

Abstract
Background & Aims
Ballooned hepatocytes in nonalcoholic steatohepatitis (NASH) generate sonic hedgehog (SHH). This observation is consistent with a cellular phenotype in which the cell death program has been initiated but cannot be executed. Our aim was to determine if ballooned hepatocytes have potentially disabled the cell death execution machinery, and if so, can their functional biology be modeled in vitro.
Methods
Immunohistochemistry was performed on human NASH specimens. In vitro studies were performed using Huh-7 cells with shRNA targeted knockdown of caspase 9 (shC9 cells) or primary hepatocytes from caspase 3−/− mice.
Results
Ballooned hepatocytes in NASH display diminished expression of the caspase 9. This phenotype was modeled using shC9 cells; these cells were resistant to lipoapoptosis by palmitate (PA) or lysophosphatidylcholine (LPC) despite lipid droplet formation. During lipid loading by either PA or LPC, shC9 cells activate JNK which via AP-1 induces SHH expression. An autocrine hedgehog survival signaling pathway was further delineated in both shC9 and caspase 3−/− cells during lipotoxic stress.
Conclusion
Ballooned hepatocytes in NASH downregulate caspase 9, a pivotal caspase executing the mitochondrial pathway of apoptosis. Hepatocytes engineered to reduce caspase 9 expression are resistant to lipoapoptosis, in part, due to a hedgehog autocrine survival signaling pathway.
Keywords: caspase 3, c-Jun-N-terminal Kinase (JNK), lipoapoptosis, lysophosphatidylcholine, palmitate
Human hepatocellular injury is ubiquitous due to the prevalence of steatohepatitis syndromes, viral hepatitis, immune mediated insults, genetic diseases, etc. [1]. In these hepatic diseases, hepatocyte fate is often modeled as a binary process - cells either survive or die to be replaced by liver regenerative processes [2]. Yet, cells may resist cell death by down regulating or inhibiting cell death programs, thereby surviving in an altered state. This new cell fate does not merely reflect an escape from death, but rather results in the formation of a unique, functional cell phenotype. The cell phenotype of nonlethal cell injury has been best characterized in model systems where it has been termed the ‘undead cell’ [3, 4]. Because cell death by proapoptotic stimuli requires activation of executioner caspases [5], the undead cell can be engineered by deletion of these downstream caspases. For example, in Drosophila melanogaster genetic deletion of the executioner caspase DrICE or Dcp-1 results in a cell type in which otherwise toxic stimuli activate alternative cell signaling cascades producing signals which enhance tissue remodeling [3, 4, 6].
A candidate undead cell in the liver is the ballooned hepatocyte in nonalcoholic steatohepatitis (NASH). Ballooned hepatocytes are thought to be important in the pathogenesis of NASH and the presence of such cells is used to score disease severity [7, 8]. Little is known about the ballooned hepatocytes other than they are enlarged cells which have lost cell polarity, frequently contain Mallory-Denk bodies, store neutral triglycerides, contain oxidized phospholipids, no longer stain for cytokeratin 18, and generate the ligand sonic hedgehog [8–10]. The seminal work by Diehl and colleagues demonstrating sonic hedgehog (SHH) generation by ballooned hepatocytes suggests this cell may, in fact, be a functional cell with an altered phenotype [8, 11]. For example, in Drosophilia melanogaster retinal cells in which the cell death program is initiated but cannot be executed also generate SHH [6]. The mechanisms by which undead cells continue to evade cell death remain elusive, but given the potency of SHH as a survival factor, it is possible that this ligand serves as an autocrine survival factor.
Herein, we demonstrate that ballooned cells have reduced expression of a potent downstream caspase, caspase 9, suggesting they may not efficiently execute cell death pathways. To understand the functional implications of this observation, we modeled lipotoxic stress in Huh-7 cells in which caspase 9 had been knocked down by short hairpin RNA technology (shC9 cells). Knockdown of caspase 9 protected hepatocytes from lipotoxicity by the saturated free fatty acid (FFA) palmitate (PA) or the phospholipid lysophosphatidylcholine (LPC). Interestingly, PA or LPC treated shC9, but not wild type cells, generate sonic hedgehog by a c-Jun-N-terminal kinase (JNK)-dependent pathway. Blockade of hedgehog signaling resulted in cell death of shC9 deficient hepatocytes following exposure to lipotoxic agents. Similar observations were confirmed in primary caspase 3−/− hepatocytes. We speculate that these engineered cells model the functional phenotype of ballooned hepatocytes, albeit not the specific morphology, and, in turn, support the concept of ballooned hepatocytes as an altered, but functional cell population which has escaped cell death.
EXPERIMENTAL PROCEDURES
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded liver sections (5 μm thick) from three patients with nonalcoholic steatohepatitis (NASH) were obtained following approval by the Institutional Review Board (IRB), Mayo Clinic, Rochester MN. In conjunction with the clinical core of P30DK084576, the sections were pre-selected by an experienced hepatopathologist for their abundance of ballooned hepatocytes. These sections were deparaffinized in xylene and rehydrated through graded alcohols. After tissue permeabilization in 0.1% Triton X100 for 2 minutes, antigen retrieval was performed by incubation in sodium citrate buffer (0.01M sodium citrate, 0.05% Tween 20; pH 6.0) for 30 minutes utilizing a vegetable steamer. Samples were cooled down, and endogenous peroxidase activity was quenched by incubation for 15 minutes in 3% H2O2 (diluted in deionized H2O). Sections were washed in changes of phosphate-buffered saline (PBS) three times for 5 minutes. The VECTASTAIN Elite ABC and ImmPACT VIP peroxidase substrate kits (Vector laboratories, Burlingame, CA) were used in further steps according to manufacturer’s instructions. After blocking non-specific binding for one hour, sections were incubated with anti-Caspase-9 p10 (F-7) mouse monoclonal primary antibodies (1:50; sc-17784) or anti-Caspase-2L (C-20) goat monoclonal primary antibodies (1:50; sc-626-G, Santa Cruz Biotechnology, Santa Cruz, CA) for 30 min at room temperature. Prior to mounting, the sections were counterstained with hematoxylin for 3 min (Sigma-Aldrich, St. Louis, MO). Samples were examined with light microscopy. Analysis of at least 20 cells of each type from each patient was conducted using Image J version 1.44 software (NIH, Bethesda, MD). Ballooned cells were confirmed in hematoxylin and eosin (H&E) stained sections.
Cells
Huh-7 cells, a human hepatocellular carcinoma cell line, were maintained in Dulbecco’s modified Eagle’s medium containing glucose (25 mM) supplemented with 10% fetal bovine serum, 100,000 IU/l penicillin and 100mg/l streptomycin. We also employed Huh-7 cells stably expressing a short-hairpin RNA (shRNA) targeting caspase 9, shC9 cells. Mouse primary hepatocytes (MPH) were isolated from C57BL/6 wild type (Jackson Laboratory, Bar Harbor, ME) and Caspase3−/− mice by collagenase perfusion and plated as primary cultures [12]. Development and characterization of the Caspase3−/− mice has been previously reported [13]. MPH were maintained in Waymouth medium supplemented with 10% fetal bovine serum, 100,000 IU/l penicillin, 100 mg/l streptomycin and 100 nM insulin.
Phase contrast, fluorescent and confocal microscopy
Huh-7 cells and shC9 cells were grown on glass bottom culture dishes for phase contrast and fluorescence microscopy, or glass coverslips for confocal microscopy. Following studies were conducted in conjunction with the microscopy core of the P30DK084567. For live cell imaging, lipid droplets were fluorescently labeled by incubation with 100 μg/ml of Bodipy 505/515 (Invitrogen, Camarillo, CA) for 1 minute at 37°C. Identical cells were viewed using phase contrast and then fluorescent optics on a Zeiss Axiovert 200 epifluorescent microscope (Zeiss, Thornwood, NJ). Fluorescence was visualized using excitation and emission wavelengths of 488 and 507 nm, respectively. Both phase contrast and fluorescent digital images were acquired and analyzed using a cooled-CCD Orca II camera (Hamamatsu Photonics, Hamamatsu City, Japan) with IPLab software (Scanalytics, Fairfax, VA). Fluorescent lipid droplet area as a percent of the total cellular area was quantified using Image J version 1.44 software (NIH, Bethesda, MD).
In separate studies, cells were fixed with 3% paraformaldehyde for 30 min at room temperature [14]. Intracellular lipid droplets were labeled with 10 μg/ml of Bodipy 505/515 for 10 min at room temperature, and mounted in Prolong Antifade mounting media (Invitrogen, Camarillo, CA). Lipid droplet fluorescence intensity per cell was quantified using an inverted phase/fluorescence microscope (Axiovert 35M; Carl Zeiss, Inc., Thornwood, NY) and Metafluor quantitative fluorescence imaging software from Universal Imaging Corp. (West Chester, PA) [15]. For confocal microscopy, cells were fixed and labeled as described above and visualized using an inverted laser-scanning confocal microscope (Zeiss LSM 510, Carl Zeiss, Jena, Germany) with excitation and emission wavelengths of 488 and 507 nm, respectively.
For SHH or ubiquitin immunocytochemistry, cells plated on coverslips were fixed with 4% paraformaldehyde in PBS for SHH staining or with cold acetone on dry ice for ubiquitin staining, respectively, and permeabilized with 0.0125% (w/v) CHAPS in PBS for both staining. The primary antisera was rabbit anti-SHH (sc-9024, Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:300 or rabbit anti-ubiquitin (Z0458, Dako, Carpinteria, CA) at a dilution of 1:500. The secondary antisera was Alexa Fluor 488-conjugated or 568-conjugated anti-rabbit IgG (Molecular Probes, Eugene, OR), and ProLong Antifade with DAPI (Molecular Probes) was used as mounting medium. Images were acquired by confocal microscopy employing excitation and emission wavelengths of 488 nm and 507 nm for SHH staining or excitation and emission wavelengths of 558 and 583 nm for ubiquitin staining. Cells with positive immunoreactivity for the active conformation of SHH or Cells with ubiquitin-punctate staining, respectively, were counted and expressed as a percentage of total cells.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as previously described by us in detail [16]. Briefly, nuclear extracts from shC9 cells were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Pierce Biotechnology, Rockford, IL,) according to the manufacturer’s instructions. For the EMSA, 20 μg of nuclear proteins were incubated at room temperature for 20 min in binding buffer (25 mM HEPES, pH 7.5, 0.1 M NaCl, 2 mM EDTA, 6% glycerol, 0.1% Triton X-100, 0.1 mM phenylmethylsulfonyl fluoride, 0.4 mM dithiothreitol, 0.5 μg/μl poly(dI-dC), 0.5 μg/μl salmon sperm) with 0.04 pmol of CY 5.5-labeled double-stranded DNA oligonucleotides containing either one of the putative Activator protein-1 (AP-1) binding sequences (BS1) or (BS2). Each sequence with the mutated putative AP-1 binding sequence (mutant BS1 or mutant BS2) was also employed as a negative control (supplemental Table 1). Protein-DNA complexes were separated from the unbound DNA probe by electrophoresis through 5% native polyacrylamide gels containing 0.5x Tris borate-EDTA. Fluorescence was visualized directly on the gel using an Odyssey fluorescent imager (Licor Biosciences, Lincoln, NE). For competition assays, a 200-fold molar excess of unlabeled double-stranded oligonucleotide was added to the reaction mixture 20 min before the addition of the fluorescent probe. In the supershift assays, nuclear cell extract was first incubated at room temperature for 25 min with 2 μg of anti-phopho-c-Jun antibody (Santa Cruz Biotechnology). The entire antibody/protein mixture was then incubated with CY 5.5-labeled probe and processed for the gel shift as described above.
Chromatin immunoprecipitaion (ChIP) assay
ChIP assays were performed employing shC9 (6–10 million cells) using a commercially available assay (Active Motif, Carlsbad, CA) according to the manufacturer’s instructions [17]. Briefly, shC9 cells were treated for 6 hours with 800 μM palmitate or for 4 hours with 85 μM LPC; the cells were then fixed with 1% formaldehyde in PBS at room temperature for 10 min. The formaldehyde crosslinking reaction was quenched with 125 mM glycine, the cells were lysed, and chromatin was sheared into small and uniform fragments by an enzymatic reaction. Following centrifugation, the DNA concentration in the supernatant was quantified by measuring absorbance at 260 nm. Samples containing 30 μg of DNA were next incubated with Protein G magnetic beads, ChIP Buffer 1, Protease Inhibitor Cocktail, the phospho-c-Jun antibody at 2 μg and dH2O at 4°C overnight. The immune complex was separated from the sample buffer by a magnetic stand and washed sequentially with ChIP Buffer1 and ChIP Buffer 2. The sample was next eluted with Elution Buffer AM2 and then added to the Reverse Cross-linking Buffer and heated at 95°C for 15 min. The proteins were digested by proteinase K, and the DNA in the ChIP sample was used as the template for quantitative real-time PCR. The primers used in this assay are depicted in supplemental Table 2.
Immunoblot analysis
Whole cell lysates were prepared as previously described[12]. Nuclear cell extracts were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Pierce Biotechnology, Rockford, IL) according to the manufacturer’s instructions. Equal amount of protein (50 to 80 μg) was resolved by SDS-PAGE on a 12.5–18% acrylamide gel, transferred to nitrocellulose membranes, and incubated with primary antibodies. Membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Biosource International, Camarillo, CA). Bound antibody was visualized using chemiluminescent substrate (ECL; Amersham, Arlington Heights, IL) and exposure to Kodak X-OMAT film (Eastman Kodak, Rochester, NY).
Materials and Methods for Treatment of hepatocytes with lipids, Quantitation of cell death, and Quantititative real-time PCR, Gli reporter assay, Antibodies and reagents, and Statistics are in the supplementary information.
RESULTS
Ballooned hepatocytes in NASH display reduced caspase 9 expression
Loss of executioner caspase function is sufficient to prevent cell death in developmental systems. Therefore, we performed immunohistochemistry in human NASH specimens for caspase 9, a potent executioner caspase. Ballooned hepatocytes, but not neighboring hepatocytes, displayed diminished caspase 9 expression (Fig. 1A). In contrast, expression of the upstream caspase 2 was readily identified in ballooned hepatocytes (Fig. 1A). Caspase 2 has a long prodomain similar to caspase 9, and therefore, served as a control for a caspase of similar size as caspase 9. These suggest ballooned hepatocytes are not merely damaged cells on the path to cellular demise, but likely are functional cells which have downregulated cell death pathways.
Figure 1. Ballooned hepatocytes display decreased caspase 9 in NASH specimens; loss of caspase 9 or 3 enhances lipid loading in vitro.

(A) Ballooned hepatocytes (BH) and nonballoned hepatocytes (NH, arrow) were identified in H&E stained sections (circled cells). Immunohistochemistry for caspase 2 and 9 was performed in sections from the same specimens. Caspase 2 and 9 staining intensity was quantified by NIH Image J software (right panel). (B) Whole cell lysates were prepared from wild type (WT) and shC9 cells and immunoblot analysis performed for caspase 9. (C) WT and shC9 cells were incubated with vehicle (Veh), palmitate (PA) or oleate (OA) at 400 μM for 12 hours. Lipid droplets were labeled with Bodipy 505/515, and cells imaged by phase contrast microscopy and fluorescent microscopy. Lipid droplet area and total cell area were measured by NIH Image J software. (D) WT and shC9 cells were incubated with PA for 12 hours at 0, 400, or 800 μM, respectively, labeled with Bodipy 505/515, and imaged by confocal microscopy. Fluorescence intensity was quantified using Metafluor quantitative software and expressed as fold change over vehicle. All data are expressed as mean ± SEM for at least three experiments; *p<0.01.
Diminished cell death in hepatocytes deficient in caspase 9 or 3 despite lipid droplet accumulation
We sought to model the ballooned hepatocyte phenotype with down regulation of caspase 9, and therefore, generated shC9 Huh-7 cells (shC9 cells) (Fig. 1B). As assessed by phase contrast morphology, the shC9 cells did not appear different from the parent cell line. The shC9 cells (Fig. 1C), however, actually demonstrated more robust lipid droplet formation as compared to wild type cells following incubation with oleate (OA) or palmitate (PA) (Fig. 1C and D). The enhanced lipid loading of shC9 cells could not be explained by decreased lipolysis as expression of the rate limiting enzyme for lipolysis[18, 19], acyl-CoA oxidase 1, palmitoyl (ACOX-1), or β-oxidation, carnitine palmitoyltransferase 1A and 2 (CPT-1A and -2), acetyl-CoA carboxylase alpha (ACACA) and acetyl-CoA carboxylase beta (ACACB) were not downregulated in these cells (supplemental Fig. 1). Rather enhanced lipid droplet accumulation by shC9 cells likely reflects their ability to resist lipoapoptosis by free fatty acids allowing further lipid accumulation over time. As the saturated free fatty acid PA induces lipoapoptosis via a metabolite lysophosphatidylcholine (LPC) [12], we quantified apoptosis in shC9 and wild type cells using both PA and LPC. Using either lipotoxic agent, shC9 cells were more resistant to PA or LPC cytotoxicity (supplemental Fig. 2A, B and C). To confirm these results in primary hepatocytes, we employed hepatocytes from wild type and caspase 3 deficient (C3−/−) mice. We posited that given the dominant role of caspase 3 as an executioner caspase downstream of caspase 9 these cells may also be resistant to lipotoxicity (supplemental Fig. 3B), and indeed, they were apoptosis resistant, despite lipid droplet accumulation (supplemental Fig. 3A). Finally, because ballooned hepatocytes in NASH liver display enhanced ubiquitin chain accumulation [20], we performed immunocytochemistry for ubiquitin chains following PA treatment. shC9 cells manifest increased punctate ubiquitin staining following PA treatment, further suggesting their phenotypic similarity to ballooned hepatocytes (supplemental Fig. 4). Taken together, these data demonstrate that knockdown or deletion of caspases downstream of mitochondrial dysfunction results in cells resistant to lipoapoptosis in spite of lipid droplet accumulation and ubiquitin chain accumulation. However, we note that these cells do not completely model the ballooned cellular morphology observed in human NASH.
PA and LPC induce SHH expression in lipoapoptosis resistant hepatocytes by a JNK-dependent pathway
Next, we examined SHH generation by shC9 cells subjected to lipotoxic stress by PA or LPC. shC9 and caspase 3−/−, but not wild type Huh-7 cells, expressed SHH at both mRNA and protein level following treatment with either PA or LPC (Fig. 2A, B and C). In Drosophila melanogaster, the undead cell phenotype requires activation of initiator caspases which induce alternative signaling pathways in the absence of downstream caspases [3, 4]. Therefore, we next explored the effect of the pan-caspase inhibitor Z-VAD-fmk on SHH generation by shC9 cells treated with PA or LPC. Z-VAD-fmk treatment blunted SHH expression in shC9 cells exposed to either PA or LPC (Fig. 3A). During lipoapoptosis, c-Jun N-terminal kinase (JNK) activation occurs by a mechanism downstream of caspase 8 activity [12, 17, 21]. Consistent with this information, Z-VAD-fmk treatment reduced phosphorylation of c-Jun, a JNK substrate, during incubation of shC9 cells with PA or LPC (Fig. 3B). As lipotoxic stress is associated with death receptor 5-dependent activation of caspase 8[22], presumably Z-VAD-fmk is inhibiting this initiator caspase. To determine whether JNK activation directly contributes to SHH expression during lipotoxic stress, we employed the pharmacological JNK inhibitor SP600125. SP600125, at a concentration which blocks c-Jun phosphorylation, reduced SHH expression in this model (Fig. 3C and D). Collectively, these observations implicate JNK activation in the generation of SHH in hepatocytes resistant to apoptosis.
Figure 2. PA- and LPC-induces SHH expression in shC9 cells and primary mouse hepatocytes from caspase 3−/− animals.

(A) Wild type (WT) and shC9 cells were incubated with vehicle (Veh), PA at 800 μM for 16 hours, or LPC at 85 μM for 6 hours. SHH mRNA was quantified by real-time PCR, normalized to 18S rRNA, and expressed as fold change over vehicle. (B) Mouse primary hepatocytes (MPH) from WT and caspase 3 deficient animals (C3−/−) were incubated with Veh, PA at 400μM for 12 hours, or LPC at 42.5 μM for 6 hours. SHH mRNA was quantified by real-time PCR. (C) WT and shC9 cells were incubated with Veh, PA at 800 μM for 8 hours or LPC at 85 μM for 4 hours, respectively. Immunofluorescence was performed for SHH and visualized by confocal microscopy; the number of SHH-positive versus total cells is depicted. All data are expressed as mean ± SEM for three experiments; *p<0.01.
Figure 3. PA- and LPC-induce SHH expression via AP-1 mediated transcription.

(A) shC9 cells incubated with PA at 800 μM for 16 hours in the absence or presence of the pancaspase inhibitor Z-VAD-fmk (ZVAD, 10 μM) and SHH mRNA expression quantified by real-time PCR. (B) Cells were treated with vehicle (Veh), PA as above or LPC at 85 μM for 8 hrs. Immunoblot analysis was performed for phospho-c-Jun (p-c-Jun) or total c-Jun using whole cell lysates. (C) shC9 cells were treated with PA as per panel A in the presence or absence of the JNK inhibitor SP600125 (50 μM) and SHH mRNA expression quantified using real-time PCR. (D) Whole cell lysates were prepared from shC9 cells treated with PA or LPC as above and immunoblot analysis performed for p–c-Jun or total c-Jun. (E) A schematic for the SHH gene is depicted along with the two putative AP-1 binding sites (BS1 and BS2). shC9 cells were incubated with PA or LPC as described above. Nuclear protein extracts were isolated and EMSA performed using a CY 5.5-labeled AP-1 probe. Anti-phospho-c-Jun antibodies were used in the supershift experiments. Protein complexes are indicated by arrows (S, shift). No specific retarded complexes were observed when adding 200-fold molar excess of the unlabeled probe or when using the CY 5.5-labeled mutated probe. CY 5.5-labeled probe only was used as a negative control. (F) shC9 cells were incubated with PA or LPC as above. Cells were fixed and lysed, DNA fragments pulled down with p-c-Jun antibody, and the collected DNA subjected to quantitative RT-PCR analysis using primers for BS1 or BS2. DNA fragments pulled down with IgG were used as negative control for the p-c-Jun specific antisera. Primers of unrelated sequence were used as negative control for the BS1 or BS2 specific primers. All data are expressed as mean ± SEM for three experiments; *p<0.01, **p<0.05.
Activator protein-1 (AP-1) mediates SHH transcription during lipotoxic stress
Phosphorylated c-Jun heterodimerizes with other proteins to form the AP-1 transcription factor complex [23]. To ascertain if AP-1 may directly up-regulate SHH transcription, computational analysis of the human SHH gene sequence was performed to identify consensus AP-1 binding sites. Two putative AP-1 binding sites were identified within the human SHH promoter; one at −4525/−4518 (Binding Sequence 1, BS1) and a second at -2608/−2601 (Binding Sequence 2, BS2) (Fig. 3E). To test if AP-1 complexes bind to these sites, an EMSA was performed using CY5.5-labeled oligonucleotide probes for each putative binding site (supplemental Table 1). Using nuclear extracts obtained from either PA or LPC treated shC9 cells, protein binding to both BS1 and BS2 was observed (Fig. 3E). This binding was no longer observed when extracts were incubated with anti-phospho-c-Jun antisera, as presumably the antisera binding to AP-1 masks the oligonucleotide binding site; an observation implicating AP-1 in formation of the protein:oligonucleotide complex. Binding was also no longer observed when extracts were incubated with a 200-fold molar excess of the unlabeled probe, or when using a mutated probe in which the critical AP-1 binding residues had been altered. Finally, to further confirm that AP-1 binds to these promoter sequences of the SHH gene, a ChIP assay was performed using the primers listed in supplemental Table 2. Immunoprecipitation of c-Jun from nuclear extracts obtained from PA- or LPC-treated shC9 cells captured both putative AP-1 binding sequences within the SHH promoter (Fig. 3F). In contrast, no binding of c-Jun to an unrelated upstream region of the SHH promoter was observed. These data provide evidence for two bonafide AP-1 binding sequences within the SHH promoter suggesting AP-1 may induce expression of this gene.
SHH provides autocrine survival signals during lipotoxic stress
As critical as caspases are for cell apoptosis, loss of caspase activity often delays but does not abrogate cell death in mammalian cells [24]. Therefore, we anticipated that SHH expression functions as an additional survival factor during lipotoxic stress given its role as a potent survival factor in other systems [6, 25–27]. To test this concept, we first looked for active hedgehog signaling in the shC9 cells. Following binding of SHH to its cognate receptor Patched, Smoothened is derepressed resulting in translocation of the Gli transcription factors into the nucleus, especially Gli2 [28, 29]. Indeed, Gli2 translocation into nuclear cell fractions was readily identified in PA and LPC treated shC9 cells (Fig. 4A). Hedgehog signaling in a feed forward manner also increases Gli2 mRNA expression [29], which was observed in shC9 cells following exposure to PA or LPC (Fig. 4B). To further examine hedgehog signaling, a Gli reporter gene assay was employed. Consistent with the above observations, PA or LPC increased Gli reporter gene activity in shC9 cells (Fig. 4C). These observations identified an autocrine hedgehog signaling pathway in shC9 cells following lipotoxic stress. To explore this concept, shC9 cells were treated either with GANT61 (a Gli1/2 inhibitor) or GDC-0449 (a Smoothened antagonist) following by exposure to PA or LPC [30]. GANT61 as well as GDC-0449 prevented PA- and LPC-induced Gli2 mRNA expression and Gli reporter gene activity (Fig. 4B and C). Because shC9 cells were generated from a human hepatoma cell line, Huh-7 cells, we confirmed the above findings in mouse primary hepatocytes. When mouse primary hepatocytes from C3−/− mice were treated either PA or LPC, Gli2 mRNA was increased 4-fold (supplemental Fig. 5A); furthermore, pretreatment with GANT51 or GDC-0449 following by exposure to PA or LPC, suppressed this increase in Gli2 mRNA expression (supplemental Fig. 5A). Finally, we sought to determine if this hedgehog autocrine signaling pathway functions as a survival pathway in both shC9 cells and C3−/− hepatocytes subjected to lipotoxic stress. PA- and LPC-induced lipoapoptosis was enhanced by concurrent treatment with either GANT61 or GDC-0449 in both shC9 cells and C3−/− hepatocytes (Fig. 4D and supplemental Fig. 5B). Taken together, these observations suggest that lipotoxic stress in hepatocytes evading cell death results in a hedgehog signaling pathway further endowing the cell with potent survival signals.
Figure 4. An autocrine SHH survival pathway in shC9 cells during lipotoxic stress.

(A) Nuclear protein extracts were isolated from shC9 cells treated with vehicle (Veh), PA or LPC as described in Fig. 2. and immunoblot analysis performed for Gli2. Lamin B was used as a loading control. (B) shC9 Huh-7 cells were pre-incubated for 24 hours with vehicle, the Gli1/2 inhibitor GANT61 at 5 μM, or the Smoothened antagonist GDC-0449 at 20 μM. After pre-incubation, shC9 Huh-7 cells were incubated with Vehicle (Veh), PA at 800 μM for 16 hours or LPC at 85 μM for 6 hours. Gli2 mRNA was quantified by real-time PCR, normalized to 18S rRNA, and expressed as fold change over vehicle. (C) Wild type (WT) and shC9 cells were transiently transfected (16 hours) with a reporter construct, containing eight consecutive consensus GLI binding sites (8x-GLI), and cotransfected with pRL-CMV. Cells were pre-incubated with/without GANT61 or GDC-0049 as described above, and then treated as indicated with PA at 800 μM for 16 hours or LPC at 85 μM for 10 hours. Cells were lysed and firefly and Renilla luciferase activities quantified; data (firefly/Renilla luciferase activity) are expressed as fold increase over vehicle-treated cells. (D) After pre-incubation with GANT61 or GDC-0449, shC9 cells were incubated for 16 hours with PA at 400 μM or LPC at 42.5 μM, respectively. Cell death was quantified as propidium iodide positive (PI) cells expressed as a percentage of total cell number. All data are expressed as mean ± SEM for three experiments; *p<0.01.
DISCUSSION
We posited that the ballooned hepatocyte in NASH represents an altered cellular phenotype which has escaped cell death analogous to the ‘undead cell’ in developmental models. Consistent with this concept, we observed a reduction of caspase 9 protein expression in ballooned hepatocytes. We sought to model this phenotype in vitro and explore its functional biology. Our results indicate that during lipotoxic stress by PA or LPC: 1) targeted knockdown of caspase 9 or genetic deficiency of caspase 3 reduces cell death despite accumulation of lipid droplets; 2) surviving cells display an altered phenotype as manifest by JNK-dependent expression of SHH; and 3) an autocrine SHH pathway contributes to cell survival. The results of this study provide new mechanistic insights regarding the phenotype of hepatocytes undergoing lipotoxic stress without succumbing to cell death, and have implications for understanding the functional biology of the ballooned hepatocyte in NASH.
Hepatocyte lipotoxicity involves a complex network of proapoptotic stimuli. Saturated free fatty acids such as palmitate promote formation of plasma membrane subdomains resulting in death receptor 5 clustering causing activation of caspase 8 [22]. In hepatocytes, caspase 8 cleaves the pro-apoptotic BH3-only protein Bid to form truncated Bid (tBid) which translocates to mitochondria [31]. JNK activation also occurs facilitating induction of the pro-apoptotic BH3-only protein PUMA [16, 17]. The mitochondrial accumulation of BH3-only proteins, tBid, PUMA, and Bim (also upregulated by toxic FFA), results in mitochondrial dysfunction with release of apoptogenic factors from the intermitochondrial membrane space into the cytosol causing caspase 9 activation. FFAs also induce loss of the anti-apoptotic Bcl-2 protein Mcl-1 [32]. Our findings suggest ballooned hepatocytes have reduced caspase 9 expression (the key caspase initiating apoptosis following mitochondrial dysfunction), and targeted knockdown of caspase 9 represses lipotoxicity in vitro. Given the intense dysregulation of Bcl-2 proteins during lipotoxic stress, it is not surprising that surviving cells have disabled the apoptotic machinery downstream of mitochondrial dysfunction. Moreover, these observations are also consistent with the observation in model systems where loss of a downstream caspase activity is cytoprotective and results in an altered cell phenotype. In our current studies, we also observed reduced cell killing despite enhanced neutral lipid accumulation in cells with reduced caspase 9 expression; this observation is consistent with evolving concepts that FFA acids are the toxic instigators of cell death whereas their esterification into neutral lipids is a detoxification process [33–35].
In a series of land mark observations, Anna Mae Diehl and co-workers have demonstrated the importance of hedgehog signaling in liver pathobiology [8, 11, 20]. For example, they have reported that hedgehog pathway activation parallels disease severity in NASH [8], whereas hedgehog signaling antagonism is antifibrogenic [11]. More importantly in a pivotal study they also identified ballooned hepatocytes as a cellular source of SHH in NASH [20]. Our in vitro model has implications for these studies. We were able to demonstrate, in cells failing to execute the cell death program during lipotoxic insults, that JNK activation induces SHH expression via stimulating the transcription factor AP-1; indeed, our studies identified two functional binding sites for AP-1 within the SHH promoter. Our results parallel those identified in developmental systems in which stressed cells that cannot fully execute the cell death program display an altered phenotype dependent upon JNK activation and aberrantly express SHH [36, 37].
The current study also identified an autocrine hedgehog survival signaling process in shC9 cells during lipotoxic stress. This observation has potential implications for NASH therapy. We speculate that inhibition of hedgehog survival signaling in ballooned hepatocytes would result in their death and elimination from the liver. If ballooned hepatocytes are the principle source of SHH in NASH [20], their elimination would also prevent pro-fibrogenic hedgehog signaling within the liver. Finally, our data may also explain the anti-fibrogenic effect of caspase inhibitors in preclinical studies of NASH since these inhibitors also block SHH expression, a profibrogenic mediator [38].
We provocatively propose that ballooned hepatocytes are cells which have initiated but cannot execute the cell death program due to loss of caspase 9 (Fig. 5). The mechanism(s) responsible for caspase 9 downregulation remain unclear and will require biochemical interrogation of this minor cell population once appropriate techniques can be validated to do so. The recent report of an animal model of NASH with ballooned hepatocytes may aid such studies [39]. Although this in vitro model does not replicate the morphology of the ballooned hepatocyte, it does duplicate at least one of the phenotypical characteristics of ballooned hepatocytes, namely SHH generation. Processes in addition to cell death avoidance apparently are required for the ballooned cell morphology.
Figure 5. A schematic model for SHH expression as an autocrine survival factor during lipotoxic stress.
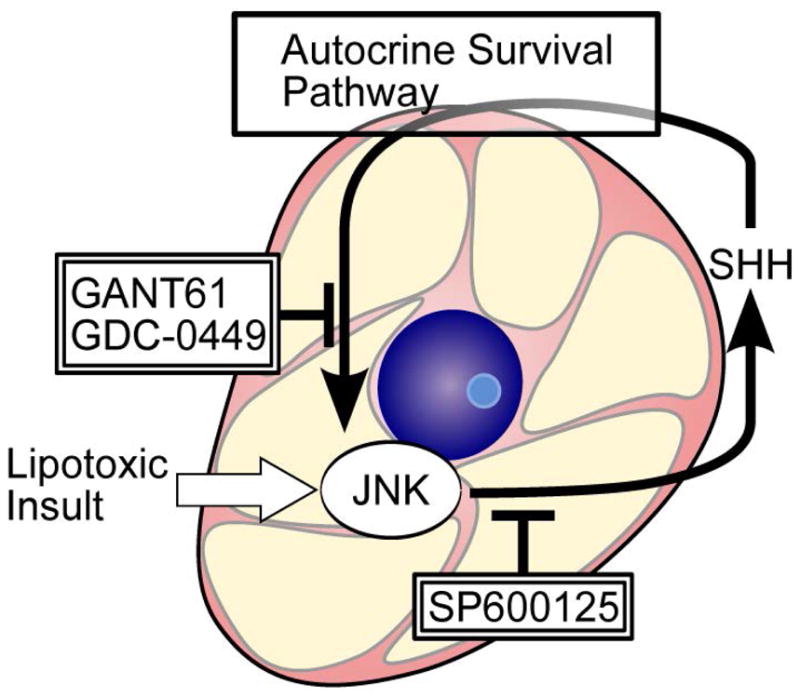
Lipotoxic insult-induced AP-1 mediates SHH expression. Blocking JNK signaling with SP600125 represses SHH generation. Inhibition of SHH pathway signaling by either GANT61 or GDC-0449 blocks this autocrine SHH survival pathway in “undead cells” resulting in their cellular demise.
Supplementary Material
Acknowledgments
This work was supported by NIH grant DK41876 and the optical and microscopy cores of P30DK084567 and the Mayo Foundation. We thank Drs. Christian D. Fingas, Maria E. Guicciardi, Justin L. Mott, and Scott H. Kaufmann for helpful discussions; Dr. Harmeet Malhi for reading the manuscript and providing insightful comments and suggestions; Eugene W. Krueger for aiding the studies employing contrast microscopy; and Courtney N. Hoover for her excellent secretarial assistance.
Abbreviations
- NASH
nonalcoholic steatohepatitis
- SHH
sonic hedgehog
- FFA
free fatty acids
- PA
palmitae
- LPC
lysophosphatidylcholine
- JNK
c-Jun N-terminal kinase
- shRNA
short-hairpin RNA
- MPH
mouse primary hepatocytes
- DAPI
4′,6-diamidine-2′-phenylindole dihydrochloride
- EMSA
Electrophoretic Mobility Shift Assay
- ChIP
Chromatin immunoprecipitaion
- OA
oleate
- C3−/−
caspase 3 genetic knockout
- AP-1
Activator protein-1
- ACOX-1
acyl-CoA oxidase 1, palmitoyl
- CPT
carnitine palmitoyltransferase
- ACACA
acetyl-CoA carboxylase alpha
- ACACB
acetyl-CoA carboxylase beta
Footnotes
The authors who have taken part in this study do not have anything to declare regarding funding from industry or other conflicts of interest with respect to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gouw AS, Clouston AD, Theise ND. Ductular reactions in human liver: diversity at the interface. Hepatology. 2011;54:1853–1863. doi: 10.1002/hep.24613. [DOI] [PubMed] [Google Scholar]
- 3.Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 4.Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M. DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol. 2006;26:7258–7268. doi: 10.1128/MCB.00183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 6.Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 2008;14:399–410. doi: 10.1016/j.devcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lackner C, Gogg-Kamerer M, Zatloukal K, Stumptner C, Brunt EM, Denk H. Ballooned hepatocytes in steatohepatitis: the value of keratin immunohistochemistry for diagnosis. J Hepatol. 2008;48:821–828. doi: 10.1016/j.jhep.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 8.Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2011 doi: 10.1002/hep.25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caldwell S, Ikura Y, Dias D, Isomoto K, Yabu A, Moskaluk C, et al. Hepatocellular ballooning in NASH. J Hepatol. 2010;53:719–723. doi: 10.1016/j.jhep.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung Y, Witek RP, Syn WK, Choi SS, Omenetti A, Premont R, et al. Signals from dying hepatocytes trigger growth of liver progenitors. Gut. 2010;59:655–665. doi: 10.1136/gut.2009.204354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology. 2011;53:106–115. doi: 10.1002/hep.23998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, et al. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. American journal of physiology Gastrointestinal and liver physiology. 2012;302:G77–84. doi: 10.1152/ajpgi.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 14.Listenberger LL, Brown DA. Fluorescent detection of lipid droplets and associated proteins. Curr Protoc Cell Biol. 2007;Chapter 24(Unit 24):22. doi: 10.1002/0471143030.cb2402s35. [DOI] [PubMed] [Google Scholar]
- 15.Higuchi H, Bronk SF, Bateman A, Harrington K, Vile RG, Gores GJ. Viral fusogenic membrane glycoprotein expression causes syncytia formation with bioenergetic cell death: implications for gene therapy. Cancer Res. 2000;60:6396–6402. [PubMed] [Google Scholar]
- 16.Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G236–243. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 19.Schreurs M, Kuipers F, van der Leij FR. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes Rev. 2010;11:380–388. doi: 10.1111/j.1467-789X.2009.00642.x. [DOI] [PubMed] [Google Scholar]
- 20.Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, et al. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol. 2011;224:401–410. doi: 10.1002/path.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 22.Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem. 2011;286:39336–39348. doi: 10.1074/jbc.M111.280420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 24.Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 25.Han ME, Lee YS, Baek SY, Kim BS, Kim JB, Oh SO. Hedgehog signaling regulates the survival of gastric cancer cells by regulating the expression of Bcl-2. Int J Mol Sci. 2009;10:3033–3043. doi: 10.3390/ijms10073033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Syn WK, Jung Y, Omenetti A, Abdelmalek M, Guy CD, Yang L, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–1488. e1478. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin K, Lee J, Guo N, Kim J, Lim A, Qu L, et al. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature. 2011;472:110–114. doi: 10.1038/nature09851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Regl G, Kasper M, Schnidar H, Eichberger T, Neill GW, Philpott MP, et al. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004;64:7724–7731. doi: 10.1158/0008-5472.CAN-04-1085. [DOI] [PubMed] [Google Scholar]
- 29.Walterhouse DO, Yoon JW, Iannaccone PM. Developmental pathways: Sonic hedgehog-Patched-GLI. Environ Health Perspect. 1999;107:167–171. doi: 10.1289/ehp.99107167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahindroo N, Punchihewa C, Fujii N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J Med Chem. 2009;52:3829–3845. doi: 10.1021/jm801420y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masuoka HC, Mott J, Bronk SF, Werneburg NW, Akazawa Y, Kaufmann SH, et al. Mcl-1 degradation during hepatocyte lipoapoptosis. J Biol Chem. 2009;284:30039–30048. doi: 10.1074/jbc.M109.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445–451. doi: 10.1586/egh.09.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6:69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan Y, Bergmann A. Apoptosis-induced compensatory proliferation. The Cell is dead Long live the Cell! Trends Cell Biol. 2008;18:467–473. doi: 10.1016/j.tcb.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol. 2006;16:1606–1615. doi: 10.1016/j.cub2006.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50:1421–1430. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 39.Yimin, Furumaki H, Matsuoka S, Sakurai T, Kohanawa M, Zhao S, et al. A novel murine model for non-alcoholic steatohepatitis developed by combination of a high-fat diet and oxidized low-density lipoprotein. Lab Invest. 2012;92:265–281. doi: 10.1038/labinvest.2011.159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.