

Abstract
HSV-1 virions contain a proteinaceous layer termed the tegument that lies between the nucleocapsid and viral envelope. The molecular mechanisms that facilitate incorporation of tegument proteins are poorly characterized. The tegument protein VP22 interacts with VP16 and the cytoplasmic tail of glycoprotein E (gE). Virion incorporation of VP22 occurs independently of interaction with VP16; however the contribution of gE binding remains undefined. Site-directed mutagenesis was used to identify VP22 mutants which abrogate interaction with gE, but retain VP16 binding. Virion incorporation assays demonstrated that failure to bind gE did not abrogate VP22 packaging. A region of VP22 which binds to both VP16 and gE failed to be packaged efficiently, with wild-type levels of incorporation only attained when residues 43–86 of VP22 were present. Mutational analysis of an acidic cluster of amino acids within this region indicates that this motif facilitates trans-Golgi network (TGN) localization and optimal virion incorporation of VP22.
INTRODUCTION
Assembly of herpes simplex virus type 1 (HSV-1) involves a intricate sequence of events, which coordinate incorporation of over 40 different viral proteins into one of three morphologically distinct virion structures: the nucleocapsid, the host-derived lipid envelope containing virally encoded glycoproteins, and the tegument, a proteinaceous region located between the nucleocapsid and envelope (Roizman and Pellett, 2001). Although, extensive studies have established that capsid assembly and packaging of the viral genome occur in the nucleus, the compartment(s) in which the tegument and envelope are acquired is less well-defined (Enquist et al., 1998; Mettenleiter, 2002; Mettenleiter, Klupp, and Granzow, 2009). The current model for HSV-1 assembly and egress suggests that nucleocapsids are exported to the cytoplasm via a budding/fusion event that occurs across the inner and outer membranes of the nucleus, respectively. Unenveloped nucleocapsids subsequently transit through the cytoplasm until they reach a trans-Golgi network (TGN)-derived vesicle, where a budding event results in concurrent acquisition of a final lipid bilayer and complement of viral glycoproteins (Sanchez et al., 2000; Skepper et al., 2001; van Genderen et al., 1994; Whealy et al., 1991; Whiteley et al., 1999). Ultimately, virion-containing vesicles follow the secretory pathway to the cell surface, where mature virus particles are released into the extracellular milieu (Mettenleiter, 2002; Mettenleiter, Klupp, and Granzow, 2009).
In contrast to nucleocapsid assembly, the molecular mechanisms that facilitate addition of tegument proteins to the nucleocapsid during the assembly pathway and the process of final envelopment itself are poorly understood (Mettenleiter, 2002; Mettenleiter, Klupp, and Granzow, 2009; Roizman and Pellett, 2001). Tegumentation of nucleocapsids can theoretically occur at various stages in the egress pathway: in the nucleus, at the nuclear membrane, in the cytoplasm or during budding at the TGN. Recent studies have demonstrated that a subset of tegument proteins are added to the capsid prior to nuclear egress; however, the mechanism(s) by which tegument proteins are selectively packaged into the assembling virion has yet to be defined (Bucks et al., 2007; Naldinho-Souto, Browne, and Minson, 2006). It is likely that a myriad of protein-protein interactions between capsid proteins, tegument proteins, and the cytoplasmic tails of virally encoded glycoproteins facilitate the process. Our studies have focused on defining the protein-protein interaction motifs of one specific HSV-1 tegument protein, VP22, and the roles these interactions play in facilitating virion incorporation of the protein.
The HSV-1 UL49 gene encodes the 301-amino-acid VP22, which is one of the most abundant proteins in the tegument region (Elliott and Meredith, 1992; Heine et al., 1974; Leslie, Rixon, and McLauchlan, 1996). Despite its abundance, the role of VP22 during HSV-1 assembly and mechanism of its incorporation are only beginning to be defined. Two VP22-null viruses have been described and demonstrate a variety of cell-specific replication defects and altered virion composition, including decreased packaging of both glycoprotein D (gD) and glycoprotein E (gE), two known binding partners of VP22 (Chi et al., 2005; Duffy et al., 2006; Elliott et al., 2005; O'Regan et al., 2007a; Pomeranz and Blaho, 2000). VP22 associates with membranes and localizes to acidic compartments of the cell including the TGN (Brignati et al., 2003). In addition to binding to cytoplasmic tails of a subset of viral glycoproteins, presumably at the TGN, VP22 is known to interact with another abundant tegument protein, VP16 (Chi et al., 2005; Elliott et al., 2005; Elliott, Mouzakitis, and O'Hare, 1995; Farnsworth, Wisner, and Johnson, 2007; O'Regan et al., 2007b; Stylianou et al., 2009). Transmission immunoelectron microscopy (TIEM) studies suggest that during the viral assembly pathway, detectable amounts of VP16 are added to the capsid in the nucleus, with additional VP16 added as the nucleocapsid moves through the cytoplasm prior to final envelopment (Miranda-Saksena et al., 2002; Naldinho-Souto, Browne, and Minson, 2006). In contrast, VP22 is packaged into virions during final envelopment, as nucleocapsids bud into TGN-derived vesicles (Miranda-Saksena et al., 2002). These findings suggest that interaction between VP22 and viral glycoproteins on the cytoplasmic face of the TGN vesicle, perhaps in concert with binding to tegument proteins located on the surface of the approaching capsid (such as VP16), may ensure accrual of VP22 in the tegument and facilitate final envelopment.
A VP22 construct deficient for VP16 binding but competent for interaction with the cytoplasmic tail of gE is still packaged into assembling HSV-1 particles (O'Regan et al., 2007b). Interestingly, simultaneous deletion of pseudorabies virus (PRV) gM and the gE/gI heterodimer, both of which are binding partners of the PRV VP22 homologue, results in capsid-bound tegument aggregates in the cytoplasm and reduced amounts of VP22 in the mature PRV particle (Brack et al., 1999; Brack et al., 2000; Fuchs et al., 2002). Furthermore, the Bartha strain of PRV, which lacks glycoproteins gI and gE, fails to package VP22 and a recent report from Stylianou and coworkers suggests that interaction with gE is a major determinant of HSV-1 VP22 incorporation (Lyman, Demmin, and Banfield, 2003; Stylianou et al., 2009). These findings suggest that binding to the cytoplasmic tail of a viral glycoprotein may be an important packaging determinant for VP22.
To ascertain the contribution of gE binding to virion packaging of HSV-1 VP22, site-directed mutagenesis was used to specifically disrupt the VP16 and gE binding activities of the protein. Assessment of these point mutants in a virion incorporation assay demonstrated that failure to bind to gE did not abrogate packaging of VP22. Furthermore, a region of VP22 which binds to both VP16 and gE, failed to be packaged to wild-type levels, with efficient levels of incorporation only attained when residues 43–86 of VP22 were present. This finding suggested the existence of additional VP22 incorporation determinants within this domain of the protein. Analysis of an acidic cluster of amino acids within this region indicates that this motif facilitates recruitment of VP22 to the TGN and is an important determinant for virion packaging.
This report provides new information on the molecular mechanism by which VP22 is incorporated into virus particles and reveals that in addition to protein-protein interactions, proper trafficking and localization of VP22 also contribute to efficient virion packaging.
RESULTS
Site-directed point mutagenesis of VP22
The gE binding activity of VP22 has been mapped to a domain encompassing residues 165 to 270, which are highly conserved among VP22 homologues of herpesviruses and also facilitate binding to VP16 (O'Regan et al., 2007a; O'Regan et al., 2007b; Stylianou et al., 2009). To ascertain the contribution of gE binding to virion incorporation of VP22, site-directed point mutagenesis was used to target conserved residues within this domain, in an attempt to abrogate gE binding while leaving the ability to bind VP16 intact.
Two tryptophan residues at positions 189 and 221 of VP22 and phenylalanine residues at amino acids 196 and 201 were the initial targets for mutagenesis (Fig. 1A). These targets were chosen due to their similarity to a “WW” domain, a well characterized protein-protein interaction motif. “WW” domains are typically 35 to 40 amino acids in length, characterized by two tryptophan residues that bookend aromatic residues with a proline residue located C-terminally of the second tryptophan (Ingham et al., 2005). Interestingly, “WW” domain ligands include proline-rich peptide motifs and phosphorylated serine/threonine-proline sites, examples of which are found in the cytoplasmic tail of gE (Harty et al., 1999). The tryptophan residue at position 189 and/or 221 was changed conservatively to a phenylalanine, creating the VP22 constructs W189F, W221F and W189F/W221F (Fig. 1A). Studies suggest that aromatic residues located between the tryptophans of a “WW” domain confer ligand specificity to this protein-protein interaction motif (Ilsley, Sudol, and Winder, 2002; Verdecia et al., 2000). Thus, phenylalanines at positions 196 and 201 of VP22 were mutated to either alanines (F196A and F201A) or more conservatively, to tryptophan residues (F196W and F201W) (Fig. 1A). Localization studies demonstrated that all mutant constructs displayed a subcellular localization reminiscent of wild-type VP22 in transfected/infected cells (data not shown). Thus, failure of any mutant to bind VP16/gE or to be incorporated into the virus particle is not due to a gross mislocalization within the cell.
Figure 1. Characterization of the ability of VP22 point mutants to bind to the cytoplasmic tail of gE in a GST pulldown assay.

(A) Point mutants of VP22 fused to GFP. Schematic representation of wild-type VP22 and amino acid substitution mutants used in this study fused to the N-terminus of the GFP protein. (B) and (C) Expression of VP22 point mutants in transfected cells. Vero cells were transfected with the indicated constructs and 20 h post-transfection, the monolayers were lysed with NP-40 lysis buffer. Expression of VP22-GFP fusion proteins was analyzed by Western blotting using a rabbit polyclonal antibody specific for GFP. (D) and (E) GST pulldown from transfected cell lysates using GST-gECT. Approximately equal amounts of purified GST fusion proteins were added to the remainder of the transfected-cell lysates. Beads were washed extensively with lysis buffer and bound proteins were separated by SDS-PAGE and transferred to nitrocellulose. Western blot analysis was performed using a rabbit polyclonal antibody raised against the GFP protein. The positions of molecular mass markers (in kilodaltons) are indicated on the left. (F) Binding efficiency of VP22 point mutants to the cytoplasmic tail of gE. Using densitometry, binding efficiency was quantitated by dividing the amount of VP22-GFP protein detected in the pulldown assay (normalized for the amount of GST-gECT present) by the amount in the cell lysate (normalized for the amount of actin present). In each experiment, the wild-type VP22-GFP construct was set at 100% binding efficiency. Error bars represent standard deviations for four replicate experiments.
Characterization of the ability of VP22 point mutants to bind to the cytoplasmic tail of gE
To determine whether mutation of the conserved tryptophan or phenylalanine residues within VP22 would abrogate binding to gE, a glutathione S-transferase (GST) pulldown assay was utilized. The cytoplasmic tail of gE fused to the C-terminus of the GST protein (GST-gECT), or GST alone were expressed in Escherichia coli cells and subsequently purified. Approximately equal amounts of GST fusion proteins were used in the pulldown assay as determined by Coomassie-blue staining (data not shown). Vero cells were transfected with plasmids encoding the green fluorescent protein (GFP) or the various mutants of VP22 fused to the N-terminus of GFP represented in Fig. 1A. The transfected monolayers were lysed with NP-40 lysis buffer and a fraction of each cell lysate was analyzed by Western blotting to verify that the GFP-tagged constructs were expressed (Fig. 1B and Fig. 1C). The remaining lysates were incubated with equivalent amounts of purified GST fusion proteins (either the cytoplasmic tail of gE fused to GST or GST alone) bound to glutathione-Sepharose beads. The beads were subsequently washed extensively with lysis buffer, and bound material was separated by SDS-PAGE and then analyzed on Western blots.
Upon analysis of transfected lysates, each of the VP22 point mutants under study demonstrated high levels of expression (Fig. 1B and Fig. 1C). Curiously, a faster migrating band was detected with the F196A construct which was not present in lysates of other point mutants (Fig. 1C). The band reacted with GFP antiserum suggesting that it is GFP-tagged and thus may represent a VP22 breakdown product that is stabilized by the F196A mutation. With regard to gE binding, VP22-GFP was efficiently pulled-down from transfected-cell lysates by GST-gECT but not by the GST alone control (Fig. 1D and Fig. 1F). In contrast, W189F or W189F/W221F (Fig. 1D and Fig. 1F), and F196A, F201A or F201W (Fig. 1E and Fig. 1F) failed to bind to gE. F196W exhibited levels of binding to GST-gECT greater than those seen with wild-type VP22 (approximately 140%) (Fig. 1F), whereas the mutant W221F although still retaining binding activity, did so at a diminished level (54% of wild-type) (Fig. 1F). Taken together, these results indicate that a variety of conservative amino acid substitutions (W189F, W189F/W221F, and F201W) are capable of inhibiting VP22's ability to bind to gE and may be useful tools in the task of deciphering the role gE binding plays in virion incorporation of VP22.
Interaction of VP22 point mutants with VP16 in a coimmunoprecipitation assay
With the identification of conservative amino acid substitutions capable of abrogating VP22's binding with gE, we were anxious to test the effect of these mutations on interaction with VP16. Our hope was to separate the two binding activities within VP22. To this end, immunoprecipitation assays from transfected/infected cell lysates were performed to ascertain if the VP22 point mutants had a deleterious effect upon interaction with VP16. The VP22 constructs represented in Fig. 1A were transfected into Vero cells and at 20 h post-transfection, cells were infected with HSV-1 at a MOI of 10. After an additional period of 10 h, the monolayers were lysed with NP-40 lysis buffer and a fraction of each cell lysate was analyzed by Western blotting to verify that the GFP-tagged truncation mutants were expressed in transfected/infected cells (Fig. 2A and Fig. 2B). [As noted earlier, a faster migrating band was detected with F196A which was not present in the lysates of other point mutants (Fig. 2B)]. The remaining lysates were then incubated with goat anti-GFP antibodies followed by Protein G-agarose beads. Immunoprecipitated material was separated by SDS-PAGE and analyzed by Western blotting using rabbit anti-VP16 antibodies to assay for immunoprecipitation of VP16 with VP22 point mutants. When the VP22 point mutants were analyzed for their ability to interact with VP16 within transfected/infected cells, VP16 coimmunoprecipitated with W221F, F196A, F196W, F201W, and the wild-type VP22 construct (Fig. 2C and Fig. 2D). W189F retained the ability to interact with VP16 (albeit poorly), however binding was abrogated with mutants F201A and W189F/W221F.
Figure 2. Coimmunoprecipitation of VP22 point mutants with VP16.

(A) and (B) Expression of VP22 point mutants represented in Fig. 1A in transfected/infected cells. Vero cells expressing GFP, VP22-GFP constructs or mock transfected cells (Mock) were infected with HSV-1, and lysed 10 h post-infection with NP-40 lysis buffer. A fraction of each cell lysate was analyzed by Western blotting using a goat polyclonal antibody specific for GFP. (C) and (D) Coimmunoprecipitation of VP16 with VP22 point mutants. The remainder of the transfected/infected cell lysate was incubated with a goat polyclonal antibody against GFP and resulting antibody-antigen complexes were collected with protein G-agarose beads. After extensive washes with lysis buffer, material that immunoprecipitated with anti-GFP antibody was separated by SDS-PAGE and transferred to nitrocellulose. Coimmunoprecipitated VP16 was detected by immunoblot using a rabbit monospecific polyclonal antibody raised against a C-terminal peptide of VP16. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
Interaction of VP22 point mutants with VP16 in the absence of additional viral proteins
The coimmunoprecipitation studies described above were performed with transfected/infected cell lysates. Thus, in addition to our mutant constructs, virally encoded VP22 is present in the experimental system. Recent studies have suggested that VP22 may possess the ability to multimerize (Mouzakitis et al., 2005; O'Regan et al., 2007b; Vittone et al., 2005). Therefore, virally expressed VP22 could theoretically act as a bridge between VP16 and the VP22 mutants under study. As several of the VP22 point mutants assayed appeared to retain the ability to bind to VP16, we sought to examine the interaction in the absence of infection in order to verify that the binding detected was authentic and not facilitated by virally encoded VP22. To attain this goal we exploited a modified version of the GST pulldown assay described above using VP16 fused to the C-terminus of GST (GST-VP16) as bait rather than the cytoplasmic tail of gE. GST-VP16 and GST alone were expressed in Escherichia coli cells and subsequently purified, with approximately equal amounts of each used in the pulldown assay as determined by Coomassie-blue staining (Fig. 3A). Vero cells were transfected with plasmids encoding GFP alone or the indicated VP22 point mutants and a fraction of each cell lysate was analyzed by Western blotting to verify that the GFP-tagged constructs were expressed (Fig. 3B). The remaining lysates were analyzed in the GST pulldown assay for binding to GST-VP16, with bound material separated by SDS-PAGE and then analyzed by Western blotting (Fig. 3C).
Figure 3. Analysis of VP22 point mutants in a GST pulldown assay.

(A) Coomassie-blue stained gel of VP16 fused to the C-terminus of the GST protein (GST-VP16) and GST alone. GST-VP16 or GST were expressed in Escherichia coli cells and purified on glutathione-Sepharose beads. Expression of VP22 point mutants represented in Fig. 1A in transfected Vero cells (B), and their ability to bind to VP16 (C) were analyzed as described in the legend to Fig. 1 with GST-VP16 used as bait rather than GST-gECT. (D) Binding efficiency of VP22 point mutants to VP16. Using densitometry, binding efficiency was quantitated by dividing the amount of VP22-GFP protein detected in the pulldown assay (normalized for the amount of GST-VP16 present) by the amount in the cell lysate (normalized for the amount of actin present). In each experiment, the wild-type VP22-GFP construct was set at 100% binding efficiency. Error bars represent standard deviations for four replicate experiments. The ability of each VP22 mutant to bind to gE (as determined in Fig. 1) is also represented. A plus indicates that the mutant construct retains binding activity, whereas a minus denotes abrogation of binding.
VP22 fused to GFP was efficiently pulled-down from transfected cell lysates, indicating that the interaction of VP22 with VP16 can occur in the absence of additional viral proteins (Fig. 3C and Fig. 3D). In agreement with the results of coimmunoprecipitation studies, W189F/W221F and F201A, point mutants which abrogated interaction with gE, failed to bind to VP16 to levels above background (Fig. 3C and Fig. 3D). In contrast, W221F and F196W, which bind to gE at levels approaching 54% and 140% of wild-type VP22 respectively, were efficiently pulled-down by GST-VP16 (Fig. 3C and Fig. 3D). Interestingly, the levels of binding to VP16 observed with these mutants parallel those seen with gE binding. W221F bound to VP16 at approximately 41% of wild-type VP22 levels, with F196W more efficient at 130% (Fig. 3D). It is appealing to speculate that the apparent repressive and enhancing effects these mutants display upon VP16 and gE binding may be due to their impact on the structure of VP22. W221F potentially masks the gE and VP16 binding sites, with F196W having the contradictory effect, exposing the binding interfaces.
Coimmunoprecipitation experiments predicted that three VP22 mutant constructs (W189F, F196A and F201W), which were unable to interact with gE, would retain the ability to bind to VP16. When these mutants were assayed for their ability to bind to GST-VP16, as expected W189F demonstrated a binding efficiency of 77% of wild-type VP22, and F196A and F201W exhibited levels of 84% and 87%, respectively (Fig. 3C and Fig. 3D). Interestingly, the faster migrating band that was observed upon expression of F196A in our previous experiments was also detected in this assay and was efficiently pulled-down by the GST-VP16 fusion protein (Fig. 3C), suggesting that this putative breakdown product of VP22 may possess the ability to bind to VP16.
Collectively, our analysis of VP22 point mutants suggests that the VP16 binding activity housed within the conserved central region of VP22 can be separated from the gE binding activity by mutation of a tryptophan residue at position 189, or one of two phenylalanine residues at positions 196 or 201.
Binding to the cytoplasmic tail of gE is not required for virion incorporation of VP22
Previously, we have shown that incorporation of VP22 into assembling virus particles is independent of interaction with VP16 (O'Regan et al., 2007b). A recent report described a VP22 construct containing an internal deletion which failed to bind to gE and demonstrated reduced virion packaging, suggesting that interaction with gE may be a major determinant of HSV-1 VP22 incorporation (Stylianou et al., 2009).
With the identification of VP22 constructs containing point mutations that abrogate gE binding yet retain the ability to interact efficiently with VP16, we now possess the tools to further examine the role gE binding plays in virion incorporation of VP22. Due to the volume of VP22 constructs assayed in this study it was impractical to examine the various mutant alleles using recombinant viruses. Thus, to accomplish our goal we utilized a transfection/infection-based packaging assay, an in vitro experimental system, which our group and others have used to identify and map numerous virion incorporation determinants (Loomis et al., 2001; Loomis, Courtney, and Wills, 2003; Loomis, Courtney, and Wills, 2006; Mukhopadhyay, Lee, and Wilson, 2006; O'Regan et al., 2007a; O'Regan et al., 2007b). Specifically, Vero cells were transfected with plasmids encoding various VP22-GFP fusion proteins and subsequently infected with either a VP22-null virus (UL49−) (Fig. 4) or wild-type HSV-1 (data not shown). The use of a VP22-null virus [a kind gift from Dr. Joel Baines, Cornell University, Ithaca (Duffy et al., 2006)] eliminates the possibility of multimerization between virally encoded VP22 and the mutant construct under study that may otherwise confound the results of the incorporation assay. At 18 h post-infection, extracellular virions were harvested and subsequently pelleted through a sucrose cushion. Pelleted virions were analyzed by Western blotting using GFP-specific antisera to detect VP22-GFP fusion proteins. To confirm that approximately equal amounts of virus were loaded with each sample, Western blots were stripped and reprobed for the major capsid protein VP5.
Figure 4. Virion incorporation of VP22 W→F and F→A/W point mutants in the absence of virally encoded VP22.

Vero cells were transfected with the indicated VP22-GFP constructs, and 20 h later, they were infected with a VP22-null virus [UL49−]. After an additional 18-h incubation, cell lysates were prepared (A) and virions were collected from the media by centrifugation through a 30% sucrose cushion (B). Cell lysates and extracellular virus were separated by SDS-PAGE and transferred to nitrocellulose. Western blot analysis was performed using a rabbit polyclonal antibody specific for GFP. As a loading control, the blot was stripped and reprobed with a rabbit monospecific polyclonal antibody raised against the HSV-1 major capsid protein VP5. The positions of molecular mass markers (in kilodaltons) are indicated on the left. (C) Packaging efficiency. Using densitometry, packaging efficiency was quantitated by dividing the amount of VP22-GFP protein detected in extracellular virus particles (normalized for VP5) by the amount in the cell lysate (normalized for VP5). In each experiment, the wild-type VP22-GFP construct was set at 100% packaging efficiency. Error bars represent standard deviations for four replicate experiments. The ability of each VP22 mutant to bind to either gE (as determined in Fig. 1) or VP16 (as determined in Fig. 2 and Fig. 3) is also represented. A plus indicates that the mutant construct retains binding activity, whereas a minus denotes abrogation of binding.
Analysis of the VP22-GFP constructs within transfected/infected cells showed that they were expressed at levels similar to the wild-type construct and migrated at the expected molecular weight (Fig. 4A). With regard to packaging, similar results were seen with both the VP22-null virus (Fig. 4B) and wild-type HSV-1 (data not shown), suggesting that multimerization with virally encoded VP22 does not facilitate incorporation of the point mutants into the virus particle. Specifically, wild-type VP22-GFP was incorporated into virus particles, whereas GFP alone was undetectable, despite high expression in transfected/infected cells indicating that GFP itself does not have a significant effect on packaging (Fig. 4B and Fig. 4C). Furthermore, none of the VP22-GFP mutant proteins were detected in media from transfected/mock-infected cells (data not shown), and prior utilization of this assay has demonstrated that many constructs fail to be packaged upon infection, indicating that incorporation is in fact a specific event and not due to aggregates that can pellet through the sucrose cushion (Loomis, Courtney, and Wills, 2006; O'Regan et al., 2007a; O'Regan et al., 2007b). The VP22 mutant constructs W189F/W221F and F201A, which abrogated binding of VP22 to both VP16 and gE failed to be packaged into virions (Fig. 4B and Fig. 4C [UL 49−]). VP22 mutants such as these, which are deficient for a variety of activities, most likely suffer from gross conformational issues as a result of the mutation itself, rather than the identification of a key residue involved in three singular activities of VP22.
The VP22 mutants W221F and F196W, which retained the ability to interact with both VP16 and gE were packaged into assembling virus particles but with markedly different efficiencies (Fig. 4B and Fig. 4C). W221F was packaged at approximately 41% of wild-type levels with a UL 49− infection, a result presumably attributable to the mutant's impaired ability to bind VP16 (41% of wild-type VP22 levels) and gE (54% of wild-type VP22 levels). In contrast, F196W which demonstrated levels of binding to both VP16 and gE in excess of wild-type (130% and 140% respectively), was packaged into assembling virus particles at approximately 120% of the full-length VP22-GFP construct with a UL 49− infection. This finding suggests that an increase in binding to one or both binding partners of VP22 may enhance incorporation of the protein into assembling virions.
The VP22 mutants of particular interest to this project, W189F, F196A, and F201W (which interact with VP16 but not gE), were incorporated into assembling virus particles albeit at levels below wild-type VP22 (Fig. 4B and Fig 4C). The reduced level of virus packaging may be a result of the impaired ability of these mutants to bind to VP16 (W189F ~77%, F196A ~84%, and F201W ~87%), possibly complemented by their failure to bind to gE. Interestingly, the faster migrating band detected upon expression of F196A, which we hypothesize may be a breakdown product of VP22 that the mutation stabilizes, was also incorporated into virus particles. These results suggest that binding to the cytoplasmic tail of gE is not required for incorporation of VP22 into assembling virus particles.
Incorporation of VP22 into virus particles is independent of interactions with gE and VP16
We have previously demonstrated that VP16 binding is not required for inclusion of VP22 into assembling virus particles (O'Regan et al., 2007b). The observation that VP22 virion incorporation also appears to be gE-independent suggests that perhaps the two binding activities act in a redundant fashion to facilitate virus packaging. Alternatively, gE and VP16 binding may be only two of a plethora of activities that ensure recruitment of VP22 into the virus particle.
To examine whether VP16 and gE binding may act in a redundant fashion to ensure virion incorporation of VP22 we utilized a previously described mutant of VP22, VP22.LI (−), which abrogates VP16 binding with minimal effects upon interaction with gE (O'Regan et al., 2007b). VP22.LI (−) was analyzed in a transfection/infection-based packaging assay with a gE/gD-null virus [vRR1097/gEβ] [a kind gift from David Johnson, The Oregon Health and Science University, Portland, Oregon (Farnsworth, Goldsmith, and Johnson, 2003)] used for infection. The gE/gD-null virus was used rather than a wild-type virus, to ensure removal of any gE from the assay which could facilitate incorporation of VP22.LI (−). The use of this virus necessitated the experiment to be performed in gD-expressing cells, as no virus particles are made in the absence of both gE and gD (Farnsworth, Goldsmith, and Johnson, 2003). gD-expressing Vero cells (VD60 cells) were transfected with plasmids encoding various VP22-GFP fusion proteins and subsequently infected with the gE/gD-null virus [vRR1097/gEβ]. At 18 h post-infection, extracellular virions were harvested and subsequently pelleted through a sucrose cushion. Pelleted virions were analyzed by Western blotting using GFP-specific antisera to detect VP22-GFP fusion proteins. To confirm that approximately equal amounts of virus were loaded with each sample, Western blots were stripped and reprobed for the major capsid protein VP5.
Analysis of the VP22 constructs within transfected/infected cells showed that VP22.LI (−) was expressed at levels similar to the wild-type construct and migrated at the expected molecular weight (Fig. 5A). With regard to packaging, despite the absence of both gE and VP16 binding, VP22.LI (−) was packaged into assembling virus particles albeit at levels below those of the wild-type construct (Fig. 5B and Fig. 5C). The levels of incorporation of VP22.LI (−) were marginally lower when the gE/gD-null virus was used for infection (28%) (Fig. 5C) rather than a wild-type (33%) or VP22 KO virus (38%) (O'Regan et al., 2007b), suggesting that the contribution of gE binding to the incorporation of VP22 may be minimal. These results indicate that VP22 can be incorporated into assembling virions in the absence of interaction with VP16 and gE, and that these binding activities do not act in a redundant fashion to facilitate inclusion of VP22 into virus particles. Furthermore, these experiments suggest the presence of additional incorporation determinants within VP22 that contribute to virion incorporation of the protein.
Figure 5. Virion incorporation of VP22 occurs independently of interactions with gE and VP16.

gD-expressing Vero cells (VD60 cells) were transfected with indicated VP22-GFP constructs, and 20 h later, they were infected with a gE/gD-null virus [vRR1097/gEβ]. After an additional 18-h incubation, cell lysates were prepared (A) and virions were collected from the media by centrifugation through a 30% sucrose cushion (B). Cell lysates and extracellular virus were separated by SDS-PAGE and transferred to nitrocellulose. Western blot analysis was performed using a rabbit polyclonal antibody specific for GFP. As a loading control, the blot was stripped and reprobed with a rabbit monospecific polyclonal antibody raised against the HSV-1 major capsid protein VP5. The blots were also probed with antibodies specific for gD and gE. The positions of molecular mass markers (in kilodaltons) are indicated on the left. (C) Packaging efficiency. Using densitometry, packaging efficiency was quantitated by dividing the amount of VP22-GFP protein detected in extracellular virus particles (normalized for VP5) by the amount in the cell lysate (normalized for VP5). In each experiment, the wild-type VP22-GFP construct was set at 100% packaging efficiency. Error bars represent standard deviations for four replicate experiments.
A VP22 construct that binds to both VP16 and gE at wild-type levels is impaired for virion packaging
Previously, we demonstrated that a domain of VP22 encompassing residues 165–270 facilitates binding to both VP16 and gE (O'Regan et al., 2007b). Curiously, despite possessing membrane association activity, in addition to high binding efficiencies to both VP16 (399% of wild-type levels) and gE (86% of wild-type levels), this domain of VP22 is only packaged to 54% of the levels seen with a full-length VP22-GFP construct (O'Regan et al., 2007b). It is attractive to propose that in order to attain wild-type levels of virion incorporation of VP22, the construct under study must also contain the heretofore unidentified incorporation determinant. In an effort to map the location of this activity within VP22, a variety of N-terminal and C-terminal extensions of the conserved core of VP22 (amino acids 165–270) were tagged with GFP, creating the fusion proteins represented in Fig. 6A. Initially, immunoprecipitation assays from transfected/infected cell lysates were performed to ascertain if the VP22 truncation mutants had a deleterious effect upon interaction with gE and/or VP16. Both binding partners, gE (Fig. 6C) and VP16 (Fig. 6D), coimmunoprecipitated with each of the VP22 truncation mutants. These results indicate that extension of the VP16 and gE binding domains either toward the carboxyl or amino terminus of the VP22 fails to abrogate VP16 or gE binding. The effect of the truncation mutants on the binding efficiency of VP22 to gE and VP16 was also analyzed using the GST-gECT and GST-VP16 pulldown assays described in Fig. 1 and Fig. 3 respectively. Analysis of the VP22 truncation mutants in these assays suggests that the gE binding activity housed within residues 165–270 of VP22 can be enhanced to levels equal to or greater than a wild-type VP22 construct by extension of the domain to either the carboxyl or amino terminus of the protein without diminishing VP16 binding to levels below those of a wild-type construct (data not shown).
Figure 6. Coimmunoprecipitation of gE and VP16 with VP22 truncation mutants.

(A) VP22 truncation mutants fused to GFP. A schematic representation of full-length and N-terminal truncation mutants of VP22 fused to the N-terminus of the GFP protein. Also represented, deletion of the region of VP22 that facilitates binding to both VP16 and gE and this domain alone fused to GFP. (B) Expression of VP22 mutants in transfected/infected cells. Vero cells were transfected with the VP22-GFP constructs represented in Fig. 6A, and 20 h later, they were infected with a VP22-null virus [UL49−]. After an additional 10-h incubation, the cells were lysed with NP-40 lysis buffer. A fraction of each cell lysate was analyzed by Western blotting using a goat polyclonal antibody specific for GFP. The remainder of the transfected/infected cell lysate was incubated with a goat polyclonal antibody against GFP and resulting antibody-antigen complexes were collected with protein G-agarose beads. After extensive washes with lysis buffer, material that immunoprecipitated with anti-GFP antibody was separated by SDS-PAGE and transferred to nitrocellulose. Coimmunoprecipitated gE (C) and VP16 (D) were detected by immunoblot using a rabbit polyclonal antibody specific for gE or a rabbit monospecific polyclonal antibody raised against a C-terminal peptide of VP16. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
To determine whether VP22 constructs that facilitate wild-type levels of binding to both VP16 and gE are packaged into virions at levels equivalent to VP22-GFP, we utilized the transfection/infection-based packaging assay described above. A wild-type HSV-1 strain was used to infect the transfected monolayers, allowing any contribution of heretofore unknown VP22 packaging determinants to be ascertained. Western blot analysis of the various VP22-GFP constructs within transfected/infected cells showed that the mutants were expressed at levels similar to wild-type VP22 and migrated at the expected molecular weight (Fig. 7A). With regard to packaging, VP22-GFP was incorporated into virus particles, whereas GFP alone was undetectable, despite high expression in the transfected/infected cells (Fig. 7B and Fig. 7C). As we had seen previously, VP22.165–270 which binds to VP16 and gE, was incorporated to approximately 49% of wild-type levels. In contrast, upon deletion of residues 165–270, VP22 virion packaging occurred at extremely low (approximately 8% of wild-type VP22) but reproducible levels, (Fig. 7B and Fig. 7C). A virion packaging signal has been described in the C-terminus of VP22 and is contained within VP22.Δ165–270, perhaps accounting for the low levels of incorporation seen with this mutant (Schlegel and Blaho, 2009).
Figure 7. Virion incorporation of VP22 truncation mutants in the presence of a wild-type HSV-1 infection.

Expression of the indicated VP22-GFP constructs in transfected/infected cells (A) and their incorporation into virus particles (B) were analyzed as described in the legend to Fig. 4 with a wild-type HSV-1 strain used for infection rather than a VP22-null virus. The positions of molecular mass markers (in kilodaltons) are indicated on the left. Packaging efficiency (C) was calculated as described in the legend to Fig. 4C, using the tegument protein VP16 as a loading control rather than VP5. Error bars represent standard deviations for four replicate experiments. Comparison of VP22.87–301 to both wild-type VP22 and VP22.44–301 indicates that the difference in incorporation levels observed is statistically significant (P < 0.001). The difference in packaging levels between VP22.44–301 and the full-length protein is not significant (P > 0.05).
VP22.87–301, which displayed wild-type binding to both VP16 and gE (Fig. 6 and data not shown), was not packaged at wild-type levels but at an efficiency comparable to VP22.165–270 (Fig. 7B and Fig. 7C). Similar results were attained with VP22.121–301 and VP22.165–301. However, upon inclusion of residues 44–86, incorporation into virus particles returned to levels resembling VP22-GFP (95%) (Fig. 7B and Fig. 7C). These results suggest that optimal VP22 incorporation levels are not facilitated by wild-type binding to both VP16 and gE alone, but require residues 44–86 of VP22.
Localization of VP22 to the TGN is facilitated by an acidic cluster of amino acids
HSV-1 virion components must traffic to the TGN for incorporation into virus particles. Some virion proteins traffic to the TGN as a multiprotein complex (e.g. nucleocapsids), whereas other structural proteins likely target to the TGN as monomers. Trafficking signals are usually short amino acid motifs within a protein that can be classified into three general categories: tyrosine-based motifs (YXXΦ), dileucine motifs, and acidic cluster motifs (Bonifacino, 2004; Kirchhausen, 1999; Kirchhausen, Bonifacino, and Riezman, 1997; Wan et al., 1998). The negatively charged amino acids within acidic clusters are thought to interact with components of the clathrin sorting machinery at the plasma membrane, as well as endosomal and Golgi compartments. A variety of herpesvirus proteins have hijacked these cellular mechanisms, including the tegument protein UL11 and glycoproteins gB, gE, and gI which contain dileucine and acidic cluster motifs that enable these proteins to traffic to the TGN (Alconada et al., 1998; Alconada et al., 1999; Loomis et al., 2001; McMillan and Johnson, 2001; Tugizov et al., 1999).
Analysis of the primary structure of amino acids 44–86 of VP22, which facilitate wild-type levels of virion incorporation, reveals a cluster of acidic amino acids between residues 61–77. To determine if the putative acidic cluster within VP22 is required for targeting to the TGN, we examined the localization of a variety of VP22 mutants that lacked this motif. Residues comprising the primary acidic cluster (amino acids 71–77) were changed to alanines (AC→Ala) or deleted (ΔAC1) (Fig. 8A). A smaller cluster of acidic amino acids is also located at residues 61–64 of VP22. To eliminate the possibility that deletion mutagenesis may enable this secondary motif to functionally substitute for the primary acidic cluster we deleted both motifs, creating the mutant ΔAC1 + ΔAC2 (Fig. 8A). Finally, if the acidic cluster of VP22 is necessary for localization to the TGN, then the localization of VP22 acidic cluster deletion mutants should be restored by insertion of other well-characterized acidic clusters into VP22. The acidic cluster of the cellular protein furin (SDSEEDE) was inserted into VP22 in the same position as the native acidic cluster (Fig. 8A).
Figure 8. Mutagenesis of the acidic cluster motifs of VP22.
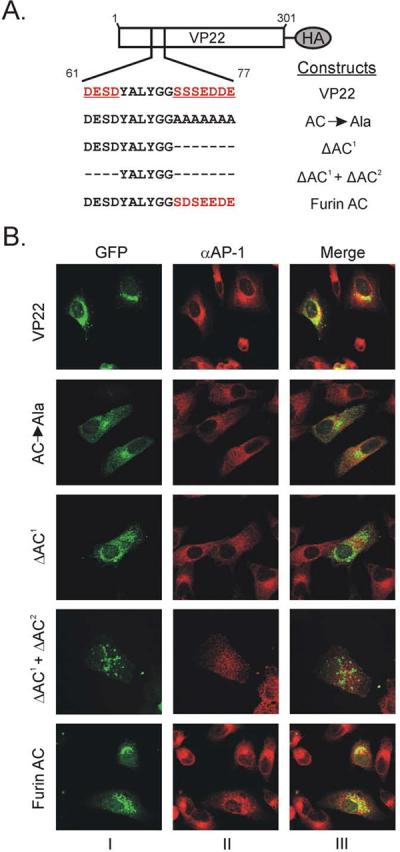
(A) Acidic cluster motif mutants of VP22. Schematic representation of HA-tagged VP22 and constructs with either, mutation of the primary acidic cluster of VP22 to alanine residues, deletion of the primary acidic cluster, or deletion of both primary and secondary acidic clusters from VP22. A VP22 HA-tagged construct in which the primary acidic cluster is replaced with the acidic cluster from the cellular protein furin is also represented. (B) Localization of VP22 acidic cluster motif mutants. Vero cells were transfected with GFP-tagged versions of the constructs represented in Fig. 8A, and 20 h later, they were infected with HSV-1. At 18 h post-infection, (38 h post-transfection), cells were fixed with paraformaldehyde and permeabilized with Triton X-100. Cells were labeled with a monoclonal antibody against AP-1, which was detected by a goat anti-mouse IgG antibody conjugated to Alexa Fluor 555. Panels within the same row are the same field viewed by confocal microscopy with the appropriate wavelength to excite GFP (column I) or Alexa Fluor 555 (column II), and these images were digitally combined to produce the image in column III.
Development of an assay to differentiate between the cellular localization of wild-type VP22 and various acidic cluster mutants was hindered due to the nature of VP22 relative to other well-characterized proteins containing acidic cluster motifs. First of all, VP22 appears to be a peripheral membrane protein (Brignati et al., 2003). Additionally, VP22 fails to accumulate at the plasma membrane, rendering classical endocytosis assays inaccessible. Therefore, we examined the localization of the VP22 acidic cluster mutants relative to that of AP-1, a major clathrin adaptor protein of the TGN using confocal microscopy (Fig. 8B).
Vero cells were transfected with GFP-tagged versions of the constructs represented in Fig. 8A, and 20 h later, they were infected with HSV-1. At 18 h post-infection, (38 h post-transfection), cells were fixed, permeabilized and then labeled with a monoclonal antibody against AP-1. Confocal microscopy indicated that inactivation of the acidic cluster by alanine mutagenesis resulted in a dispersed punctate fluorescence relative to the concentrated punctate fluorescence which colocalizes with AP-1 that wild-type VP22 exhibits. Deletion of the primary acidic cluster resulted in an intermediate phenotype with some dispersed puncta reminiscent of AC→Ala, and modest colocalization with AP-1. Interestingly ΔAC1 + ΔAC2 exhibited a dispersed punctate fluorescence with minimal colocalization with AP-1, suggesting that the secondary acidic cluster may be able to functionally substitute upon deletion of the primary cluster. Similar to wild-type VP22, the acidic cluster from furin efficiently localized VP22 to a similar cellular compartment as AP-1 (Fig. 8B), suggesting that the acidic cluster motif of VP22 is functionally equivalent to other well-characterized acidic clusters. Collectively these results indicate that the acidic cluster of VP22 facilitates localization to the TGN.
Deletion of the acidic cluster of VP22 does not abrogate interaction with VP16 or gE
Prior to examining whether the packaging determinant that, in tandem with optimal gE and VP16 binding, facilitates wild-type levels of VP22 incorporation is in fact the acidic cluster motif, we needed to ensure that mutagenesis of the acidic clusters did not have a detrimental effect on binding to gE and VP16. The acidic cluster mutants were assayed for binding in a GST pulldown assay using VP16 (Fig. 9) or the cytoplasmic tail of gE (Fig. 10) as bait.
Figure 9. Characterization of the ability of VP22 acidic cluster motif mutants to bind to VP16 in a GST pulldown assay.

Expression of VP22 acidic cluster motif mutants in transfected Vero cells (A) and (C), their ability to bind to VP16 (B) and (D) and binding efficiency (E) were analyzed as described in the legend to Fig. 3, with Western blot analysis performed with a HA-specific antibody rather than one specific for GFP. The positions of molecular mass markers (in kilodaltons) are indicated on the left. Error bars represent standard deviations for four replicate experiments.
Figure 10. Characterization of the ability of VP22 acidic cluster motif mutants to bind to the cytoplasmic tail of gE in a GST pulldown assay.

Expression of VP22 acidic cluster motif mutants in transfected Vero cells (A) and (C), their ability to bind to the cytoplasmic tail of gE (B) and (D) and binding efficiency (E) were analyzed as described in the legend to Fig. 1, with Western blot analysis performed with a HA-specific antibody rather than one specific for GFP. The positions of molecular mass markers (in kilodaltons) are indicated on the left. Error bars represent standard deviations for four replicate experiments.
Expression analysis of the various acidic cluster mutants within transfected cells showed that each was expressed and migrated at the expected molecular weight (Fig. 9A and Fig. 9C). With regards to VP16 binding, VP22-HA was efficiently pulled-down from transfected cell lysates by GST-VP16; however, AC→Ala failed to bind, suggesting that the structural conformation of VP22 may be sensitive to alanine mutagenesis (Fig. 9B and Fig. 9E). In contrast, deletion of one or both of the acidic clusters from VP22 had little impact upon interaction with VP16 (Fig. 9B and Fig. 9E). Also, substitution of the furin acidic cluster had minimal effects on VP16 binding (Fig. 9D and Fig. 9E).
Similar results were seen with GST-gECT, with VP22-HA and the acidic cluster deletion mutants binding efficiently, whereas AC→Ala abrogated interaction with gE (Fig. 10). These results suggest that mutation of the acidic cluster does not have a detrimental effect on binding of VP22 to gE or VP16.
An acidic cluster is required to facilitate wild-type levels of VP22 virion packaging
To elucidate the role of the acidic cluster in virion incorporation of VP22, we utilized the packaging assay described above. Western blot analysis of the acidic cluster mutants of VP22 within transfected/infected cells showed that the mutants were expressed at levels similar to wild-type VP22 and migrated at the expected molecular weight (Fig. 11A and Fig. 11C). VP22-HA was packaged into assembling virus particles; however, AC→Ala failed to be incorporated, presumably due to structural defects ensuing from insertion of seven consecutive alanines (Fig. 11E). Upon deletion of the primary acidic cluster (residues 71–77 of VP22), an intermediate phenotype was observed (Fig. 11B and Fig. 11E). ΔAC1 failed to attain wild-type levels of incorporation (77%), yet was more efficiently packaged than VP22.87–301 (51%) (Fig. 7C). However, upon deletion of both acidic clusters from VP22 (ΔAC1 + ΔAC2), packaging was reduced to levels similar to VP22.87–301. This finding further suggests that the secondary acidic cluster may partially substitute for the absence of the primary motif. In addition, the furin acidic cluster substitution mutant was incorporated into virus particles at near wild-type levels (Fig. 11E), indicating that a foreign acidic cluster can functionally substitute for the role of the VP22 acidic cluster in virion packaging. Collectively, these results suggest that an acidic cluster of amino acids is required to ensure localization of VP22 at the TGN and to facilitate efficient incorporation of VP22 into the virion. To our knowledge this is the first description of a VP22 trafficking signal with functional significance for virion incorporation.
Figure 11. Virion incorporation of VP22 acidic cluster motif mutants.

Expression of the indicated VP22-HA constructs in transfected/infected cells (A) and (C), and their incorporation into virus particles (B) and (D) were analyzed as described in the legend to Fig. 4, with Western blot analysis performed with a HA-specific antibody rather than one specific for GFP. The positions of molecular mass markers (in kilodaltons) are indicated on the left. (C) Packaging efficiency. Using densitometry, packaging efficiency was quantitated by dividing the amount of VP22-HA protein detected in extracellular virus particles (normalized for VP5) by the amount in the cell lysate (normalized for VP5). Error bars represent standard deviations for four replicate experiments. Comparison of ΔAC1 and ΔAC1 + ΔAC2 to each other and to wild-type VP22 indicates that the difference in incorporation levels observed is statistically significant (P < 0.001). The difference in packaging levels between wild-type VP22 and a construct in which the primary acidic cluster of VP22 is replaced with that of furin is not significant (P > 0.05).
DISCUSSION
The HSV-1 tegument protein VP22 is packaged into virions during final envelopment as nucleocapsids bud into TGN-derived vesicles (Miranda-Saksena et al., 2002). This observation suggests that VP22 traffics to the TGN where interactions with viral glycoproteins, perhaps in concert with binding to tegument proteins located on the surface of cytoplasmic capsids ensures tegumentation of the protein.
In support of this model, VP22 binds to the viral envelope proteins gM, gD, gE and US9, while the VP22 homologue of the related alphaherpesvirus, PRV, interacts with the cytoplasmic tails of gE and gM (Brignati et al., 2003; Chi et al., 2005; Fuchs et al., 2002; O'Regan et al., 2007a; O'Regan et al., 2007b; Stylianou et al., 2009). Furthermore, VP22 interacts with VP16, whose presence on the surface of capsids undergoing secondary envelopment may facilitate packaging of VP22 into the tegument (Elliott, Mouzakitis, and O'Hare, 1995; Miranda-Saksena et al., 2002; Naldinho-Souto, Browne, and Minson, 2006). A previous study demonstrated that the gE binding domain of VP22 (residues 165–270) which also facilitates binding to VP16, is packaged into assembling virions (O'Regan et al., 2007a). Interestingly, VP16 binding is not essential for incorporation of VP22 into virions (O'Regan et al., 2007b). Thus, to further elucidate the mechanism by which VP22 is packaged into assembling virus particles, our studies have focused on defining the functional significance of gE binding on incorporation of VP22 into the tegument.
To abrogate the VP22-gE interaction with minimal disruption to the overlapping VP16 binding activity, residues 165–270 of VP22 were subjected to extensive point mutagenesis. A variety of point mutants which abrogated gE binding, but not that of VP16, were packaged into assembling virus particles, albeit at reduced levels. It is unclear whether the observed reduction in packaging is a result of the inability of these mutants to bind to gE or is due to the impaired VP16 binding that occurs upon mutation of VP22. Nevertheless these results indicate that gE binding is not absolutely required for virion incorporation of VP22.
In contrast to our findings, a recent report from Stylianou and coworkers described an internal deletion of 14 amino acids within VP22 which abrogated binding to gE and dramatically reduced packaging of VP22 into the virus particle, suggesting that gE is a major determinant of VP22 incorporation (Stylianou et al., 2009). Throughout our studies of VP22 we have noted that the protein seems to be exquisitely sensitive to mutation, with even single point mutations resulting in loss of multiple activities. Interestingly, the VP22 construct described by Stylianou and colleagues, although retaining the ability to bind to VP16, does exhibit decreased binding to the cytoplasmic tail of gM. Thus, we hypothesize that the internal deletion within VP22 in addition to abrogating gE binding, may have inadvertently disrupted unidentified activities of VP22 which facilitate virion incorporation; activities presumably unaffected by the VP22 point mutants characterized in this study.
The potential contribution of other activities of VP22 to its virion incorporation was further highlighted when we examined the possible redundancy that VP16 and gE binding may play in VP22 virion packaging. As addition of VP22 to the tegument is also independent of interaction with VP16, we speculated that both binding activities (gE and VP16) could act in a redundant, or perhaps additive fashion, to facilitate virion packaging of VP22. Functional redundancy is a common theme across herpesvirology, at least for virus growth in cell culture (Brack et al., 1999; Brack et al., 2000; Fuchs et al., 2002). However, studies examining the incorporation of a VP22 construct deficient in VP16 binding into a gE-null virus indicate that gE and VP16 binding do not act in a redundant fashion. This is not overly surprising, as VP22 has additional binding partners including gM, gD, US9, and the membrane associated protein pUL46 (Chi et al., 2005; Lee et al., 2008; Stylianou et al., 2009). Furthermore, it has also been suggested that the C-terminus of VP22 may contain an incorporation determinant of VP22 (Hafezi et al., 2005; Schlegel and Blaho, 2009). However, mutants which harbor this region, in addition to maintaining gE and VP16 binding activities, are still packaged at reduced levels.
In support of the concept that additional incorporation determinants exist, a VP22 construct (VP22.87–301), which binds to both VP16 and gE at wild-type levels, is not incorporated into virus particles at wild-type levels; however, wild-type levels of incorporation were attained when amino acids 44–301 of VP22 were examined (Fig. 7). This result suggests that the additional incorporation determinants for VP22 are putatively located within residues 44–86. The domains of VP22 which interact with gD, US9 and pUL46 have not been characterized; however, a conserved acidic cluster of amino acids is found within this region of VP22 encompassing residues 61–77.
Acidic clusters are well characterized trafficking motifs, shuttling proteins to and from the Golgi apparatus. Considering the dynamic nature of the TGN, one would anticipate that a resident protein such as VP22 would contain sorting sequences that facilitate its vesicular trafficking and retrieval (Brignati et al., 2003). Many herpesvirus proteins utilize such motifs to localize to the TGN (Alconada et al., 1998; Alconada et al., 1999; Loomis et al., 2001; McMillan and Johnson, 2001; Tugizov et al., 1999). Furthermore, an acidic cluster of amino acids appears to be required for virion incorporation of the HSV-1 tegument protein UL11; however, this motif is not sufficient, as UL11 constructs which localize to the TGN and associate with membranes are not packaged into virus particles (Loomis, Courtney, and Wills, 2006).
Mutational analysis of the acidic cluster of VP22 demonstrated that the motif is required to facilitate both TGN localization and wild-type levels of incorporation of VP22 into the virion (Fig. 8 and Fig. 11). Furthermore, upon deletion of the primary acidic cluster from VP22, a secondary group of acidic amino acids, encompassing residues 61–64, could in some measure, functionally substitute. Deletion of the acidic cluster motif had no detrimental effect on the gE and VP16 binding activities of VP22 (Fig. 9 and Fig. 10); however, the effect on other VP22 binding activities was not determined. Thus, although one cannot rule out the possible contribution of gD, US9 or pUL46 binding, or other unidentified activities which may map to this region of VP22, the results suggest that the acidic cluster facilitates efficient incorporation of VP22 into the virion.
Interestingly, two VP22 point mutants (W189F/W221F and F201A) which are both unable to interact with either VP16 or gE (Figs. 1–3) localize to a subcellular compartment reminiscent of the TGN (data not shown). These constructs harbor the acidic cluster motif and residues 165–225 (which facilitate membrane association of VP22), however neither VP22 mutant was packaged into virus particles [Fig. 4 and (O'Regan et al., 2007b)]. This finding suggests that in a similar fashion to UL11, localization to the putative site of final envelopment is not sufficient to facilitate virus packaging of VP22, and additional determinants are required (presumably protein-protein interactions which are disrupted by the point mutations present in these VP22 constructs).
A logical model would suggest that VP22 must first associate with membranes through the actions of the membrane association domain and then localize to the TGN via the acidic cluster motif. The converse is unlikely to occur because acidic cluster motifs are membrane trafficking motifs and would require membrane association for functionality. After localization to the TGN, protein-protein interactions ensure retention of VP22 in the assembly pathway with resultant incorporation into assembling virus particles. In absence of an acidic cluster, protein interactions with VP16 and gE (or other binding partners) may only recruit a portion of VP22 into the assembly pathway (perhaps through encounters with VP16-coated capsids, or with the cytoplasmic tail of gE as VP22 traffics through the TGN).
MATERIALS AND METHODS
Cells and viruses
Vero (ATCC CCL-81) and A7 (human melanoma) cells, a gift from Gary Thomas (The Oregon Health and Science University, Portland, Oregon) were grown in Dulbecco's modified Eagles medium (DMEM) (GIBCO) supplemented with 10% fetal bovine serum (FBS), 2.25% sodium bicarbonate, 25 mM HEPES buffer, glutamine (300 μg/ml), penicillin (100 μg/ml), and streptomycin (131 mg/ml). gD-expressing Vero cells (VD60 cells), a kind gift from David Johnson [The Oregon Health and Science University, Portland, Oregon (Farnsworth, Goldsmith, and Johnson, 2003)] were grown in media lacking histidine supplemented with 1.2 mM histidinol. Infected cells were grown in DMEM supplemented with 2% FBS, 25 mM HEPES buffer, glutamine (300 μg/ml), penicillin (100 μg/ml), and streptomycin (131 mg/ml). The viruses used in this study were the KOS strain (Smith, 1964), a VP22-null virus (UL49−), a kind gift from Joel Baines [Cornell University, Ithaca, New York (Duffy et al., 2006)] and a gE/gD-null virus [vRR1097/gEβ], a kind gift from David Johnson [The Oregon Health and Science University, Portland, Oregon (Farnsworth, Goldsmith, and Johnson, 2003)].
Construction of VP22-GFP and VP22-HA chimeras
Plasmids encoding VP22 fused to GFP, N-terminal truncations of VP22 fused to GFP, internal deletions of VP22 (pVP22.Δ165–270-GFP and pVP22.165–270-GFP) and a dileucine motif mutant of VP22 (pVP22.LI(−)-GFP) were described previously (Brignati et al., 2003; Murphy et al., 2008; O'Regan et al., 2007a; O'Regan et al., 2007b).
To make plasmids encoding the various point mutant variants of VP22, the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) was used according to the manufacturer's instructions. Using pVP22-GFP as a template (Brignati et al., 2003), mutagenic primers were designed which changed the nucleotide sequence of codon 189 of VP22 to encode for a phenylalanine residue rather than a tryptophan; thereby creating pVP22.W189F-GFP. A similar strategy was used to create pVP22.W221F-GFP, pVP22.F196A-GFP, pVP22.F196W-GFP, pVP22.F201A-GFP and pVP22.F201W-GFP. To construct plasmids encoding the double point mutant of VP22, W189F/W221F, the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) was used with pVP22.W189F-GFP acting as a template.
The plasmid encoding VP22 fused to the hemagglutinin epitope (pVP22-HA), has been described previously (O'Regan et al., 2007b). To construct pVP22.AC→Ala-HA, the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) was used according to the manufacturer's instructions. Using pVP22-HA as a template, mutagenic primers were designed which changed the nucleotide sequence of codons 71–77 of VP22 to encode for alanines rather than the residues of the acidic cluster motif. This QuikChange XL Site-Directed Mutagenesis methodology was also utilized to make pVP22.ΔAC1-HA. Mutagenic primers were designed complementary to 15-bp immediately upstream of codon 71 and 15-bp immediately downstream of codon 77, essentially looping out the sequence encoding amino acids 71–77 of VP22 in pVP22-HA. Using pVP22.ΔAC1-HA as a template, a similar approach was used to construct pVP22.ΔAC1+ΔAC2-HA.
The furin acidic cluster was introduced into VP22 through QuikChange mutagenesis. Mutagenic primers were designed to encode the acidic cluster of furin (TCC GAC TCG GAA GAA GAC GAA) and flank both the upstream and downstream sequence of the VP22 acidic cluster with 12 complementary nucleotides.
To create GFP-tagged versions of the acidic cluster motif mutants of VP22, DNA was amplified from the respective HA-tagged acidic cluster constructs by using a forward primer containing a BglII site 92-bp upstream of the start codon of VP22 and a reverse primer containing a HindIII site immediately downstream of codon 301 of VP22. This product was digested with BglII and HindIII and ligated into the vector pEGFP-N2 (Clontech) digested with the same restriction enzymes, to produce pVP22.AC→Ala-GFP, pVP22.ΔAC1-GFP, pVP22.ΔAC1+ΔAC2-GFP and pVP22.FurinAC-GFP.
All constructs were sequenced to confirm the identity of VP22 and to ensure that the gene encoding VP22 (or mutated forms) was in frame with the gene encoding the GFP protein or the sequence that encodes the HA tag.
Construction and purification of GST fusion proteins
Construction of the plasmid encoding VP16 fused to the C-terminus of the GST protein (GST-VP16) was described previously (O'Regan et al., 2007b). The vector encoding the cytoplasmic tail of gE fused to GST (GST-gECT) was a kind gift from David Johnson (The Oregon Health and Science University, Portland, Oregon).
To express and purify the various GST fusion proteins, plasmids encoding the respective constructs were transformed into BL21 competent cells (Stratagene) according to the manufacturer's instructions and overnight cultures were prepared. Approximately 10 ml of these cultures were used to inoculate fresh 100 ml cultures which were subsequently grown at 37 °C to A600=0.4. To induce expression, 100 μL of 1 M IPTG (GIBCO) was added and the cultures were grown at 30 °C for 3–4 h, followed by centrifugation at 10,000 × g for 10 min at 4 °C. The bacterial pellet was resuspended in 10 ml of lysis buffer (100 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1 mM CHAPS, 400 mM NaCl, 1% Triton X-100, Complete Mini protease inhibitors [Roche]). The suspension was sonicated and clarified by centrifugation at 27,000 × g for 10 min at 4 °C. A volume of 133 μl of glutathione-Sepharose 4B beads (Pharmacia) that had been washed twice with phosphate-buffered saline (PBS) was added to the supernatant and incubated overnight at 4 °C. Beads were washed thoroughly with PBS and the yield of each purified GST fusion protein was determined by SDS-PAGE and subsequent staining with Coomassie blue to detect the recombinant protein.
GST pulldown assay
To analyze the VP22-gE and VP22-VP16 interaction within transfected cells, a GST pulldown assay was used. Confluent monolayers of Vero cells grown in 100-mm plates were transfected with the indicated constructs by using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. At 20 h post-transfection, monolayers were washed twice with PBS and scraped into 10 ml of cold PBS. A 1 ml sample of the cell suspension was removed for analysis of expression of the VP22-GFP or VP22-HA fusion proteins, and the remaining cells were pelleted by centrifugation (1000 × g for 5 min at 4 °C) and lysed in NP-40 lysis buffer (1% NP-40, 200 mM NaCl, 50 mM Tris-HCl [pH 7.4], 2mM MgCl2). Transfected-cell lysates were precleared overnight at 4 °C with glutathione-Sepharose 4B beads that had been previously washed twice with PBS, and the samples were then incubated with approximately equal amounts of purified GST fusion proteins on glutathione-Sepharose beads (as determined by Coomassie blue-stained gel) for 3 h at 4 °C. Beads were washed three times with NP-40 lysis buffer and once with 10 mM Tris-HCl (pH 7.4). GFP or HA-tagged VP22 fusion proteins bound to GST constructs were detected by Western blotting using a rabbit polyclonal antibody raised against the GFP protein (Santa Cruz) or a rabbit polyclonal antibody specific for the hemagglutinin epitope (Sigma), a goat anti-rabbit antibody conjugated to horseradish peroxidase (Sigma), ECL reagents (Pharmacia), and autoradiography on Kodak BioMax XAR film. To further control for the quantities of GST fusion proteins used in the pulldown, nitrocellulose membranes were stripped (60 mM Tris-HCl [pH 8.0], 2% SDS, 0.75% β-mercaptoethanol [β-ME] for 45 min at 55 °C) and reprobed with a goat polyclonal antibody raised against the GST protein (Rockland). To detect expression of the VP22-GFP or VP22-HA fusion proteins in transfected cells, proteins in the 1 ml cell suspension sample were analyzed by Western blotting using a rabbit polyclonal antibody against GFP or the hemagglutinin epitope, a goat anti-rabbit antibody conjugated to horseradish peroxidase, ECL reagents, and autoradiography on Kodak BioMax XAR film. To ensure equal loading, nitrocellulose membranes were stripped as described above and reprobed with a goat polyclonal antibody raised against actin (Santa Cruz). Using densitometry, binding efficiency was quantitated by dividing the amount of VP22-GFP or VP22-HA fusion protein detected in the pulldown assay (normalized for the amount of GST-VP16 or GST-gECT present) by the amount in the cell lysate (normalized for the amount of actin present).
Immunoprecipitation-Western assay
Confluent monolayers of Vero cells grown in 100-mm plates were transfected with the indicated constructs by using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. At 20 h post-transfection, the monolayers were infected with HSV-1 KOS strain or a VP22-null virus [UL 49−] at a MOI of 10. At 10 h post-infection (30 h post-transfection), cells were washed twice with PBS and scraped into 10 ml of cold PBS. A 1 ml sample of the cell suspension was removed for analysis of expression of the VP22-GFP fusion proteins, and the remaining cells were pelleted by centrifugation (1000 × g for 5 min at 4 °C). The 9 ml samples were lysed in NP-40 lysis buffer containing Complete Mini protease inhibitors as described above and precleared overnight at 4 °C with protein G-agarose beads (Roche) that had been washed twice in lysis buffer. The lysates were then incubated with a goat polyclonal antibody raised against GFP (Rockland) for 1 h at 4 °C, and immune complexes were collected with protein G-agarose beads that had been washed twice with lysis buffer. Beads were washed three times with NP-40 lysis buffer and once with 10 mM Tris-HCl (pH 7.4). Coimmunoprecipitated VP16 and gE were detected by immunoblot using a rabbit monospecific polyclonal antibody raised against a C-terminal peptide of the HSV-1 tegument protein VP16 (Clontech), or a rabbit polyclonal antibody specific for gE (a kind gift from Harvey Friedman, University of Pennsylvania, Philadelphia, Pennsylvania), a goat anti-rabbit antibody conjugated to horseradish peroxidase, ECL reagents, and chemiluminescence autoradiography on Kodak BioMax XAR film. To detect expression of the VP22-GFP fusion proteins in transfected/infected cells, proteins in the 1 ml cell suspension sample were analyzed by Western blotting using a goat polyclonal antibody against GFP, a rabbit anti-goat antibody conjugated to horseradish peroxidase, ECL reagents, and autoradiography on Kodak BioMax XAR film.
Virion incorporation assay
Confluent monolayers of Vero cells or gD-expressing Vero cells (VD60 cells) grown in 100-mm plates were transfected with the indicated constructs using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. At 20 h post-transfection, cells were infected with either HSV-1 KOS strain, a VP22-null virus (UL 49−), or a gE/gD-null virus [vRR1097/gEβ] at a MOI of 10, or mock infected. At 18 h post-infection (38 h post-transfection), the medium was removed by pipetting and centrifuged at 1000 × g for 10 min at 4 °C to remove cellular debris. The supernatant was retained and extracellular virions were purified by centrifugation (115,000 × g for 1 h in a Beckman SW41 rotor) through a 30% (wt/vol) sucrose cushion (1.7 ml) in NTE buffer (100 mM NaCl, 10 mM Tris, 1 mM EDTA, [pH 7.4]). Pelleted virions and infected cells were disrupted in 1X sample buffer (50 mM Tris-HCl [pH 6.8], 10% glycerol, 2% SDS, 5% β-ME). GFP or HA-tagged proteins were detected by Western blotting using a goat polyclonal antibody raised against the GFP protein (Rockland) or a rabbit polyclonal antibody specific for the hemagglutinin epitope (Sigma), followed by the appropriate secondary antibody conjugated to horseradish peroxidase, ECL reagents, and chemiluminescence autoradiography on Kodak BioMax XAR film. As a loading control, nitrocellulose membranes were stripped as described above and reprobed with either a rabbit polyclonal antibody raised against the major capsid protein VP5 or a rabbit monospecific polyclonal antibody raised against a C-terminal peptide of the HSV-1 tegument protein VP16 (Clontech). As an additional control when using the gE/gD-null virus [vRR1097/gEβ], blots were probed for gD and gE. Using densitometry, packaging efficiency was quantitated by dividing the amount of VP22-GFP or VP22-HA protein detected in extracellular virus particles by the amount in the cell lysate (both normalized for either VP5 or VP16).
Confocal microscopy
Vero cells were transfected with the indicated constructs using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. At 20 h post-transfection, cells were infected with HSV-1 KOS strain at a MOI of 10 or mock infected. At 18 h post-infection (38 h post-transfection), cells were washed twice in Tris-buffered saline (TBS), fixed with 3% paraformaldehyde in PBS and permeabilized with 0.1% Triton-X-100 in PBS. Cell monolayers were incubated for 1 h with a monoclonal antibody raised against AP-1 (Sigma). The cells were then reacted with a goat anti-mouse IgG antibody conjugated to Alexa Fluor 555 (Molecular Probes) and mounted on slides using SlowFade antifade reagent with DAPI (Molecular Probes). Intrinsic GFP fluorescence was observed using a Leica TSC SP2 AOBS confocal microscope using the 488 nm laser line, while immunofluorescence was detected using the 543 nm laser line. Images were formatted in Adobe Photoshop 7.0.
Statistical analysis
For all statistical considerations, the data were normalized to the control prior to analysis. Statistical calculations were performed using GraphPad Prism version 5 software. One way analysis of variance (ANOVA) was used with Bonferroni post test analysis. Statistical significance was set to at least p-value < 0.05.
ACKNOWLEDGEMENTS
We extend special thanks to David G. Meckes Jr. and Nicholas L. Baird for discussions and careful review of the manuscript. We also thank E. Aaron Runkle for help with statistical analysis and Noel Gallagher and Chris Martin for musical accompaniment.
This work was supported by National Institutes of Health (NIH) grant CA42460.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alconada A, Bauer U, Baudoux L, Piette J, Hoflack B. Intracellular transport of the glycoproteins gE and gI of the varicella-zoster virus. gE accelerates the maturation of gI and determines its accumulation in the trans-Golgi network. J Biol Chem. 1998;273(22):13430–6. doi: 10.1074/jbc.273.22.13430. [DOI] [PubMed] [Google Scholar]
- Alconada A, Bauer U, Sodeik B, Hoflack B. Intracellular traffic of herpes simplex virus glycoprotein gE: characterization of the sorting signals required for its trans-Golgi network localization. J Virol. 1999;73(1):377–87. doi: 10.1128/jvi.73.1.377-387.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol CellBiol. 2004;5(1):23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- Brack AR, Dijkstra JM, Granzow H, Klupp BG, Mettenleiter TC. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J Virol. 1999;73(7):5364–72. doi: 10.1128/jvi.73.7.5364-5372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AR, Klupp BG, Granzow H, Tirabassi R, Enquist LW, Mettenleiter TC. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J Virol. 2000;74(9):4004–16. doi: 10.1128/jvi.74.9.4004-4016.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignati MJ, Loomis JS, Wills JW, Courtney RJ. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J Virol. 2003;77(8):4888–98. doi: 10.1128/JVI.77.8.4888-4898.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucks MA, O'Regan KJ, Murphy MA, Wills JW, Courtney RJ. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology. 2007;361(2):316–24. doi: 10.1016/j.virol.2006.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi JH, Harley CA, Mukhopadhyay A, Wilson DW. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J Gen Virol. 2005;86(Pt 2):253–61. doi: 10.1099/vir.0.80444-0. [DOI] [PubMed] [Google Scholar]
- Duffy C, Lavail JH, Tauscher AN, Wills EG, Blaho JA, Baines JD. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J Virol. 2006;80(17):8664–75. doi: 10.1128/JVI.00498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott G, Hafezi W, Whiteley A, Bernard E. Deletion of the herpes simplex virus VP22-encoding gene (UL49) alters the expression, localization, and virion incorporation of ICP0. J Virol. 2005;79(15):9735–45. doi: 10.1128/JVI.79.15.9735-9745.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott G, Mouzakitis G, O'Hare P. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J Virol. 1995;69(12):7932–41. doi: 10.1128/jvi.69.12.7932-7941.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott GD, Meredith DM. The herpes simplex virus type 1 tegument protein VP22 is encoded by gene UL49. J Gen Virol. 1992;73(Pt 3):723–6. doi: 10.1099/0022-1317-73-3-723. [DOI] [PubMed] [Google Scholar]
- Enquist LW, Husak PJ, Banfield BW, Smith GA. Infection and spread of alphaherpesviruses in the nervous system. Adv Virus Res. 1998;51:237–347. doi: 10.1016/s0065-3527(08)60787-3. [DOI] [PubMed] [Google Scholar]
- Farnsworth A, Goldsmith K, Johnson DC. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J Virol. 2003;77(15):8481–94. doi: 10.1128/JVI.77.15.8481-8494.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth A, Wisner TW, Johnson DC. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J Virol. 2007;81(1):319–31. doi: 10.1128/JVI.01842-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W, Klupp BG, Granzow H, Hengartner C, Brack A, Mundt A, Enquist LW, Mettenleiter TC. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J Virol. 2002;76(16):8208–17. doi: 10.1128/JVI.76.16.8208-8217.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi W, Bernard E, Cook R, Elliott G. Herpes simplex virus tegument protein VP22 contains an internal VP16 interaction domain and a C-terminal domain that are both required for VP22 assembly into the virus particle. J Virol. 2005;79(20):13082–93. doi: 10.1128/JVI.79.20.13082-13093.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, Paragas J, Sudol M, Palese P. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J Virol. 1999;73(4):2921–9. doi: 10.1128/jvi.73.4.2921-2929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine JW, Honess RW, Cassai E, Roizman B. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J Virol. 1974;14(3):640–51. doi: 10.1128/jvi.14.3.640-651.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilsley JL, Sudol M, Winder SJ. The WW domain: linking cell signalling to the membrane cytoskeleton. Cell Signal. 2002;14(3):183–9. doi: 10.1016/s0898-6568(01)00236-4. [DOI] [PubMed] [Google Scholar]
- Ingham RJ, Colwill K, Howard C, Dettwiler S, Lim CS, Yu J, Hersi K, Raaijmakers J, Gish G, Mbamalu G, Taylor L, Yeung B, Vassilovski G, Amin M, Chen F, Matskova L, Winberg G, Ernberg I, Linding R, O'Donnell P, Starostine A, Keller W, Metalnikov P, Stark C, Pawson T. WW domains provide a platform for the assembly of multiprotein networks. Mol Cell Biol. 2005;25(16):7092–106. doi: 10.1128/MCB.25.16.7092-7106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T. Adaptors for clathrin-mediated traffic. Annu Rev Cell Dev Biol. 1999;15:705–32. doi: 10.1146/annurev.cellbio.15.1.705. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T, Bonifacino JS, Riezman H. Linking cargo to vesicle formation: receptor tail interactions with coat proteins. Curr Opin Cell Biol. 1997;9(4):488–95. doi: 10.1016/s0955-0674(97)80024-5. [DOI] [PubMed] [Google Scholar]
- Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology. 2008;378(2):347–54. doi: 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- Leslie J, Rixon FJ, McLauchlan J. Overexpression of the herpes simplex virus type 1 tegument protein VP22 increases its incorporation into virus particles. Virology. 1996;220(1):60–8. doi: 10.1006/viro.1996.0286. [DOI] [PubMed] [Google Scholar]
- Loomis JS, Bowzard JB, Courtney RJ, Wills JW. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J Virol. 2001;75(24):12209–19. doi: 10.1128/JVI.75.24.12209-12219.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis JS, Courtney RJ, Wills JW. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J Virol. 2003;77(21):11417–24. doi: 10.1128/JVI.77.21.11417-11424.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis JS, Courtney RJ, Wills JW. Packaging determinants in the UL11 tegument protein of herpes simplex virus type 1. J Virol. 2006;80(21):10534–41. doi: 10.1128/JVI.01172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman MG, Demmin GL, Banfield BW. The attenuated pseudorabies virus strain Bartha fails to package the tegument proteins Us3 and VP22. J Virol. 2003;77(2):1403–14. doi: 10.1128/JVI.77.2.1403-1414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan TN, Johnson DC. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J Virol. 2001;75(4):1928–40. doi: 10.1128/JVI.75.4.1928-1940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC. Herpesvirus assembly and egress. J Virol. 2002;76(4):1537–47. doi: 10.1128/JVI.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC, Klupp BG, Granzow H. Herpesvirus assembly: an update. Virus Res. 2009;143(2):222–34. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Miranda-Saksena M, Boadle RA, Armati P, Cunningham AL. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J Virol. 2002;76(19):9934–51. doi: 10.1128/JVI.76.19.9934-9951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzakitis G, McLauchlan J, Barreca C, Kueltzo L, O'Hare P. Characterization of VP22 in herpes simplex virus-infected cells. J Virol. 2005;79(19):12185–98. doi: 10.1128/JVI.79.19.12185-12198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Lee GE, Wilson DW. The amino terminus of the herpes simplex virus 1 protein Vhs mediates membrane association and tegument incorporation. J Virol. 2006;80(20):10117–27. doi: 10.1128/JVI.00744-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MA, Bucks MA, O'Regan KJ, Courtney RJ. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology. 2008;376(2):279–89. doi: 10.1016/j.virol.2008.03.018. [DOI] [PubMed] [Google Scholar]
- Naldinho-Souto R, Browne H, Minson T. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J Virol. 2006;80(5):2582–4. doi: 10.1128/JVI.80.5.2582-2584.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Regan KJ, Bucks MA, Murphy MA, Wills JW, Courtney RJ. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE) Virology. 2007a;358(1):192–200. doi: 10.1016/j.virol.2006.08.024. [DOI] [PubMed] [Google Scholar]
- O'Regan KJ, Murphy MA, Bucks MA, Wills JW, Courtney RJ. Incorporation of the herpes simplex virus type 1 tegument protein VP22 into the virus particle is independent of interaction with VP16. Virology. 2007b;369(2):263–80. doi: 10.1016/j.virol.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Pomeranz LE, Blaho JA. Assembly of infectious Herpes simplex virus type 1 virions in the absence of full-length VP22. J Virol. 2000;74(21):10041–54. doi: 10.1128/jvi.74.21.10041-10054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Pellett PE. The family herpesviridae: A brief introduction. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott-Raven; Philadelphia: 2001. pp. 2381–2387. [Google Scholar]
- Sanchez V, Greis KD, Sztul E, Britt WJ. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol. 2000;74(2):975–86. doi: 10.1128/jvi.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel EF, Blaho JA. A conserved carboxy-terminal domain in the major tegument structural protein VP22 facilitates virion packaging of a chimeric protein during productive herpes simplex virus 1 infection. Virology. 2009;387(2):449–58. doi: 10.1016/j.virol.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skepper JN, Whiteley A, Browne H, Minson A. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment --> deenvelopment --> reenvelopment pathway. J Virol. 2001;75(12):5697–702. doi: 10.1128/JVI.75.12.5697-5702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KO. Relationship between the Envelope and the Infectivity of Herpes Simplex Virus. Proc Soc Exp Biol Med. 1964;115:814–6. doi: 10.3181/00379727-115-29045. [DOI] [PubMed] [Google Scholar]
- Stylianou J, Maringer K, Cook R, Bernard E, Elliott G. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 occurs via glycoprotein E-specific recruitment to the late secretory pathway. J Virol. 2009;83(10):5204–18. doi: 10.1128/JVI.00069-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugizov S, Maidji E, Xiao J, Pereira L. An acidic cluster in the cytosolic domain of human cytomegalovirus glycoprotein B is a signal for endocytosis from the plasma membrane. J Virol. 1999;73(10):8677–88. doi: 10.1128/jvi.73.10.8677-8688.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Genderen IL, Brandimarti R, Torrisi MR, Campadelli G, van Meer G. The phospholipid composition of extracellular herpes simplex virions differs from that of host cell nuclei. Virology. 1994;200(2):831–6. doi: 10.1006/viro.1994.1252. [DOI] [PubMed] [Google Scholar]
- Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7(8):639–43. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol. 2005;79(15):9566–71. doi: 10.1128/JVI.79.15.9566-9571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Molloy SS, Thomas L, Liu G, Xiang Y, Rybak SL, Thomas G. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998;94(2):205–16. doi: 10.1016/s0092-8674(00)81420-8. [DOI] [PubMed] [Google Scholar]
- Whealy ME, Card JP, Meade RP, Robbins AK, Enquist LW. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J Virol. 1991;65(3):1066–81. doi: 10.1128/jvi.65.3.1066-1081.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley A, Bruun B, Minson T, Browne H. Effects of targeting herpes simplex virus type 1 gD to the endoplasmic reticulum and trans-Golgi network. J Virol. 1999;73(11):9515–20. doi: 10.1128/jvi.73.11.9515-9520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]