNA polymerase. J. Biol. Chem. 283, 15339–15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence R. A.; Kati W. M.; Anderson K. S.; Johnson K. A. (1995) Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science 267, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton R. L.; Chen S.; Xu X. L.; Grant G. A. (2009) Transient kinetic analysis of the interaction of l-serine with Escherichia colid-3-phosphoglycerate dehydrogenase reveals the mechanism of V-type regulation and the order of effector binding. Biochemistry 48, 12242–12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkeviciene J.; Luo Y.; Graciani N. R.; Combs A. P.; Copeland R. A. (2001) Mechanism of inhibition of β-site amyloid precursor protein-cleaving enzyme (BACE) by a statine-based peptide. J. Biol. Chem. 276, 23790–23794. [DOI] [PubMed] [Google Scholar]
- Champagne K. S.; Piscitelli E.; Francklyn C. S. (2006) Substrate recognition by the hetero-octameric ATP phosphoribosyltransferase from Lactococcus lactis. Biochemistry 45, 14933–14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H.; Pietrak B. L.; Grubmeyer C. (2002) Quinolinate phosphoribosyltransferase: Kinetic mechanism for a type II PRTase. Biochemistry 41, 3520–3528. [DOI] [PubMed] [Google Scholar]
- Gross J. W.; Rajavel M.; Grubmeyer C. (1998) Kinetic mechanism of nicotinic acid phosphoribosyltransferase: Implications for energy coupling. Biochemistry 37, 4189–4199. [DOI] [PubMed] [Google Scholar]