


Abstract
Recent genome-wide mapping of the mammalian replication origins has suggested the role of transcriptional regulatory elements in origin activation. However, the nature of chromatin modifications associated with such trans-factors or epigenetic marks imprinted on cis-elements during the spatio-temporal regulation of replication initiation remains enigmatic. To unveil the molecular underpinnings, we studied the human lamin B2 origin that spatially overlaps with TIMM 13 promoter. We observed an early G1-specific occupancy of c-Myc that facilitated the loading of mini chromosome maintenance protein (MCM) complex during subsequent mid-G1 phase rather stimulating TIMM 13 gene expression. Investigations on the Myc-induced downstream events suggested a direct interaction between c-Myc and histone methyltransferase mixed-lineage leukemia 1 that imparted histone H3K4me3 mark essential for both recruitment of acetylase complex HBO1 and hyperacetylation of histone H4. Contemporaneously, the nucleosome remodeling promoted the loading of MCM proteins at the origin. These chromatin modifications were under the tight control of active demethylation of E-box as evident from methylation profiling. The active demethylation was mediated by the Ten-eleven translocation (TET)-thymine DNA glycosylase-base excision repair (BER) pathway, which facilitated spatio-temporal occupancy of Myc. Intriguingly, the genome-wide 43% occurrence of E-box among the human origins could support our hypothesis that epigenetic control of E-box could be a molecular switch for the licensing of early replicating origins.
INTRODUCTION
Complete and accurate DNA replication is crucial for genetic integrity of all organisms and is initiated at hundreds of chromosomal elements called origins of replication (1). The competence of origin to initiate DNA replication during the G1 phase of cell cycle is orchestrated by the ordered assembly of pre-replicative complex (pre-RC) and pre-initiation complex (pre-IC) (2). Formation of pre-RC via sequential binding of origin recognition complex (ORC), Cdc6, Cdt1 and mini chromosome maintenance 2-7 (MCM 2-7) helicase licenses the origins for subsequent firing after maturing into pre-IC (3). The key determinant of replication at only once per cell cycle is separation of pre-RC formation and replication initiation into two mutually exclusive phases (4). The assembly of pre-RC ensued by targeting of ORC to specific replication origins relies on local chromatin structure and DNA topology (5).
The role of strategically positioned nucleosomes in initiation of replication is evident from (i) compromised pre-RC formation following perturbation of ARS1 nucleosomal configuration (6) and (ii) impaired loading of MCM complex due to alterations in the nucleosome arrangement of an ectopic human c-Myc replicator (7). Further, chromatin modifications mediated by histone acetyl transferases (HATs), histone deacetylases (HDACs) and ATP-dependent chromatin remodelers also have been reported in the regulation of replication origins (8–10). It was found that chromatin remodeling had an important role in the functioning of yeast chromosomal origin (11) and licensing of OriP viral origin (12). However, the chromatin architecture of any endogenous mammalian replicator and its contribution in replication initiation is elusive till date.
Chromatin remodeling could be a key regulatory mechanism utilized by transcription factors to activate DNA replication (13,14). Beyond the transcriptional-dependent functions, c-Myc is reported to have a direct role in triggering the number of replication origins, which is perhaps attributed by Myc-dependent chromatin modifications (15). Myc forms heterodimers with other members of Myc family and binds to DNA sequence CACGTG called E-box. The DNA-bound heterodimers then associate with effector protein complexes to induce histone modifications and chromatin remodeling (16).
The control of replication origins by a combination of trans-factors seems to confer specificity in origin selection as the origins lack consensus initiation sites (17). Further, binding of transcription factors to chromatin in G1 phase could facilitate efficient recruitment of pre-RC components. However, a recent study in Xenopus egg extract suggests that c-Myc controls a replication step in G1 phase that is independent of pre-RC formation but precedes origin activation (18). Albeit, the execution point for c-Myc in mammalian origins remains an enigma and is of paramount importance because c-myc is a frequently deregulated proto-oncogene in many cancers.
Among the 30 well-mapped human replication origins so far (17), the lamin B2 origin has been studied in detail for its dynamic molecular and topological transactions at nucleotide level (19). However, the chromatin structure and epigenetic regulation of lamin B2 origin by DNA methylation and histone modifications remain obscure. Herein, we report that cell cycle-regulated epigenetic control of E-box is mediated by active DNA demethylation via ten-eleven translocation (TET) and thymine DNA glycosylase (TDG) pathway. This further drives the temporally tuned c-Myc occupancy and its downstream histone cross talk relay events that specify the licensing of lamin B2 origin by nucleosome remodeling.
MATERIALS AND METHODS
Cell culture, reagents and antibodies
The human embryonic kidney HEK293 (ATCC CRL-1573) and human epithelial cervical HeLa (ATCC CCL2) cell lines were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen), whereas human promyelocytic leukemic cell line HL-60 (ATCC CCL-240TM) was maintained as a suspension culture in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum (FBS). G1-staged HEK293 cells were obtained by serum stimulation of 48 h starved cells, and the samples were harvested every 3 h till 12 h of stimulation for fluorescence-activated cell sorting (FACS) analysis. HeLa cells were arrested in M phase by nocodazole (Sigma-Aldrich) at 50 ng/ml concentration, and mitotic shake-off was performed 16 h later. For RNA interference studies, 2 μg of shRNA (M-T) (20) was transfected using Fugene 6 (Invitrogen). Thirty-six hours post-transfection, cells were subjected to starvation for 24 h and then serum stimulated as described above to obtain G1-staged cells. Enforced expression of c-Myc was accomplished by transfecting 2 µg of pCGN-Myc expression vector (21) using Lipofectamine (Invitrogen) as per manufacturer’s protocol. For DNMT inhibitor study, 200 µM of RG108 (Sigma-Aldrich) was added throughout the starvation and post-serum stimulation till harvesting of HEK293 cells. To induce differentiation of HL-60 cells, 1.5% vol/vol dimethyl sulfoxide was added for 48 h.
Antibodies were procured from the following sources: Santa Cruz Biotechnology for c-Myc, Max, ORC2, Cdc6, Cdt1, MCM4, GCN5, p300, HBO1, MLL1, TRRAP, TET2, Phospho-Rb, cyclin E, cyclin D1, GAPDH and IgG; Upstate Biotechnology for histone H3 K9Ac and H4 tetra-Ac (K5, K8, K12 and K16) and H3 K4me3; and TDG from Abcam.
Establishment of HEK293-MycERTam inducible cell line
The c-MycERTam chimeric gene was excised as EcoRI fragment from pBabe puro construct (22) and subcloned into the pIRES2-EGFP expression vector (BD Biosciences Clontech). The c-MycERTam-pIRES2-EGFP bicistronic plasmid was stably transfected into HEK293 cells using Fugene 6 (Invitrogen) to establish HEK293-MycERTam cell line using G418 selection. Cells grown in phenol red-free DMEM (Invitrogen) supplemented with 10% charcoal-stripped FBS were subjected to 24 h starvation and induced by 2 µM/ml tamoxifen citrate (Tam) (Calbiochem) for the indicated time points (23).
MNase-Southern hybridization assay
Nuclei isolation was carried out as described elsewhere (24). MNase digestion and Southern hybridization were performed as described earlier (25). Southern hybridization probes were generated by polymerase chain reaction (PCR) amplification of specific primers (Supplementary Table S1), and end labeling of probes was performed using [γ-32P] ATP and T4 polynucleotide kinase (Fermentas) as per supplier’s protocol.
MNase chromatin accessibility assay based on real-time PCR assay
Mononucleosomes obtained after complete digestion with MNase were resolved on agarose gel (1.8%) and eluted using QIAquick Gel extraction kit (Qiagen). The genomic DNA thus obtained was used to perform SYBR green real-time quantitative PCR (qPCR) in triplicates with chromatin accessibility assay based on real-time PCR (CHART-PCR) primers (Supplementary Table S1). The Ct-values were converted to DNA concentrations as described previously (24) using the standard curve of corresponding primer set and normalized for input variations. Standard curve for each primer set was generated using serial dilutions of genomic DNA (26). The results were expressed as relative nucleosome occupancy.
Chromatin immunoprecipitation-qPCR assay
This was carried out as per the manufacturer’s instructions (Upstate Biotechnology). The immunoprecipitated chromatin was purified using QIAquick PCR purification kit (Qiagen), and the eluted genomic DNA was subjected to SYBR green real-time qPCR with indicated primer sets (Supplementary Table S1). Data obtained were normalized with input DNA and expressed as fold DNA enrichment over mock (25).
Nascent strand abundance assay by qPCR
The nascent strand abundance assay was performed according to Romero and Lee (27). The short nascent DNA strands were resolved by alkaline gel electrophoresis, isolated and quantitated by real-time qPCR using primers given in Supplementary Table S1.
RNA isolation and quantitative RT-PCR assay
Total RNA was isolated from cells using TRIzol reagent as per the supplier’s instructions (Invitrogen). Reverse transcriptase–PCR (RT–PCR) was performed with M-MuLV reverse transcriptase (Fermentas) according to the manufacturer’s guidelines. The real-time qPCR was carried out using specific primers (Supplementary Table S2) as described previously (28).
Immunoprecipitation and western blot analysis
The methods for immunoprecipitation (IP) and western blotting have been described earlier (29).
Methylation-sensitive restriction analysis
Genomic DNA digested with PmlI was quantitated by qPCR as described above using E-box primers (30). The results were expressed as percent PmlI uncut of each time point with reference to late G1 phase.
Methylation profiling by bisulfite sequencing
The EZ DNA MethylationTM Kit (Zymo Research) was used as per supplier’s instructions for bisulfite treatment of genomic DNA isolated from G0 and G1 phases of HEK293 cells. The modified genome was subjected to PCR to amplify the lamin B2 origin-bearing E-box element using the following primer set: E-box BS-F 5′-gtagttagtgtaaataggatttagg-3′, E-box BS-R 5′-aaaaaaaaccctaacttaacc-3′. As a control, the untreated genomic DNA was subjected to PCR with the following primer set to amplify the same region of origin: E-box F 5′-aacaggacccaggcgatg-3′, E-box R2 5′-ctgccgttacctacacgagctac-3′. The eluted PCR product was cloned into pGEM-T Easy vector (Promega) according to the manufacturer’s protocol. Plasmid DNA from individual clones was sequenced using 3730xl DNA analyzer (Macrogen Inc.).
Detection and quantitation of 5-hmC by qPCR
The Quest 5-hmC Detection Kit™-Lite (Zymo Research) was used for a sequence-specific detection of 5-hydroxymethylcytosine (5-hmC) as per supplier’s instructions. Briefly, DNA extracted from each time point was treated with 5-hmC glucosyltransferase (GT). This enzyme specifically adds a glucose moiety only to 5-hmC but not to 5-methylcytosine (5-mC) to yield glucosyl-5-hydroxymethylcytosine (glucosyl-5-hmC). Exploitation of restriction enzymes that possess sensitivity to glucosyl-5-hmC could facilitate the effective distinction of 5-hmC from 5-mC pool. This is due to their efficiency to cleave only 5-mC and 5-hmC but not the modified glucosyl-5-hmC. As a result only the modified 5-hmC would be amplified by the downstream qPCR. Because GlaI restriction enzyme (‘ACGT’) is specific for E-box element (CACGTG), we performed GlaI digestion of each sample either before or after the treatment with GT as described by supplier’s protocol. After digestion, the spin column DNA clean-up procedure was used to elute the DNA and subjected to SYBR green real-time qPCR with E-box primer. The results were expressed as 5-hmC level relative to G0 phase.
Electrophoretic mobility shift assay
For showing specific complex formation of c-Myc with lamin B2 E-box in vitro, nuclear extract was prepared from HEK293 cells transfected with either vector control or Myc shRNA (M-T). Nuclear extracts (15 µg) were incubated with 50 fmoles of radiolabeled double-stranded lamin B2 E-box oligo in binding buffer (10 mM Tris–HCl (pH 7.5), 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, 4% glycerol and 0.1% Nonidet P-40) along with 0.25 µg of poly (dI:dC) for 60 min on ice (28). The free and protein-bound oligonucleotide probes were separated by electrophoresis on 5% polyacrylamide gel. Subsequently, the gels were dried and the bands were visualized by Typhoon scanner. For methylation sensitive assay, the consensus and lamin B2 E-box oligos (Supplementary Table S3) were methylated by dimethyl sulphate treatment for 5 min, and the binding reactions were carried out with 10 µg of nuclear extract as described above.
Flow cytometry (fluorescence-activated cell sorting)
Flow cytometry of HEK293 and HeLa cells was performed as described previously (29), and the analysis of FACS data was done manually.
Statistical analysis
Data were expressed as mean ± SD. Means were compared by one-factor analysis of variance followed by Fisher protected least significant difference to assess specific group differences. Data were considered significant at P < 0.05 and highly significant at P < 0.01.
RESULTS
c-Myc recruitment to TIMM 13 promoter does not induce TIMM 13 gene expression
Recent large-scale studies have elucidated the interplay between the two fundamental nuclear processes—DNA replication and transcription—by sharing common cis-regulatory elements (31). To better illustrate the functional significance of spatial overlapping between replicator and promoter, we initiated the locus-specific analysis at lamin B2 origin, which is located between 3′ UTR of lamin B2 and promoter of ppv1 (TIMM 13) genes (32). The organization of 1.2 kb human lamin B2 replicator along with canonical E-box element is shown schematically (Figure 1A). In vivo genomic foot printing analysis of lamin B2 origin has suggested the binding of a cluster of transcription factors (33) and persistent binding of c-Myc through S phase was shown afterward (34). Nonetheless, the precise role of c-Myc in regulation of pre-RC formation is yet to be investigated (35). Our protein–DNA interaction studies by electrophoretic mobility shift assay (EMSA) confirmed c-Myc binding to lamin B2 E-box in vitro, and the specificity of interaction was ensured from impaired protein–DNA complex formation by RNA interference (Figure 1B). Next, we analyzed the c-Myc occupancy of lamin B2 origin in different stages of G1-phased HEK293 cells (Supplementary Figure S1A). Each stage of the cell cycle was monitored with the help of phase-specific molecular markers (Supplementary Figure S1B). As evident from the chromatin immunoprecipitation (ChIP) results, c-Myc recruitment to E-box region was specific to early G1 phase just as its dimerization partner Max (Figure 1C). Further, like the endogenous c-Myc, Myc-ER chimera also bound to the E-box region on stimulation by tamoxifen in the HEK293-MycERTam cells (Supplementary Figure S1C).
Figure 1.

Recruitment of c-Myc to TIMM 13 promoter and its role in the regulation of TIMM 13 gene. (A) Schematic representation of 1.2 kb lamin B2 origin (GenBankTM accession number: M94363) flanked by lamin B2 and TIMM 13 genes. The OPR (3910–4007) marks the position of ORI region and the OBR indicates the origin of bidirectional replication. The position of Myc E-box binding element is represented along with the primers specific for ORI and E-box regions. (B) EMSA for showing specific complex formation (marked by arrow head) of lamin B2 E-box with nuclear extract of either vector control (VC) or Myc shRNA-transfected HEK293 cells. FP, free probe. (C) ChIP-qPCR analysis of HEK293 cells either starved or serum stimulated to show the recruitment kinetics of c-Myc and Max over IgG control. (D and E) qRT-PCR analysis to measure the ODC and TIMM 13 transcripts of either serum-stimulated HEK293 cells (D) or Tam induced (6 h) HEK293-MycERTam cells (E). Data shown in (C–E) are mean ± SD of three independent experiments. Number sign indicates statistically significant difference at P < 0.01.
As c-Myc recruitment was observed in lamin B2 origin that overlaps with the promoter region of TIMM 13 gene, next we asked whether TIMM 13 gene was a transcriptional target of c-Myc. Our reverse transcriptase qPCR results showed the activation of ornithine decarboxylase (ODC) transcripts peaking at mid G1 phase following serum stimulation. However, unlike bona fide Myc target ODC, TIMM 13 gene could not be activated by Myc (Figure 1D). Further, these results could be recapitulated by both Tam induction of Myc-ER chimera (Figure 1E) and enforced expression of c-Myc (Supplementary Figure S1D). Together, these results suggested that early G1 occupancy of c-Myc to lamin B2 origin was not involved in the stimulation of TIMM 13 gene.
Myc recruitment during early G1 facilitates locus-specific loading of MCM proteins
As Myc binding to lamin B2 was not associated with TIMM 13 gene expression, we then speculated its role in the formation of pre-RC. We monitored the assembly of pre-RC components (ORC2, Cdc6, Cdt1 and MCM4 of MCM2-7 helicases) to the ORI region during early G1 phase. Although there was no much change in the enrichment of most factors during G1, MCM4 levels selectively peaked during mid-G1 and specified licensing of replication origin (Figure 2A). When we extended our study till S phase, the chromatin occupancy of pre-RC components except ORC2 declined extensively in consistent with earlier reports (Supplementary Figure S2A). As a control, we used non-ORI LB2-P region (∼2.5 kb upstream from LB2 ORI), which did not show any enrichment of pre-RC components (Supplementary Figure S2B). Next, we confirmed the involvement of c-Myc in the recruitment of pre-RC components by applying the Myc knockdown strategy. As expected, Myc occupancy of E-box region during early G1 phase was dramatically reduced (Supplementary Figure S2C). Intriguingly, the c-Myc-silenced cells that were allowed to progress till mid-G1 phase showed no apparent change in the occupancy of early G1-specific replication proteins like ORC2 and Cdt1. However, the mid-G1-specific peak of MCM4 was selectively impaired on c-Myc knockdown (Figure 2B) without affecting MCM4 levels (Supplementary Figure S2D). To further confirm the Myc-dependent MCM4 loading, we performed ChIP assay in HL-60 cells that carry nearly 15–30-fold genomic amplification of c-Myc locus (36). The recruitment of Myc was seen only in proliferating cells but not in replication-ceased differentiated cells. As the levels of Myc recruitment to LB2 ORI region reduced on differentiation, the loading of MCM4 also reduced significantly (Figure 2C). These results imply that the early G1 occupancy of Myc to E-box region may trigger a downstream relay event involving mid-G1-specific MCM4 loading to lamin B2 origin.
Figure 2.

Myc occupancy and kinetics of MCM4 helicase loading at lamin B2 origin. (A) ChIP-qPCR analysis of G1-staged HEK293 cells showing the recruitment kinetics of indicated proteins over IgG control for LB2 ORI region. (B) Recruitment kinetics measured by ChIP-qPCR for ORC2, Cdt1 and MCM4 proteins during mid-G1 phase of HEK293 cells transfected either with vector control or Myc shRNA. (C) ChIP-qPCR analysis of either proliferating or differentiating HL-60 cells showing the fold DNA enrichment of Myc, ORC2 and MCM4 proteins. Data shown are mean ± SD of three independent experiments. The asterisk and number signs indicate statistically significant difference at P < 0.05 and P < 0.01, respectively.
To know whether ‘E-box’ element (CACGTG) was essential for Myc-dependent MCM4 loading, the Myc recruitment to lamin B2 (having E-box element) and MCM4 origin (lacking E-box element) was analyzed by ChIP-qPCR using inducible Myc-ERTam. As shown in Figure 3A, Myc binding to LB2 ORI peaked within 4 h of Tam induction followed by a progressive increase in MCM4 loading. Note that under these conditions, no Myc recruitment or increase in the basal loading of MCM4 was observed at MCM4 ORI (Figure 3B). Interestingly, the LB2 ORI and E-box regions show almost equal enrichment for c-Myc occupancy (Figure 3A and Supplementary Figure S1C) due to their close proximity at lamin B2 origin. Further, like endogenous Myc (Figure 2A), Myc-ERTam had no influence over ORC2 recruitment (Figure 3A). Thus, pre-occupancy of c-Myc seems to trigger subsequent loading of MCM helicases and seems to be a locus-specific event regulated through E-box.
Figure 3.

Locus-specific loading of MCM4 helicase protein in the presence of c-Myc. (A and B) HEK293-MycERTam cells were induced for the indicated time points, and fold DNA enrichment of c-Myc, ORC2 and MCM4 over IgG control was measured for either LB2 ORI (A) or MCM4 ORI (B) by ChIP-qPCR. (C and D) The nascent DNA template of 1.0 kb (C) and 1.5 kb (D) were isolated from either vector control or Myc shRNA transfected HEK293 cells and subjected to qPCR with indicated primers. The values obtained were normalized with non-ORI primer amplification. MCM4 ORI was used as positive control. Data shown are mean ± SD of three independent experiments. The asterisk and number sign indicate statistically significant difference at P < 0.05 and P < 0.01, respectively.
As the loading of MCM complex specifies the licensing of origin, then we used the nascent strand abundance assay (27) to demonstrate the Myc-dependent origin activation. The nascent DNA strands of 1.0 kb (Figure 3C) and 1.5 kb size (Figure 3D) were isolated from the cells transfected with either control or Myc shRNA, and their relative abundance was determined by qPCR using primer pairs corresponding to LB2 ORI, MCM4 ORI and non-ORI LB2-P regions. As shown in Figure 3C and D, the nascent strands were five to seven times more abundant for the LB2 ORI and MCM4 ORI regions than for the non-ORI LB2-P region. Further, c-Myc silencing abrogated the nascent strand abundance of LB2 ORI but not that of MCM4 ORI. As a control, the MCM4 non-ORI region (∼4.0 kb downstream to MCM4 ORI) was also studied, which did not show any enrichment of nascent strands (Supplementary Figure S2E). Thus, these results corroborated our earlier observation that the origin regulatory function of c-Myc is locus specific and controlled by the E-box element present in lamin B2 origin.
The lamin B2 origin is nucleosomal during early G1 and is remodeled following c-Myc recruitment
Because binding of transcription factors to ‘cis’ elements is well known to signal a cascade of events that facilitate gene expression (37), we wondered whether Myc-mediated chromatin remodeling was the underlying mechanism of MCM loading. To address this, we first analyzed the nucleosomal status of LB2 ORI and E-box regions by MNase-Southern assay using nuclei isolated from the nutrient-deprived and serum-stimulated cells. The partially digested nuclei showed a physiologically spaced nucleosomal ladder of DNA fragments with ∼160 bp periodicity (38) when hybridized with the LB2 ORI probe (Figure 4A), whereas the complete MNase digestion showed positioned mononucleosomes in Southern blot (Figure 4B). Because beads-on-string nucleosome array was observed at LB2 ORI, we analyzed the nucleosomal status of adjacent E box region and found this also to be nucleosomal (Figure 4C and D). Although MNase-Southern assay provided some insight into the nucleosomal status of lamin B2 origin, the quantitative data on chromatin remodeling were obtained using MNase CHART-PCR assay (24). When G0 cells were stimulated with serum, the nucleosomes assembled on LB2 ORI and E-box regions underwent an enhanced remodeling during early to mid-G1 progression and again re-assembled by late G1 phase (Figure 4E). The corresponding raw dataset of CHART-PCR amplification obtained using LB2 ORI and E-box primers has been shown in Supplementary Figures S3A and B. This temporally regulated nucleosome remodeling was abolished when c-Myc was down-regulated by shRNA (Figure 4F and Supplementary Figure S2C). These results suggest the role of early G1-specific c-Myc recruitment in triggering the nucleosome remodeling during early to mid-G1 phase to facilitate MCM4 loading.
Figure 4.

Nucleosome assembly of lamin B2 origin and its remodeling mediated by c-Myc recruitment. (A–D) HEK293 cells either starved or serum-stimulated were subjected to MNase-Southern assay. Ethidium bromide stained gel (left) and the corresponding hybridized blot (right) with probes LB2 ORI (A and B) and E-box (C and D) showing either the nucleosome array generated by partial MNase digestion (A and C) or mononucleosomal DNA obtained after complete MNase digestion (B and D). M-DNA marker lane. (E) MNase CHART-PCR analysis of HEK293 cells at different phases of cell cycle for both LB2 ORI and E-box regions. (F) HEK293 cells transfected with either vector control or Myc shRNA were subjected to MNase CHART-PCR analysis during G1 phase. Data shown in (E and F) are mean ± SD of three independent experiments. The asterisk and number sign indicate statistically significant difference at P < 0.05 and P < 0.01, respectively.
When proliferating cells enter into the quiescent state (G0), they gradually lose their replication licensing capacity. However, their licensing competency can be reversed during G0–G1 transition by serum stimulation (39). So, we next asked whether the chromatin remodeling observed at lamin B2 origin was exclusive to cells released from quiescent state or otherwise. To address this, mitotic shake-off was performed by re-seeding the M phase-arrested HeLa cells with fresh medium. The M to G1 transiting cells were harvested at different stages of G1 phase (Supplementary Figure S3C) and were subjected for MNase CHART-PCR analysis. Each stage of G1 phase synchronization was ensured by monitoring cell cycle phase-specific markers (Supplementary Figure S3D). The CHART-PCR analysis of mononucleosomes isolated from M-G1 transiting cells also showed chromatin remodeling of LB2 ORI and E-box regions during mid-G1 phase (Supplementary Figure S3E). Thus, the M-G1 transiting cells followed a remodeling kinetics similar to that of G0–G1 transiting cells. Together, these results suggested that c-Myc-dependent accessibility of chromatin at mid-G1 phase may be the underlying molecular mechanism associated with the loading of MCM complex.
c-Myc-mediated mid-G1 nucleosome remodeling involves hyperacetylation of histone H4
As HATs are critical for nucleosome remodeling, then we analyzed the occupancy of nuclear HATs at lamin B2 origin. Among the HAT complexes, major forms in human are GCN5/PCAF, Tip60 and HBO1. The GCN5 subclass of GNAT-acetyl transferases are represented by two closely related proteins, GCN5 and p300/CBP-associated factors. The MYST family of HATs includes Tip60 and HBO1 (40). To understand the role played by these HAT complexes in the nucleosome remodeling, we used ChIP-qPCR to study the occupancy of GCN5, p300, HBO1 and TRRAP (the large subunit of Tip60 complex) on LB2 ORI region. As shown in Figure 5A, GCN5 did not seem to be involved in chromatin remodeling, whereas p300 was recruited exclusively during early G1 phase, and TRRAP occupancy was observed only at late G1 phase. Interestingly, HBO1 recruitment was observed throughout the G1 phase with a mid-G1 peak and appeared to be in concert with nucleosome remodeling of lamin B2 origin (Figure 5A). Neither HBO1 nor other HATs were enriched on non-ORI LB2-P control region (Supplementary Figure S4A). Analysis of histone tail modifications revealed no H3K9 acetylation (Ac), whereas tetra-acetylation of histone H4, that is K5, K8, K12 and K16, was observed throughout G1 phase with a peak at mid-G1 phase (Figure 5B). The mid-G1 HBO1 occupancy and histone H4 hyperacetylation are consistent with the fact that this acetylation is dependent on HBO1 (41). Interestingly, a significant enrichment of histone H3K4 trimethylation (H3K4me3) mark was also observed (Figure 5B) at ORI region. In contrast, no significant histone tail modifications were observed at non-ORI LB2-P control region (Supplementary Figure S4B). Note that as c-Myc knockdown impaired chromatin remodeling of lamin B2 origin, we also observed the abolishment of HBO1 recruitment and histone H4 hyperacetylation under the same conditions (Figure 5C). However, the expression levels of HBO1 remained unaffected on c-Myc silencing (Supplementary Figure S4C). Thus, an increased accessibility of chromatin during mid-G1 phase seems to be the consequence of Myc-dependent hyperacetylation relay event that could specify the licensing of lamin B2 origin.
Figure 5.

Kinetics of acetylase recruitment and histone tail modifications at lamin B2 origin. (A and B) The G0/G1-staged HEK293 cells were subjected to ChIP-qPCR, and the fold DNA enrichment of LB2 ORI region over IgG control was measured for GCN5, p300, HBO1 and TRRAP proteins (A) as well as for histone H3 K9Ac, H4 tetra-Ac and H3 K4me3 (B). (C) ChIP-qPCR showing the HBO1 recruitment and histone H4 tetra-Ac during mid G1 phase of HEK293 cells transfected either with vector control or Myc shRNA. (D and E) HEK293-MycERTam cells were induced for the indicated time points and fold DNA enrichment of HBO1, and histone H4 tetra-Ac over IgG control was measured by ChIP-qPCR for both LB2 ORI (D) and MCM4 ORI (E). Data shown are mean ± SD of three independent experiments. The asterisk sign indicates statistically significant difference at P < 0.05.
Next, we investigated whether Myc-mediated replication licensing was locus specific or a global event, by comparing the Myc-dependent downstream relay events at both LB2 ORI and MCM4 ORI. On induction of Myc-ERTam, the HBO1 recruitment and histone H4 hyperacetylation peaked within 8 h at LB2 ORI (Figure 5D), whereas only a basal recruitment of acetylase and acetylated status was observed at MCM4 ORI until 12 h (Figure 5E). Besides just as LB2 ORI, the E-box region also showed the enrichment of HBO1 and histone H4 hyperacetylation that peaked during 8 h of Myc-ER induction by Tam (Supplementary Figure S4D). Altogether, these results implied that the ‘E-box’ element in lamin B2 origin is critical for origin licensing via Myc-dependent histone H4 hyperacetylation relay events.
The histone methylase mixed-lineage leukemia 1 (MLL1) interacts with c-Myc and specifies the Myc-dependent hyperacetylation relay events
As c-Myc occupancy of lamin B2 origin favored the recruitment of HBO1, we speculated a direct interaction between the two proteins. However, this possibility was ruled out as our IP studies consistently showed the Myc interaction with its dimerization partner Max but not HBO1 (Figure 6A). According to a recent report, HBO1 exists in two forms with respect to methylation status of histone H3K4 (42). When in complex with ING5 proteins, the HAT activity of HBO1 is greatly stimulated by H3K4me3 mark owing to the higher affinity of ING PHD fingers for the trimethylation signature (43). Accordingly, we also observed a synergy between histone H3K4me3 mark at LB2 ORI and HBO1 recruitment followed by histone H4 hyperacetylation (Figure 5A and B) during G1 phase. Thus, it is likely that the H3K4me3 mark observed at LB2 ORI promotes the recruitment of HBO1.
Figure 6.

Kinetics of MLL1 occupancy at lamin B2 origin and its interaction with c-Myc. (A and C) Cell extracts prepared from HEK293 cells transfected with either PCGN or PCGN-Myc expression plasmids were proceeded for IP with mouse monoclonal anti-c-Myc antibody followed by western blotting for HBO1, MLL1 and Max (used as positive control). One-tenth of the cell extract was also probed for total HBO1, MLL1, c-Myc and GAPDH. Single and double asterisks indicate the expression of either endogenous or HA-tagged c-Myc, respectively. (B and D) Fold DNA enrichment of MLL1 over IgG control was measured by ChIP-qPCR for both LB2 ORI and E-box regions of either G0/G1 transiting HEK293 cells (B) or vector control and c-Myc shRNA transfected HEK293 cells (D). Data shown in (B) and (D) are mean ± SD of three independent experiments. The asterisk and number signs indicate statistically significant difference at P < 0.05 and P < 0.01, respectively.
As the H3K4me3 mark at lamin B2 origin seems to play an important role in origin licensing, next we addressed the underlying mechanism of establishment of this mark. Our kinetic study for the occupancy of mixed-lineage leukemia 1 (MLL1) histone methyl transferase showed an exclusive enrichment on both LB2 ORI and E-box regions during early G1 phase (Figure 6B), whereas the non-ORI LB2-P control region has failed to do so (Supplementary Figure S5A). Interestingly, the contemporaneous occupancy of MLL1 and c-Myc proteins on the origin was confirmed by IP blotting studies that showed a direct interaction between the two proteins (Figure 6C). Next we probed whether H3K4me3 signature was a cause or consequence of Myc binding. As shown in Figure 6D and Supplementary Figure S5B, both MLL1 recruitment and H3K4me3 mark for ORI and E-box regions were abolished on Myc silencing. However, the MLL1 expression remained unaffected during Myc knockdown (Supplementary Figure S5C). Note that Myc-dependent recruitment of MLL1 is observed only during early G1 phase (Figure 6B), whereas the MLL1-specific H3K4me3 mark showed its peak during mid-G1 phase (Figure 5B). These results clearly indicated that MLL1-specific H3K4me3 mark is a consequence of c-Myc recruitment.
To better understand the sequential order of molecular events at both ORI and E-box regions, we performed kinetic studies with closer time points while chasing early to mid-G1 phases. As showed in Supplementary Figure S5D and E, the MLL1 recruitment peaked during post-early G1 phase was followed by an overlapping kinetics of H3K4me3 mark. As cells progressed to mid-G1 phase, there was no MLL1 occupancy, because it was c-Myc dependent. However, the H3K4me3 mark was persistent until mid-G1 phase, suggesting that once it is established, the methyl transferase activity is no longer required. Further, as stated earlier, the H3K4me3 mark established is likely to promote the peak of HBO1 recruitment during mid-G1 phase. Thus, our results suggest that the direct interaction of c-Myc and MLL seems to regulate the downstream histone cross talk events that specify the origin licensing.
Epigenetic control of E-box element signals the temporally programmed licensing of lamin B2 origin
From the preceding results, we wondered whether E-box-mediated licensing of lamin B2 origin could be true for other origins as well. To address this, we performed ‘Short Match’ search for the E-box element ‘CACGTG’ in the 283 human origins mapped recently by genome-wide high-throughput studies using ENCODE consortium (17). In silico analysis of these origins revealed that ∼43% of origins possess E-box element (Supplementary Table S4). However, the regulation of origins by c-Myc occupancy could be under the tight temporal control as evident from ChIP-on-chip data reported recently (44). To further elucidate the molecular underpinnings of temporally programmed c-Myc occupancy, we used methylation-sensitive restriction analysis (MSRA) for the E-box element as it is well known that methylated E-box (mE-box) could not recruit c-Myc. Further, if E-box is unmethylated, it can be cut by PmlI restriction enzyme, whereas mE-box is insensitive to cleavage (30). Thus, percent uncut DNA by PmlI should be directly proportional to mE-box. The MSRA of E-box region suggested an exclusive active demethylation event during early G1 phase (Figure 7A) coinciding with c-Myc occupancy (Figure 1C).
Figure 7.

Epigenetic control of E-box element of lamin B2 origin during cell cycle. (A) MSRA-qPCR analysis of lamin B2 E-box region during G0 and G1 phases of HEK293 cells by PmlI digestion. (B) EMSA was performed to show the specific complex formation (indicated by arrow head) by incubating asynchronous nuclear extract of HEK293 cells with radiolabeled consensus and lamin B2 E-box probes either unmethylated (E-box) or methylated (mE-box). (C and D) HEK293 cells either untreated or treated with RG108 were harvested at G0 and G1 phases and subjected to the following experiments. MSRA-qPCR analysis was performed by PmlI digestion (C). ChIP-qPCR was performed with E-box primer to measure the c-Myc occupancy over IgG control during G0 and G1 phases of HEK293 cells (D). (E) ChIP-qPCR was performed with E-box primer to measure the TET2 and TDG occupancy over IgG control during G0 and G1 phases of HEK293 cells. (F) The DNA extracted from each time point was subjected to GlaI digestion either before or after the treatment with glucosyl transferase, and the relative 5-hmC levels were quantified by qPCR with E-box primer. Except (B), the rest are shown as mean ± SD of three independent experiments. The asterisk and number signs indicate statistically significant difference at P < 0.05 and P < 0.01, respectively.
Next, we carried out in vitro E-box binding by EMSA to confirm the methylation sensitivity of c-Myc occupancy as it is reported earlier that c-Myc does not bind mE-box (45). Our EMSA results clearly indicated that c-Myc binding to both consensus and lamin B2 E-box was abolished when E-box was methylated (Figure 7B). To confirm whether the genomic occupancy of c-Myc was also influenced by methylation status of E-box, we used a specific DNMT inhibitor RG108 that targets its catalytic domain (46). RG108 treatment of cells rescued the unmethylated state of E-box throughout the G1 phase (Figure 7C) that allowed persistent occupancy of c-Myc (Figure 7D).
More recently, active DNA demethylation is reported in somatic cells at specific genomic loci in response to inducible signals and further studies unearthed the involvement of oxidases of the TET family or deaminases of the AID/APOBEC family. The catalytic activity of TDG is required downstream to deaminase-catalyzed conversion of 5-mC into thymine and/or TET-mediated 5-hydroxyl methyl cytosine (5-hmC) into 5-hmU (47). Interestingly, our in vivo kinetic study suggested the occupancy of TET2 followed by TDG in the active DNA demethylation of E-box during G0 to early G1 phase (Figure 7E). As a control, the MCM4 ORI lacking E-box showed neither TET2 nor TDG occupancy (Supplementary Figure S5F). TET2 is known for the oxidation of 5-mC to 5-hmC, which is the first step toward TET-mediated active DNA demethylation. Indeed, we also observed the kinetic coherence between TET2 occupancy with immediate downstream peak of 5-hmC (Figure 7E and F). Together, these results suggest the role of TET and TDG pathway in cell cycle regulated active demethylation of E-box element.
Because active demethylation acted as an epigenetic control of E-box element that ultimately drove the licensing of lamin B2 origin, we next examined the methylation profile of E-box region by bisulfite sequencing using genomic DNA isolated from G0 and early G1 phase cells. As shown in Figure 8, the methylated E-box observed during G0 phase underwent a demethylation event during early G1 phase as evident from the conversion of ‘CG’ to ‘TG’ after bisulfite treatment. Intriguingly, this demethylation event appeared exclusive to E-box because the adjacent CGs were not converted to TGs under similar conditions (Figure 8 and Supplementary Figure S6). Hence, in vivo methylation profiling of lamin B2 origin by bisulfite sequencing corroborated our earlier observations of MSRA and TET–TDG–BER pathway-mediated active DNA demethylation. Further, methylation profiling indicates that E-box is epigenetically controlled by dynamic cycles of methylation and demethylation, which apparently controls the licensing of lamin B2 origin through a temporally programmed Myc-dependent downstream relay events.
Figure 8.
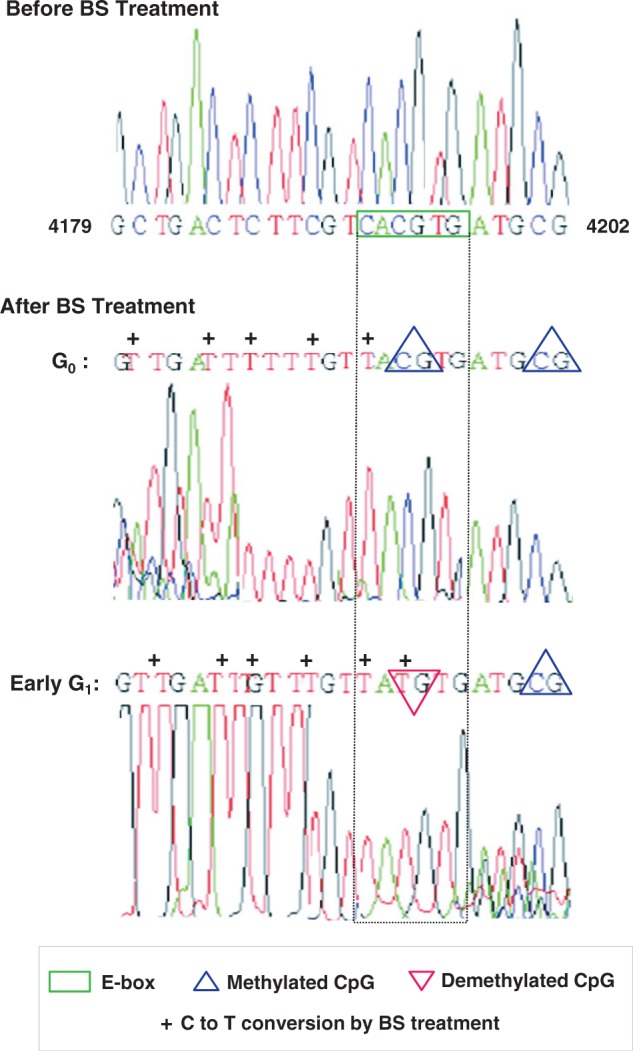
Methylation profiling of lamin B2 origin bearing E-box element. The genomic DNA isolated from G0 and G1 phases of HEK293 cells were processed for bisulphite (BS)-mediated base conversion as described in Materials and Methods, and DNA sequence information was obtained. The native DNA sequence (before BS treatment) corresponding to lamin B2 origin encompassing E-box (top: nucleotide 4179 to 4202, GenBankTM accession number: M94363) was compared with sequence data of modified lamin B2 origin (after BS treatment). The unmethylated cytosines (Cs) got converted to Thymines (Ts) on BS treatment and marked as ‘+’, whereas the methylated Cs remained unchanged due to resistance to such conversion. The methylated CpG is denoted by upward triangle (blue). As cells progressed from G0 to early G1 phase, the E-box region (green box flanked by dotted lines) underwent a demethylation event (downward triangle in red) confirming the demethylated status of this CpG.
DISCUSSION
Myc preferentially associates with sites in genomic DNA with a high CpG dinucleotide content, which is called as CpG islands (48). CpG islands are generally associated with promoters that are transcriptionally active (49), and active promoters are typically overlapped with early replicating origins (50). Coalescence of these facts hints that early firing origin can be specified by binding of c-Myc where the molecular underpinnings are unclear. However, based on our work, a new model can be proposed that accurately depicts the molecular mechanisms by which c-Myc occupancy regulates the early replicating origin (Figure 9). When cells are arrested at G0, methylated E-box of lamin B2 origin recruits the DNA demethylase TET2. Subsequently, the oxidation of 5-mC to 5-hmC predominates to set out the TET–TDG–BER pathway of active DNA demethylation. As cells progress through early G1, TDG being recruited to origin that denotes the removal of methylation mark on E-box, which ultimately favors the c-Myc occupancy. Concurrently, the pre-RC components including ORC, Cdc6 and Cdt1 are recruited to the ORI region during early G1 phase; however, the loading of MCM helicases is hindered by nucleosome assembly. The c-Myc occupancy during early G1 phase favors the recruitment of histone H3K4 methylase MLL1 during the same time window and imparts the H3K4me3 mark at the origin. As cells progressed to mid-G1 phase, the histone H3K4me3 mark promoted the binding of HBO1 acetylase and facilitated histone H4 hyperacetylation. By this time, the E-box switches to methylated state and strips off c-Myc and the associated MLL1 from the origin, whereas the c-Myc-induced hyperacetylation favors nucleosome remodeling that facilitates the loading of MCM proteins. Together, the temporally programmed epigenetic modification of E-box involving TET–TDG–BER pathway seems to drive the Myc-dependent cross-talk of histone modifications that specifies the licensing of lamin B2 origin.
Figure 9.
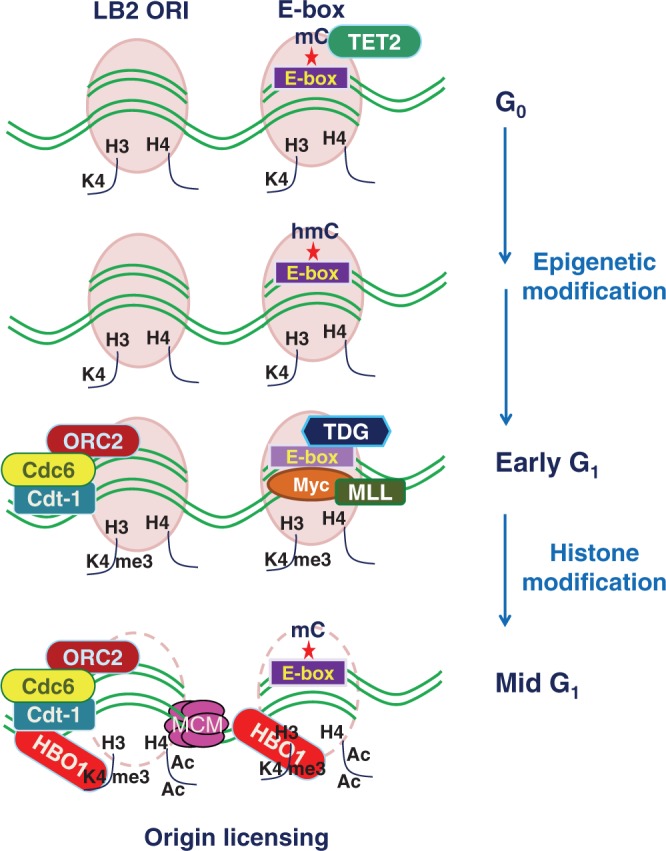
Model depicting the molecular underpinnings of lamin B2 origin licensing. Cell cycle regulated active DNA demethylation involves TET2 and TDG pathway that signals the occupancy of c-Myc to demethylated E-box during early G1 phase. The subsequent Myc-dependent cross talk of histone modifications during mid G1 phase favors the remodeling of nucleosomes at the origin and specifies its licensing by facilitating the loading of MCM proteins.
As many potential origins are set between the end of mitosis and early G1 phase, the replication origin decision point in mid-G1 phase selects the origins to be used for subsequent firing event (39). Loading of MCMs during mid-G1 phase is considered to be the first step toward the specification of origins that undergo subsequent firing (51). Extrapolation of these facts with our kinetic studies could help to set out the hypothesis that Myc-dependent loading of MCM complex to origins is a finely tuned temporal program which may confer the early replication timing of these origins. Intriguingly, the temporal regulation of Myc-dependent MCM loading, in turn, is controlled by epigenetic modifications of E-box (Figures 7 and 8) suggesting the possible role of DNA demethylases and DNMTs in specifying the early firing origins. Consistently, cell cycle-regulated expression of DNMTs has been reported recently (52). However, it is likely complex to explore the hierarchy of DNMTs in the epigenetic regulation of lamin B2 origin due to their dual role in both DNA methylation and demethylation. Further studies are required in near future to characterize the DNA methylases that act on lamin B2 origin.
Recent findings have raised the possibility that regulation by DNA methylation may be quite dynamic rather being static (47) that led to the emergence of enzymes capable of mediating DNA demethylation in mammalian cells (53). The involvement of DNA demethylation under certain conditions such as extrinsic signals, in early stages of development and in highly specialized post-mitotic cells has been reported recently. Active DNA demethylation has been studied as a rapid response to signal transduction in interleukein-2 stimulation of T lymphocytes (54) and estrogen stimulation of breast cancer cells (55,56). As active DNA demethylation is considered to play a major role in gene transcription, our study explores its novel role in the regulation of replication origin. Because TET and AID/APOBEC enzymes have been recently identified as active regulators of DNA demethylation along with TDG, BER glycosylases (57–59), it is important to investigate their role in origin licensing. Our kinetic studies on lamin B2 origin suggested TET2-mediated oxidation of 5-mC to 5-hmC and the downstream TDG-base excision repair (BER) pathway in the active demethylation of E-box (Figure 7E and F). Further, this report is the first in declaring the role of cell cycle-regulated epigenetic changes in DNA replication in contrast to the conventional differentiation-induced epigenetic control of gene transcription.
A recent report has suggested serum-induced hyperacetylation of histone H4 and HBO1 recruitment at MCM4 ORI (10). However, we did not observe a Myc-dependent hyperacetylation relay event at the MCM4 ORI that lacks an E-box element. Nevertheless, this implied that origin licensing mediated by hyperacetylated histone H4 is a global event stimulated by mitogens but the effector molecules transducing the mitogenic cues to replication origins are locus specific and perhaps regulated by ‘cis’ elements present in the origins. Accordingly, MCM4 ORI has binding sites for many transcription factors including E2F (60), which may confer the hyperacetylation of this ORI just as c-Myc does to LB2 ORI. Further, in this study, we also found that c-Myc interacts with MLL1 protein to regulate the down-stream events. Consistent with this observation, the association of c-Myc with menin, a subunit of MLL1 methyltransferase, has already been reported (61). Besides, c-Myc may interact with MLL1 via p300/CBP complex as the latter also shows a binding kinetics similar to c-Myc (Figures 1C and 5A). In support of this view, the interaction between c-Myc and p300 (62) as well as CBP and MLL (63) have been reported earlier.
It is now well established that Cdt1 functioning requires chromatin remodeling activity, which is achieved by its interaction with HBO1 (10). Herein, we show yet another mechanism of HBO1 recruitment to origins involving Myc-specific E-box element. Thus, it is possible to achieve HBO1 targeting to chromatin by different complex formation and perhaps in specific chromatin context. Consistent with this, the H3K4me3 mark not only favors HBO1 recruitment (42) but also c-Myc binding (64). Future studies in this line may shed light on the characteristics of precise HBO1–HAT complexes involving in the regulation of replication origin activity. Besides, the role of other MYST HATs like Tip60 needs further investigation as this could be involved in the later stages of origin activation but not during the licensing step (Figure 5A).
It is well known that Myc recruits distinct complexes to specific subsets of genomic targets and probably different complexes depending on the specific cellular context (65). In this view, this study not only elucidates a novel epigenetic control mechanism for Myc-mediated chromatin modifications and replication licensing but also paves the way for exploring the involvement of other effector molecules that causally link mitogenic signaling to locus-specific replication initiation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4 and Supplementary Figures 1–6.
FUNDING
International Centre for Genetic Engineering and Biotechnology, New Delhi; Senior Research Fellowships from Indian Council of Medical Research, New Delhi (to M.S.) and Council of Scientific and Industrial Research, New Delhi (to A.K.S.). Funding for open access charge: Extramural research [BT/PR133228/MED/29/157/2009] from the Department of Biotechnology, Government of India.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr William P. Tansey (Cold Spring Harbor Laboratory) for the generous gift of pCGN-Myc expression vector and Dr. Madhulika Srivastava (National Institute of Immunology, New Delhi) for designing bisulphite sequencing experiments. Ravinder Kumar helped in cell culture work.
REFERENCES
- 1.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 2.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu. Rev. Genet. 2007;42:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mechali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010;11:728–738. doi: 10.1038/nrm2976. [DOI] [PubMed] [Google Scholar]
- 4.Machida YJ, Hamlin JL, Dutta A. Right place, right time, and only once: replication initiation in metazoans. Cell. 2005;123:13–24. doi: 10.1016/j.cell.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Rampakakis E, Arvanitis DN, Di Paola D, Zannis-Hadjopoulos M. Metazoan origins of DNA replication: regulation through dynamic chromatin structure. J. Cell Biochem. 2009;106:512–520. doi: 10.1002/jcb.22070. [DOI] [PubMed] [Google Scholar]
- 6.Lipford JR, Bell SP. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol. Cell. 2001;7:21–30. doi: 10.1016/s1097-2765(01)00151-4. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh M, Kemp M, Liu G, Ritzi M, Schepers A, Leffak M. Differential binding of replication proteins across the human c-myc replicator. Mol. Cell. Biol. 2006;26:5270–5283. doi: 10.1128/MCB.02137-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimoto N, Yugawa T, Iizuka M, Kiyono T, Fujita M. Chromatin remodeler sucrose nonfermenting 2 homolog (SNF2H) is recruited onto DNA replication origins through interaction with Cdc10 protein-dependent transcript 1 (Cdt1) and promotes pre-replication complex formation. J. Biol. Chem. 2011;286:39200–39210. doi: 10.1074/jbc.M111.256123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox CA, Weinreich M. Beyond heterochromatin: SIR2 inhibits the initiation of DNA replication. Cell Cycle. 2008;7:3330–3334. doi: 10.4161/cc.7.21.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol. Cell. 2010;37:57–66. doi: 10.1016/j.molcel.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flanagan JF, Peterson CL. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999;27:2022–2028. doi: 10.1093/nar/27.9.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou J, Chau CM, Deng Z, Shiekhattar R, Spindler MP, Schepers A, Lieberman PM. Cell cycle regulation of chromatin at an origin of DNA replication. EMBO J. 2005;24:1406–1417. doi: 10.1038/sj.emboj.7600609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu YF, Hao ZL, Li R. Chromatin remodeling and activation of chromosomal DNA replication by an acidic transcriptional activation domain from BRCA1. Genes Dev. 1999;13:637–642. doi: 10.1101/gad.13.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danis E, Brodolin K, Menut S, Maiorano D, Girard-Reydet C, Mechali M. Specification of a DNA replication origin by a transcription complex. Nat. Cell Biol. 2004;6:721–730. doi: 10.1038/ncb1149. [DOI] [PubMed] [Google Scholar]
- 15.Cole MD, Cowling VH. Transcription-independent functions of MYC: regulation of translation and DNA replication. Nat. Rev. Mol. Cell Biol. 2008;9:810–815. doi: 10.1038/nrm2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levens DL. Reconstructing MYC. Genes Dev. 2003;17:1071–1077. doi: 10.1101/gad.1095203. [DOI] [PubMed] [Google Scholar]
- 17.Cadoret JC, Meisch F, Hassan-Zadeh V, Luyten I, Guillet C, Duret L, Quesneville H, Prioleau MN. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl Acad. Sci. USA. 2008;105:15837–15842. doi: 10.1073/pnas.0805208105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lebofsky R, Walter JC. New Myc-anisms for DNA replication and tumorigenesis? Cancer Cell. 2007;12:102–103. doi: 10.1016/j.ccr.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 19.Falaschi A, Abdurashidova G, Sandoval O, Radulescu S, Biamonti G, Riva S. Molecular and structural transactions at human DNA replication origins. Cell Cycle. 2007;6:1705–1712. doi: 10.4161/cc.6.14.4495. [DOI] [PubMed] [Google Scholar]
- 20.Hung L, Kumar V. Specific inhibition of gene expression and transactivation functions of hepatitis B virus X protein and c-myc by small interfering RNAs. FEBS Lett. 2004;560:210–214. doi: 10.1016/S0014-5793(04)00113-9. [DOI] [PubMed] [Google Scholar]
- 21.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18:717–726. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greasley PJ, Bonnard C, Amati B. Myc induces the nucleolin and BN51 genes: possible implications in ribosome biogenesis. Nucleic Acids Res. 2000;28:446–453. doi: 10.1093/nar/28.2.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao S, Procko E, Shannon MF. Chromatin remodeling, measured by a novel real-time polymerase chain reaction assay, across the proximal promoter region of the IL-2 gene. J. Immunol. 2001;167:4494–4503. doi: 10.4049/jimmunol.167.8.4494. [DOI] [PubMed] [Google Scholar]
- 25.Singh AK, Swarnalatha M, Kumar V. c-ETS1 facilitates G1/S-phase transition by up-regulating cyclin E and CDK2 genes and cooperates with hepatitis B virus X protein for their deregulation. J. Biol. Chem. 2011;286:21961–21970. doi: 10.1074/jbc.M111.238238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overbergh L, Valckx D, Waer M, Mathieu C. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine. 1999;11:305–312. doi: 10.1006/cyto.1998.0426. [DOI] [PubMed] [Google Scholar]
- 27.Romero J, Lee H. Asymmetric bidirectional replication at the human DBF4 origin. Nat. Struct. Mol. Biol. 2008;15:722–729. doi: 10.1038/nsmb.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janbandhu VC, Singh AK, Mukherji A, Kumar V. p65 negatively regulates transcription of the cyclin E gene. J. Biol. Chem. 2010;285:17453–17464. doi: 10.1074/jbc.M109.058974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukherji A, Janbandhu VC, Kumar V. HBx-dependent cell cycle deregulation involves interaction with cyclin E/A-cdk2 complex and destabilization of p27Kip1. Biochem. J. 2011;401:247–256. doi: 10.1042/BJ20061091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perini G, Diolaiti D, Porro A, Della Valle G. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proc. Natl Acad. Sci. USA. 2005;102:12117–12122. doi: 10.1073/pnas.0409097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maric C, Prioleau MN. Interplay between DNA replication and gene expression: a harmonious coexistence. Curr. Opin. Cell Biol. 2010;22:277–283. doi: 10.1016/j.ceb.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Paixao S, Colaluca IN, Cubells M, Peverali FA, Destro A, Giadrossi S, Giacca M, Falaschi A, Riva S, Biamonti G. Modular structure of the human lamin B2 replicator. Mol. Cell Biol. 2004;24:2958–2967. doi: 10.1128/MCB.24.7.2958-2967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimitrova DS, Giacca M, Demarchi F, Biamonti G, Riva S, Falaschi A. In vivo protein-DNA interactions at human DNA replication origin. Proc. Natl Acad. Sci. USA. 1996;93:1498–1503. doi: 10.1073/pnas.93.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, Dalla-Favera R. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–451. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 35.Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: where, when, and how? Annu. Rev. Biochem. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- 36.Collins S, Groudine M. Amplification of endogenous myc-related DNA sequences in a human myeloid leukaemia cell line. Nature. 1982;298:679–681. doi: 10.1038/298679a0. [DOI] [PubMed] [Google Scholar]
- 37.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 38.Nedospasov SA, Shakhov AN, Georgiev GP. Analysis of nucleosome positioning by indirect end-labeling and molecular cloning. Methods Enzymol. 1989;170:408–420. doi: 10.1016/0076-6879(89)70059-8. [DOI] [PubMed] [Google Scholar]
- 39.Wu JR, Gilbert DM. The replication origin decision point is a mitogen-independent, 2-aminopurine-sensitive, G1-phase event that precedes restriction point control. Mol. Cell Biol. 1997;17:4312–4321. doi: 10.1128/mcb.17.8.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 41.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Saksouk N, Avvakumov N, Champagne KS, Hung T, Doyon Y, Cayrou C, Paquet E, Ullah M, Landry AJ, Cote V, et al. HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol. Cell. 2009;33:257–265. doi: 10.1016/j.molcel.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cadoret JC, Prioleau MN. Genome-wide approaches to determining origin distribution. Chromosome Res. 2010;18:79–89. doi: 10.1007/s10577-009-9094-2. [DOI] [PubMed] [Google Scholar]
- 45.Prendergast GC, Lawe D, Ziff EB. Association of Myn, the murine homolog of max, with c-Myc stimulates methylation-sensitive DNA binding and ras cotransformation. Cell. 1991;65:395–407. doi: 10.1016/0092-8674(91)90457-a. [DOI] [PubMed] [Google Scholar]
- 46.Tang M, Xu W, Wang Q, Xiao W, Xu R. Potential of DNMT and its epigenetic regulation for lung cancer therapy. Curr. Genomics. 2009;10:336–352. doi: 10.2174/138920209788920994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antequera F, Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr. Biol. 1999;9:R661–R667. doi: 10.1016/s0960-9822(99)80418-7. [DOI] [PubMed] [Google Scholar]
- 50.Sequeira-Mendes J, Díaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gómez M. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 2009;5:e1000446. doi: 10.1371/journal.pgen.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cimbora DM, Groudine M. The control of mammalian DNA replication: a brief history of space and timing. Cell. 2001;104:643–646. [PubMed] [Google Scholar]
- 52.Robertson KD, Keyomarsi K, Gonzales FA, Velicescu M, Jones PA. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000;28:2108–2113. doi: 10.1093/nar/28.10.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nabel CS, Kohli RM. Molecular biology. Demystifying DNA demethylation. Science. 2011;333:1229–1230. doi: 10.1126/science.1211917. [DOI] [PubMed] [Google Scholar]
- 54.Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat. Immunol. 2003;4:235–240. doi: 10.1038/ni887. [DOI] [PubMed] [Google Scholar]
- 55.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 56.Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 57.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ladenburger EM, Keller C, Knippers R. Identification of a binding region for human origin recognition complex proteins 1 and 2 that coincides with an origin of DNA replication. Mol. Cell. Biol. 2002;22:1036–1048. doi: 10.1128/MCB.22.4.1036-1048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bres V, Yoshida T, Pickle L, Jones KA. SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol. Cell. 2009;36:75–87. doi: 10.1016/j.molcel.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, Martinez E. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol. Cell.. Biol. 2005;25:10220–10234. doi: 10.1128/MCB.25.23.10220-10234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell. Biol. 2001;21:2249–2258. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall' Olio V, Zardo G, Nervi C, Bernard L, Amati B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell. Biol. 2006;8:764–770. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 65.Amente S, Lania L, Majello B. Epigenetic reprogramming of Myc target genes. Am. J. Cancer Res. 2011;1:413–418. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.