

Abstract
Our goal is to obtain a comprehensive description of molecular processes occurring at cellular membranes in different biological functions. We aim at characterizing the complex organization and dynamics of the plasma membrane at single-molecule level, by developing analytic tools dedicated to Single-Particle Tracking (SPT) at high density: Multiple-Target Tracing (MTT)1. Single-molecule videomicroscopy, offering millisecond and nanometric resolution1-11, allows a detailed representation of membrane organization12-14 by accurately mapping descriptors such as cell receptors localization, mobility, confinement or interactions.
We revisited SPT, both experimentally and algorithmically. Experimental aspects included optimizing setup and cell labeling, with a particular emphasis on reaching the highest possible labeling density, in order to provide a dynamic snapshot of molecular dynamics as it occurs within the membrane. Algorithmic issues concerned each step used for rebuilding trajectories: peaks detection, estimation and reconnection, addressed by specific tools from image analysis15,16. Implementing deflation after detection allows rescuing peaks initially hidden by neighboring, stronger peaks. Of note, improving detection directly impacts reconnection, by reducing gaps within trajectories. Performances have been evaluated using Monte-Carlo simulations for various labeling density and noise values, which typically represent the two major limitations for parallel measurements at high spatiotemporal resolution.
The nanometric accuracy17 obtained for single molecules, using either successive on/off photoswitching or non-linear optics, can deliver exhaustive observations. This is the basis of nanoscopy methods17 such as STORM18, PALM19,20, RESOLFT21 or STED22,23, which may often require imaging fixed samples. The central task is the detection and estimation of diffraction-limited peaks emanating from single-molecules. Hence, providing adequate assumptions such as handling a constant positional accuracy instead of Brownian motion, MTT is straightforwardly suited for nanoscopic analyses. Furthermore, MTT can fundamentally be used at any scale: not only for molecules, but also for cells or animals, for instance. Hence, MTT is a powerful tracking algorithm that finds applications at molecular and cellular scales.
Keywords: Physics, Issue 63, Single-particle tracking, single-molecule fluorescence microscopy, image analysis, tracking algorithm, high-resolution diffusion map, plasma membrane lateral organization
Protocol
In this video, we present a full single particle tracking experiment, using quantum-dots targeted to a specific membrane receptor. The main goal of this experiment consists in discriminating different types of molecular diffusion behaviors measured within the plasma membrane of live cells. Indeed, molecular movements arising at the membrane can typically deviate from Brownian diffusion by being linearly directed or confined within nanodomains26-29, for instance. We aim at simultaneously following as many receptors as technically possible, to provide a snapshot of the variety arising in the dynamics occurring within the membrane of a live cell. This is ultimately expected to allow deciphering of the mechanisms regulating cell surface receptor signaling.
1. Cell Culture
Prepare the cellular sample: use adherent COS-7 cells which endogenously express the epidermal growth factor receptor (EGFR)30. When working with live cells without antibiotics make sure to have no latent sub-detectable contamination by using proper sterile techniques at all times during the preparation.
Grow cells in complete medium (DMEM with 10% calf serum, 1% glutamine, 1% HEPES and 1% sodium pyruvate, see table of specific reagents below), at 37 °C with 7% CO2, taking care to have them in sub-confluent, exponential growth before spreading them in Lab-Tek.
On the day before the experiment, spread 5,000 cells/well in 8-well chambers (Lab-Tek) and incubate overnight. Counting cells ensures a constant cell density, hence a reproducible ratio of quantum-dots per cell.
2. Cell Labeling
Prepare quantum-dots with a specific coating. Quantum-dots are fluorescent nanoparticles composed of semiconductors. These nanoparticles present a high interest because they are very bright and photostable compared to classical fluorescent probes31,32, which allows achievement of an appropriate signal to noise ratio (SNR) for single molecule imaging.
Before the experiment, produce Fab fragments against EGFR from hybridoma cell line (mAb 108, ATCC HB-9764), by digestion with papain as previously described21.
Conjugate Fab with biotin, as per manufacturer's instructions (EZ-link sulfo-NHS-LC-biotinylation Kit; Thermo Scientific, Fig. 1A).
Use quantum-dots functionalized with streptavidin (Invitrogen) and emitting at 605 nm (optimal emission wavelength for brilliance, detection and separation from cellular autofluorescence).
Prepare the labeling solution in complete medium (see table of specific reagents), in order to saturate the streptavidins present around the quantum-dots, as well as to prevent aggregation and non-specific binding to the cells and to the coverslip.
Prepare two intermediate solutions: one with quantum-dots-streptavidin and another one with biotinylated Fab, each at 20 nM.
Mix an equal volume of these two solutions: mix the quantum-dots with Fab in 1:1 ratio to obtain a final working concentration of 10 nM Fab:quantum-dots complex. Using an equimolar ratio favors the formation of mono-functionalized complexes composed of one quantum-dot and one biotinylated Fab fragment. This favors monovalent labeling, limiting artifact observations due to receptor cross-linking33,34.
Incubate for 15 minutes at 25 °C, using a shaker at 1,200 rotations per minute to prevent aggregation. The mix is then ready to use on living cells.
Incubate cells in 100 μl of mix for 5 minutes at 37 °C with 7% CO2.
Wash cells with non-autofluorescent imaging medium (HBSS buffer, 1% HEPES, see table of specific reagents) pre-warmed at 37 °C.
Carefully remove the excess of labels to prevent unnecessary spoiling the SNR by unbound, out of focus quantum-dots, by extensively washing each well with imaging medium, typically 5 times at room temperature, with at least 5 minutes delay before the last wash.
3. Optical Setup
The video-microscopy setup is composed of four major parts:
An inverted microscope with a specific fluorescence cube (FF01-457/50 excitation, FF495-Di02 dichroic, and FF01-617/73 emission filters, Semrock) and a high numerical aperture (1.3 or 1.49) oil-immersion 100x objective.
A 100 W mercury lamp (coupled to the microscope through an optic fiber, avoiding interfering with thermoregulation).
A 512 x 512 pixels high sensitivity EMCCD camera to achieve a sufficient SNR.
An incubator to keep biological samples at 37 °C during the experiment.
4. Acquisition
Select isolated and well-spread cells. This favors the extension of nice, flat lamellipodia, best adapted to look for planar motion of membrane molecules.
Evaluate the cellular physiological status by its appearance both in transmitted light and in fluorescence: intense vesicular traffic, absence of necrotic or apoptotic signs, low autofluorescence, and average-strong quantum-dot labeling (typically up to 1,000 quantum-dots per cell, see Fig. 1B, C).
Select a high labeling density, which is critical for simultaneously monitoring as many receptors as possible. This is actually limited both by physiological (surface expression, epitope accessibility, lateral motion) and algorithmic parameters (non-overlapping point-spread functions (PSF), even taking into account blurring due to motion, to allow proper detection and reconnection).
For each cell, first acquire a brightfield field image (preferentially using DIC, if available) that can further allow checking for the cell aspect and spatial limits of the lamellipodia.
Save this image using the same name as the video-stack (such as cell1.tif & cell1.stk for instance), in a sub-folder named dic, since, with these conventions, the algorithm can automatically find back the matching image for each stack.
Acquire 1 to 3 videos per cell, continuously, typically at 36-ms rate, the fastest rate achievable in full frame with this camera. However one can acquire at higher frequencies, up to 1-ms rate, using dedicated CCD with increased sensitivity and/or less pixels. The frame-transfer technology provides negligible delay between frames.
Electron multiply enhancement should always be set at maximum (just below saturation, if relevant) to allow reaching single molecule sensitivity, with a sufficient SNR, at least above 20 dB (for efficient peak detection1), typically around 25-30 dB.
Typically acquire 300 frames per video, since trajectories are reconstructed over ~100 frames on average, mostly limited by long blinking events. Video frequency and length can be adapted for a given measure, which could require longer traces for instance.
5. MTT Analysis
Select the path to the directory containing the video files to evaluate a given dataset with Matlab or Octave.
To start the fully automatized analysis1,35, type the command detect_reconnex23 in Octave, or MTT23i in Matlab. This program stands for "version 2.3, with user interface" and is available for download, together with the previous version 2.2. It first displays a graphic interface listing all the parameters used, as shown in Fig. 2.
6. Representative Results
MTT automatically analyzes each recorder video, to deliver traces of detected and estimated targets, complemented by further investigations, such as confinement detection. This ultimately allows mapping traces over cell images (Fig. 1C & 3).
MTT Description
The core MTT analysis is performed over each frame, invoking 3 main tasks (Fig. 3):
Detection of the presence or absence of a target (quantum-dot), within a sliding sub-region successively centered around each pixel, where two hypotheses are compared: presence either of a signal, with the PSF modelized as a bi-dimensional Gaussian peak, or only noise, using a threshold insuring low enough false alarm, with less than one spurious detection per frame. This leads to a map of detection probability. Each local maximum is considered as a putative target, insuring that its probability level is higher than a minimum value, set according to the desired Probability of False Alarm (PFA). This limit is set low enough to efficiently discriminate signals from noise, avoiding at best spurious detections (PFA of 10-6 by default, to insure less than one error in 512 x 512 pixel images), while still allowing a sufficiently high probability of detection, reaching the theoretically predicted optimum1. Note that the requirement of using a sub-region implies that the image borders (3 pixels, for a default window of 7 x 7 pixels) cannot be evaluated.
Estimation, for each detected target, of the relevant parameters, such as sub-pixel position and signal intensity. For every detected target, a least-square Gauss-Newton fit is next performed to estimate the position, width and height of the detected Gaussian. This provides notably the sub-pixel position of the dye (10 to 20 nm accuracy for typical SNR values and immobile or slowly diffusing dyes, increasing up to ~100 nm for dyes diffusing at 0.1 μm2/s).
Reconnection of the new targets with the traces already built over the previous frames. The set of new targets is matched with the set of previous traces. For this purpose, in order to assign each target to a trace, if possible, all available statistical information obtained from the detection step is used, not only position, but also intensity, width, blinking and associated statistics. Hence, targets are not just assigned to the nearest trace: in case of crossing traces, intensity, speed, width and blinking will be considered. This delivers the statistically optimal reconnection score. This strategy avoids, when possible, biasing the reconnection toward the nearest neighbors.
Detected peaks can be a posteriori rejected if their estimation or reconnection fails. A special test handles the detection of new peaks, which would initiate new traces. This test uses a more stringent PFA (10-7), since reconnecting a peak to a trace can be interpreted de facto as a validation of its relevance (this criterion being by definition not applicable for new peaks).
Trajectory Analysis
Possible transient confinement is next evaluated by a function inversely related to local diffusion24-29. Applying a threshold allows to define confined or not episodes. By iterating these over all traces, we can map membrane dynamics, in terms of transient confinement/slow down events. This can be alternatively represented using either the binary or discrete values of this confinement index.
By default, MTT automatically performs those tasks, saving 8 peak parameters in a text file: frame number, i and j position, signal intensity, radius, offset and blink, for each video frame (group of 7 rows) and trace (column). These output parameters can be reloaded in Matlab or Octave using the fread_data_spt script for further analyzing i.e. traces or signal intensities, as exemplified in the MTT_example script in appendix.
Further analyses lead to map traces over each cell (Fig. 1C & 3) and to provide histogram distribution for relevant parameters (such as peak intensities, SNR or local diffusion values). For each file, mean and standard deviation of each parameter are saved in a text file, together with an image of the histograms. Logarithmic distributions, such as for square displacements r2, lead to geometric mean. The diffusion coefficient D is computed from a linear fit over the five first points of the MSD curve. These values provide an overview of an experiment involving for example kinetics of cellular reactions or pharmaceutical/enzymatic treatments affecting membrane organization. Since MTT is an open-source code, this aspect may be readily adapted to any dedicated investigation.
 Figure 1. Monitoring membrane receptors dynamics by MTT. (A) Membrane components, such as the EGFR, are tagged with quantum-dots coupled to biotinylated Fab fragments (schematic drawing with approximately correct scaling of each molecule). (B) Typical fluorescent image acquired from a live COS-7 cell, with 36-ms exposure time, depicting diffraction-limited peaks corresponding to individually labeled receptor. (C) Output of the MTT analysis displaying the reconstituted trajectories of the receptors, overlaid on the brightfield image of the cell.
Figure 1. Monitoring membrane receptors dynamics by MTT. (A) Membrane components, such as the EGFR, are tagged with quantum-dots coupled to biotinylated Fab fragments (schematic drawing with approximately correct scaling of each molecule). (B) Typical fluorescent image acquired from a live COS-7 cell, with 36-ms exposure time, depicting diffraction-limited peaks corresponding to individually labeled receptor. (C) Output of the MTT analysis displaying the reconstituted trajectories of the receptors, overlaid on the brightfield image of the cell.
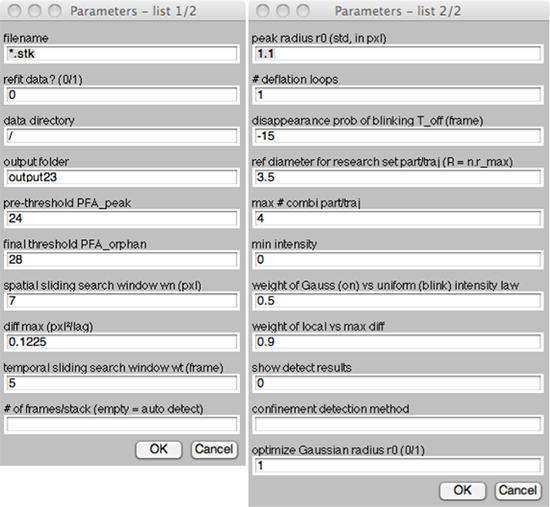 Figure 2. MTT input parameters. Running MTT23i opens a graphic user interface listing all input parameters, names and default values, as described in our previous publication1. In the algorithm, space and time parameters (search windows, peak radius, maximum diffusion and blinking) are in dimensionless standard units, pixels and frames. Calibrations can be applied a posteriori to convert output results. Default values, corresponding to a Cascade 512BFT with 100x magnification, are pixel size: 156 nm/pxl and frame delay: 36 ms/frame.
Figure 2. MTT input parameters. Running MTT23i opens a graphic user interface listing all input parameters, names and default values, as described in our previous publication1. In the algorithm, space and time parameters (search windows, peak radius, maximum diffusion and blinking) are in dimensionless standard units, pixels and frames. Calibrations can be applied a posteriori to convert output results. Default values, corresponding to a Cascade 512BFT with 100x magnification, are pixel size: 156 nm/pxl and frame delay: 36 ms/frame.
Investigators should optimize a few critical parameters, such as the maximum expected diffusion coefficient ("Diff max") and maximum blinking disappearance ("Toff"). These two space and time limits are almost the only ones that need to be reconsidered for a given experimental condition, the others being set to robust default values. For instance, the number of false alarms is directly set to ensure less than one error per million pixels, hence less than one per frame, which is satisfactory in most cases. All parameters are saved in a text file in the output folder, allowing users to verify afterwards which settings were used for analysis.
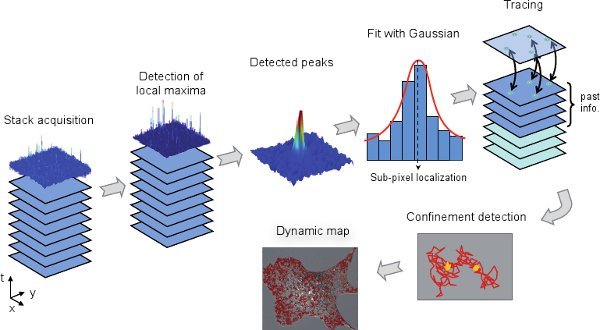 Figure 3. Major steps of the MTT analysis. Starting from an experimental stack of fluorescence images, peaks are sequentially and automatically detected, estimated by a Gaussian fit and reconnected over frames (first range of operations, upper part of the flowchart). Traces can further be analyzed for putative confinement, for instance, ultimately leading to a dynamic map of relevant descriptors (second range of operations, lower part).
Figure 3. Major steps of the MTT analysis. Starting from an experimental stack of fluorescence images, peaks are sequentially and automatically detected, estimated by a Gaussian fit and reconnected over frames (first range of operations, upper part of the flowchart). Traces can further be analyzed for putative confinement, for instance, ultimately leading to a dynamic map of relevant descriptors (second range of operations, lower part).
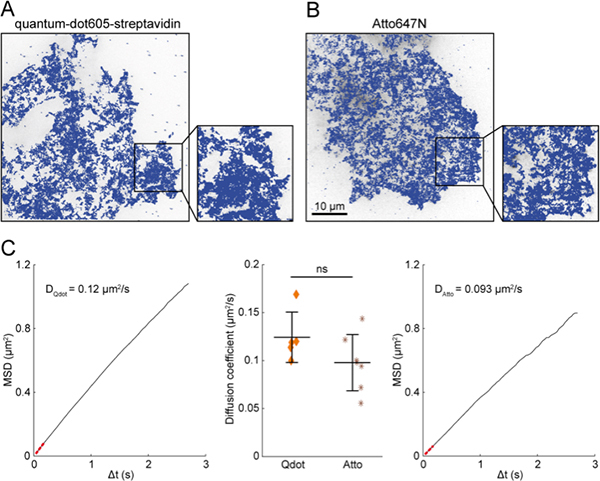 Figure 4. Labeling valency does not impact MTT. To evaluate the possible bias introduced by artefactual multivalent labeling, MTT analysis was performed to track endogenous EGFR tagged using 2 different schemes, to generate and analyze maps of trajectories. (A) Receptors were tagged with biotinylated Fab and quantum-dots605-streptavidin, as described in the protocol. In this case, the multi-valency of quantum-dots and streptavidins may result in coupling several receptors to a single dye. (B) Receptors were tagged with Fab directly coupled to an organic dye, Atto647N. In this case, one Fab, hence one receptor, can be coupled to more than one dye. (C) Mean square displacement (MSD) curve was computed for all traces for each cell, labeled either with quantum-dot or Atto dyes (left and right graphs, respectively). Diffusion coefficients were computed by linear fit over the five first points of the MSD (red dotted line). Each scheme of labeling led to similar diffusion values (central graph). Qdot: quantum-dots605 (n = 5 cells), Atto: Atto647N (n = 7 cells), ns: not significant (Student's t-test p value > 0.05).
Figure 4. Labeling valency does not impact MTT. To evaluate the possible bias introduced by artefactual multivalent labeling, MTT analysis was performed to track endogenous EGFR tagged using 2 different schemes, to generate and analyze maps of trajectories. (A) Receptors were tagged with biotinylated Fab and quantum-dots605-streptavidin, as described in the protocol. In this case, the multi-valency of quantum-dots and streptavidins may result in coupling several receptors to a single dye. (B) Receptors were tagged with Fab directly coupled to an organic dye, Atto647N. In this case, one Fab, hence one receptor, can be coupled to more than one dye. (C) Mean square displacement (MSD) curve was computed for all traces for each cell, labeled either with quantum-dot or Atto dyes (left and right graphs, respectively). Diffusion coefficients were computed by linear fit over the five first points of the MSD (red dotted line). Each scheme of labeling led to similar diffusion values (central graph). Qdot: quantum-dots605 (n = 5 cells), Atto: Atto647N (n = 7 cells), ns: not significant (Student's t-test p value > 0.05).
Discussion
In single-particle tracking, beside the cell and microscopy aspects, the analysis represents a substantial part of the work. This addresses the algorithm used to perform the three main tasks: detecting, estimating and reconnecting peaks over each frame. But the consequent aspect of this work resides in elaborating the algorithm itself, which may need to be adapted for any new dedicated investigation, essentially for the last, extra steps (such as deciphering modes of motion, interactions or stoichiometry).
However, once the algorithm is fully developed, running it is straightforward, especially since we paid attention at keeping the number of input parameters at minimum (Fig. 3). This renders MTT very robust and easy to implement. When elaborating MTT, we aimed at entirely reconsidering the analytical options used for each task. We wanted to optimize the process along two challenging axes:
Dealing with high densities of labeling provides detailed spatial information in term of cell surface sampling. Ideally, one would try to follow a nearly complete molecular population simultaneously, in order to obtain a comprehensive observation. However, such exhaustive labeling would lead to an image similar to that classically obtained by immunofluorescence, without single-molecule resolution, since all labels strongly overlap due to diffraction. For typical molecular number (i.e. ~105 receptors by cell30) and cell size (i.e. ~30 μm), if we assume homogeneous distribution, the average inter-molecule distance is ~100 nm. Diffusion during acquisition also contributes to spreading the PSF to a comparable extent: typically ~100 nm for Brownian motion at 0.1 μm2/s during 30 ms. Noteworthy, this spreading is isotropic on average and thus still generates a Gaussian-like PSF. Hence up to 10% of the population could be theoretically detected, since this would lead to an average distance of ~300 nm, compatible with diffraction and motion limits. However, this needs to be modulated by accounting for inhomogeneity in the spatial distribution, labeling limitations (such as steric constraints, epitope accessibility, etc.), increasing and even unsolvable conflicts due to crossing trajectories, and overall detection efficiency. Experimentally, we could reach and accurately monitor a substantial fraction of the overall population, with densities of ~1000 labels within a field of view of 80 μm width. This upper limit results from a compromise between the need for individual detections and the goal of an exhaustive spatial measurement (see Fig. 1C to appreciate the spatial sampling of the cell surface).
Handling the weakest possible SNR allows imaging either at low illumination, which is beneficial for cells viability, and/or at high speed, which provides better temporal information. However, this implies lower signals per image, due to lower number of collected photons. Note that the shortest timing achievable by EMCCD cameras (1 ms) appears to correspond to the lowest acceptable SNR (20 dB) for proper detection and estimation by MTT.
Labeling valency is a recurrent issue in single molecule studies. Indeed, direct measure of the dye/target ratio is highly challenging34. However, an alternate way for investigating the putative bias introduced by artefactual multivalent labeling consists in comparing measures with either quantum-dot tags (with putatively several targets per dye, inducing artefactual crosslinking) or organic dye tags (with, at contrary, putatively several dyes per target, biasing only signal intensity and bleaching characteristics). We and others6,36,37 have observed comparable behavior when using either quantum-dots or organic dyes (atto647N in our case, Fig. 4), in terms of diffusion and transition between modes of motion. Variation in diffusion values between the two conditions is lower than the cell-to-cell disparity (Fig. 4C). This shows that labeling valency, even if not strictly monovalent, does not significantly affect SPT results.
Confinement and Molecular Interactions
Transient or stable confinement can be interpreted as the signature of preferential affinity or interaction24-29. Biomembranes are indeed strongly inhomogeneous, with local assemblies dictated by structural features such as hydrophobicity, transmembrane size and interfacial tension. These driving forces lead to spatiotemporal remodeling of functional assemblies with critical roles for i.e. signaling, adhesion or trafficking. Mapping and quantifying such events is expected to provide a key to decipher how a cell integrates continuously presented stimuli. Indeed, for a cell, exhibiting an adequate adaptation through an accurate response requires tuning interactions among signaling partners and their environment. Membrane receptors may interact either with the sub-membrane cytoskeleton fences, membrane structures such as endocytic pits or focal adhesions, or also proteic/lipid partners, involved in signaling domains for instance. In our representation, such events can be investigated through confinement strength and its variations over space and time. This further strengthens the importance of working at the highest achievable space-time resolution, keeping in mind the constraints associated to short timings and high densities, as discussed above.
Complementary Techniques
It is a good practice to compare such dynamic measurements with other approaches. For instance other dynamic microscopy techniques, like FRAP and FCS, provide complementary sensitivity and resolution4,38,39. FRAP provides an ensemble measurement of molecular diffusion, while FCS can reach single-molecule sensitivity, with a very good time resolution. However, both methods are inherently restricted to a local measure (typically within a confocal spot). This motivated us to take advantage and push the limits the spatial possibilities of SPT: investigating a large field of view, to encompass a whole cell at once, together with a locally relevant spatiotemporal accuracy.
Further Developments & Investigations
According to the range of biological questions that may be addressed using single-molecule measurements, MTT can be extended along various directions, at each level: detection, estimation, reconnection and further analyses. For instance, one may consider dealing with more than one molecular population, calling for multicolor detection – as can be performed using dedicated optic or analytic schemes. Estimation can include new relevant descriptors, such as any spectral or polarization information, or the axial z position, for extending tracing into 3D40.
For reconnection, the Brownian and blinking assumptions can be fully reconsidered for a specific problem. Interestingly enough, single-molecule measurements have been extended over the last decade to the so-called nanoscopic regime17,22,23. Although MTT is directly addressing single-molecule localization, it is of crucial importance to consider the case of a fixed sample, which provides a safe solution for an exhaustive localization of a molecular population. However, in such a case, the Brownian assumption is no longer valid. Replacing it by accurately handling a nanoscopic measure, only limited by SNR, is critical for reaching the best nanometric resolution.
Along such developments, based on either multicolor, 3D and/or exhaustive measures, future work is expected to deliver a comprehensive view of molecular static and dynamic data. This will have a direct relevance for cell biology studies, such as deciphering the subtle modalities regulating extra and intracellular signaling.
Downloading MTT
The source code of MTT is available as open-source software for academic research. It can be downloaded from our web page, at ciml.univ-mrs.fr, by navigating to our team's page, He & Marguet Lab, which provides a link for software description and download. Please note that we ask you for your name and institution only for information, for instance in case of further collaboration or to inform you about a new release or update.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank members of our team, particularly MC Blache for technical assistance, as well as M Irla and B Imhof, for their support and fruitful discussions. Figures for deflation and confinement reproduced courtesy of Nature Methods. This project is supported by institutional grants from the CNRS, INSERM and Marseille University, and by specific grants from the Région Provence-Alpes-Côte-d'Azur, Institut National du Cancer, Agence Nationale de la Recherche (ANR-08-PCVI-0034-02, ANR 2010 BLAN 1214 01) & Fondation pour la Recherche Médicale (Equipe labélisée FRM-2009). VR is supported by a fellowship from the Ligue Nationale Contre le Cancer.
References
- Serge A, Bertaux N, Rigneault H, Marguet D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat. Methods. 2008;5:687–694. doi: 10.1038/nmeth.1233. [DOI] [PubMed] [Google Scholar]
- Schmidt T, Schutz GJ, Baumgartner W, Gruber HJ, Schindler H. Imaging of single molecule diffusion. Proc. Natl. Acad. Sci. U S A. 1996;93:2926–2929. doi: 10.1073/pnas.93.7.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommerse PH. Single-molecule imaging of the H-ras membrane-anchor reveals domains in the cytoplasmic leaflet of the cell membrane. Biophys. J. 2004;86:609–616. doi: 10.1016/S0006-3495(04)74139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marguet D, Lenne PF, Rigneault H, He HT. Dynamics in the plasma membrane: how to combine fluidity and order. EMBO. J. 2006;25:3446–3457. doi: 10.1038/sj.emboj.7601204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton MJ, Jacobson K. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- Dahan M. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- Harms GS. Single-molecule imaging of l-type Ca(2+) channels in live cells. Biophys. J. 2001;81:2639–2646. doi: 10.1016/S0006-3495(01)75907-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino R, Koyama I, Kusumi A. Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface. Biophys. J. 2001;80:2667–2677. doi: 10.1016/S0006-3495(01)76236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako Y, Minoghchi S, Yanagida T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- Schutz GJ, Kada G, Pastushenko VP, Schindler H. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. Embo. J. 2000;19:892–901. doi: 10.1093/emboj/19.5.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger G. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science. 2001;294:1929–1932. doi: 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]
- Jacobson K, Sheets ED, Simson R. Revisiting the fluid mosaic model of membranes. Science. 1995;268:1441–1442. doi: 10.1126/science.7770769. [DOI] [PubMed] [Google Scholar]
- Saffman PG, Delbruck M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. U S A. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Papoulis A. Probability, Random Variables and Stochastic Process 277. McGraw Hill; 2001. [Google Scholar]
- Van Trees HL. Detection, Estimation, and Modulation Theory, Wiley Inter-Science. 1968.
- Moerner WE. Single-molecule mountains yield nanoscale cell images. Nat. Methods. 2006;3:781–782. doi: 10.1038/nmeth1006-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Manley S. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- Andrew SM. Enzymatic digestion of monoclonal antibodies. Methods Mol. Med. 2000;40:325–331. doi: 10.1007/978-1-59259-076-6_25. [DOI] [PubMed] [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Klar TA, Hell SW. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999;24:954–956. doi: 10.1364/ol.24.000954. [DOI] [PubMed] [Google Scholar]
- Meilhac N, Guyader LLe, Salome L, Destainville N. Detection of confinement and jumps in single-molecule membrane trajectories. Phys. Rev. E. Stat. Nonlin. Soft. Matter Phys. 2006;73:011915. doi: 10.1103/PhysRevE.73.011915. [DOI] [PubMed] [Google Scholar]
- Saxton MJ. Single-particle tracking: effects of corrals. Biophys. J. 1995;69:389–398. doi: 10.1016/S0006-3495(95)79911-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serge A, Fourgeaud L, Hemar A, Choquet D. Receptor activation and homer differentially control the lateral mobility of metabotropic glutamate receptor 5 in the neuronal membrane. J. Neurosci. 2002;22:3910–3920. doi: 10.1523/JNEUROSCI.22-10-03910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simson R, Sheets ED, Jacobson K. Detection of temporary lateral confinement of membrane proteins using single-particle tracking analysis. Biophys. J. 1995;69:989–993. doi: 10.1016/S0006-3495(95)79972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K, Dietrich C. Looking at lipid rafts. Trends Cell Biol. 1999;9:87–91. doi: 10.1016/s0962-8924(98)01495-0. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Sako Y, Yamamoto M. Confined lateral diffusion of membrane receptors as studied by single particle tracking (nanovid microscopy). Effects of calcium-induced differentiation in cultured epithelial cells. Biophys. J. 1993;65:2021–2040. doi: 10.1016/S0006-3495(93)81253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh E. Large deletions in the cytoplasmic kinase domain of the epidermal growth factor receptor do not affect its laternal mobility. J. Cell Biol. 1986;103:327–331. doi: 10.1083/jcb.103.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Quantum dot bioconjugates for imaging, labelling and. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- Wu X, Bruchez MP. Labeling cellular targets with semiconductor quantum dot conjugates. Methods Cell Biol. 2004;75:171–183. doi: 10.1016/s0091-679x(04)75007-4. [DOI] [PubMed] [Google Scholar]
- Mohammadi M. Aggregation-induced activation of the epidermal growth factor receptor protein tyrosine kinase. Biochemistry. 1993;32:8742–8748. doi: 10.1021/bi00085a004. [DOI] [PubMed] [Google Scholar]
- Howarth M. Monovalent, reduced-size quantum dots for imaging receptors on living cells. Nat. Methods. 2008;5:397–399. doi: 10.1038/nmeth.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertaux N, Marguet D, Rigneault H, Sergé A. Multiple-target tracing (MTT) algorithm probes molecular dynamics at cell surface. Protocol Exchange. 1038.
- Groc L. Surface trafficking of neurotransmitter receptor: comparison between single-molecule/quantum dot strategies. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:12433–12437. doi: 10.1523/JNEUROSCI.3349-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B. One at a time, live tracking of NGF axonal transport using quantum dots. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13666–13671. doi: 10.1073/pnas.0706192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HT, Marguet D. Detecting nanodomains in living cell membrane by fluorescence correlation spectroscopy. Annu. Rev. Phys. Chem. 2011;62:417–436. doi: 10.1146/annurev-physchem-032210-103402. [DOI] [PubMed] [Google Scholar]
- Cebecauer M, Spitaler M, Serge A, Magee AI. Signalling complexes and clusters: functional advantages and methodological hurdles. J. Cell. Sci. 2010;123:309–320. doi: 10.1242/jcs.061739. [DOI] [PubMed] [Google Scholar]
- Kao HP, Verkman AS. Tracking of single fluorescent particles in three dimensions: use of cylindrical optics to encode particle position. Biophys. J. 1994;67:1291–1300. doi: 10.1016/S0006-3495(94)80601-0. [DOI] [PMC free article] [PubMed] [Google Scholar]