


Abstract
Small RNA-directed DNA methylation (RdDM) is an important epigenetic pathway in Arabidopsis that controls the expression of multiple genes and several developmental processes. RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) and DICER-LIKE 3 (DCL3) are necessary factors in 24-nt small interfering RNA (siRNA) biogenesis, which is part of the RdDM pathway. Here, we found that Increase in BONSAI Methylation 1 (IBM1), a conserved JmjC family histone demethylase, is directly associated with RDR2 and DCL3 chromatin. The mutation of IBM1 induced the hypermethylation of H3K9 and DNA non-CG sites within RDR2 and DCL3, which repressed their expression. A genome-wide analysis suggested that the reduction in RDR2 and DCL3 expression affected siRNA biogenesis in a locus-specific manner and disrupted RdDM-directed gene repression. Together, our results suggest that IBM1 regulates gene expression through two distinct pathways: direct association to protect genes from silencing by preventing the coupling of histone and DNA methylation, and indirect silencing of gene expression through RdDM-directed repression.
INTRODUCTION
DNA methylation is a major epigenetic mechanism of transcriptional repression and gene silencing in eukaryotic development and reproduction (1). Compared with animals, in which cytosine is methylated exclusively at symmetric CG sites in somatic cells, DNA methylation in plants occurs in all sequence contexts (e.g. CG, CHG and CHH) (2,3). In Arabidopsis, the maintenance of DNA methylation is primarily dependent on three methyltransferases: METHLYTRANSFERASE1 (MET1), a DNMT1 homolog that is responsible for maintaining CG methylation after DNA replication (4,5); DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2), a DNMT3 homolog that catalyzes de novo DNA methylation (6,7); and CHROMOMETHYLASE3 (CMT3), a plant-specific DNA methyltransferase that functions in non-CG methylation maintenance redundantly with DRM2 (6,7).
How particular sequences are recognized or eliminated as substrates of DNA methyltransferases and in transcriptional silencing is a major question in the epigenetic control of genes via DNA methylation. DNA methylation is guided by small interfering RNAs (siRNAs) through the recognition of complementary DNA sequences in the genome, in a process known as RNA-directed DNA methylation (RdDM (8,9).
In Arabidopsis, several components function in the biogenesis of 24-nt siRNAs, which function specifically in chromatin silencing, including NUCLEAR RNA POLYMERASE IV A (NRPD1A), RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) and DICER-LIKE 3 (DCL3) (10,11). RDR2 is responsible for the production of dsRNA precursors from single-stranded RNA transcripts, which are produced by RNA polymerase IV (Pol IV) and which correspond to the silenced genes or elements (12,13). dsRNAs are subsequently cleaved by the Dicer-like ribonuclease DCL3 to yield 24-nt siRNAs, which are captured by ARGONAUTE 4 (AGO4) and localized to Cajal bodies in the nucleus (14–16). Although these enzymes are not associated with the initiation of silencing, they are necessary for the accumulation of siRNAs and maintenance of RdDM; thus, they may function in siRNA biogenesis as part of a putative feedback reinforcement mechanism (11,17).
Although RdDM typically participates in the suppression of transposable and repetitive sequences and in genome stability, the expression of a large number of endogenous genes is regulated by DNA methylation and siRNAs (2–4). For instance, an F-box protein-coding gene, SUPPRESSOR OF drm1 drm2 cmt3 (SDC), is highly methylated at non-CG cytosines in tandem repeats within its promoter and is transcriptionally silenced in wild-type (WT) plants (18). SDC misexpression is responsible for the multiple developmental defects seen in drm1 drm2 cmt3 (ddc) triple mutants (18). Genetic analysis has shown that siRNAs generated from repeated sequences are required to maintain the DNA methylation of SDC, indicating the significance of RdDM in the regulation of protein-coding genes during development (18).
Besides the interaction between siRNAs and DNA methylation, histone H3K9 methylation, an epigenetic marker of transcriptional inactivation, is highly associated with DNA methylation in plants and animals (19). In Arabidopsis, three Set-domain methyltransferases responsible for H3K9 dimethylation, SU(VAR)3-9 HOMOLOGUE4 (SUV4, also known as KRYPTONITE), SUV5 and SUV6, are necessary to maintain the non-CG methylation caused by CMT3 (20–22). Further, a genome-wide analysis has shown a connection between H3K9 and DNA methylation (23).
It has also been reported that the SRA domain in SUV4 preferentially binds double-stranded oligonucleotides containing methylated cytosines in a non-CG context, which suggests that this histone modifier can read DNA methylation marks directly (24). Furthermore, the chromodomain of CMT3 can recognize and bind K9 dimethylated H3 tails (25). The interdependent recruitment of CMT3 and SUV4 indicates that the maintenance of non-CG methylation and H3K9 dimethylation is mediated by a positive feedback loop (24,25).
Increase in BONSAI methylation 1 (IBM1) encodes a JmjC domain-containing histone demethylase that catalyzes the removal of H3K9 monomethylation and dimethylation in Arabidopsis (26). The mutation of IBM1 produces multiple developmental defects, including small and narrow leaves, pollen grain abortion, floral organ and embryo abnormalities, and decreased reproduction (27). JMJ706, the rice (Oryza sativa) homolog of IBM1, also encodes an H3K9 demethylase that participates in the regulation of floral organ formation (28).
A loss-of-function of IBM1 causes ectopic H3K9 methylation at the BONSAI locus, leading to KYP- and CMT3-dependent non-CG DNA hypermethylation and gene silencing (27). These results suggest that IBM1 protects protein-coding genes from repression via H3K9 and non-CG DNA methylation arising from flanking transposable elements (27). Genome-wide profiling has shown that a large number of genes are hypermethylated at non-CG cytosines and histone H3K9 in ibm1 (26,29). Further, it has been shown by genetic analysis that known components of the RdDM pathway, including NRPD1A, RDR2 and AGO4, are dispensable in ibm1-induced DNA hypermethylation and the transcriptional repression of BONSAI, indicating that de novo DNA methylation of the direct targets of this gene in ibm1 is subject to siRNA-independent regulation (26,29).
In the present study, we found that RDR2 and DCL3, two necessary components of the RdDM pathway, are direct targets of IBM1. IBM1 associates with their chromatin and removes the methyl group(s) from H3K9. The hypermethylation of H3K9 in ibm1 was accompanied by an increase in non-CG DNA methylation at RDR2 and DCL3, which inhibited the expression of these genes. This reduction in RDR2 and DCL3 expression affected the biogenesis of 24-nt siRNAs in a gene-specific manner. We also found that the decrease in siRNAs homologous to their target sequences was accompanied by an increase in transcription and decrease in DNA methylation of their targets in ibm1. These results demonstrate that IBM1 regulates gene expression through two distinct pathways: direct association to protect genes from silencing by preventing the coupling of histone and DNA methylation, and indirect silencing of gene expression through RdDM-directed repression.
MATERIALS AND METHODS
Plant materials and growth
The lines used in this study, ibm1-3 (SALK_023533) and ibm1-4 (SALK_035608), were described previously (27); ibm1-5 (SALK_128403) is a new allele. All plant materials were obtained from the ABRC (http://abrc.osu.edu/). All mutations were confirmed by PCR and sequencing.
Seeds were sterilized in 2.25% bleach, washed with sterilized water, kept for 3 days at 4°C, and then dispersed on Murashige and Skoog (MS) medium (Sigma-Aldrich) containing 1% sucrose and 0.3% Phytagel (Sigma-Aldrich). After 10 days of growth under a cool white fluorescent light (160 µmol m-2 s-1) and long-day (LD) conditions (16 h of light, 22°C/8 h of dark, 18°C) in a growth chamber (Percival CU36L5), the plants were transplanted to soil and grown in a growth room with coincident conditions at 50% relative humidity.
Plant transformation
The 35 S::YFP::IBM1 construct consisted of the Cauliflower Mosaic Virus 35 S (CaMV35) promoter, the gene encoding yellow fluorescence protein (YFP), and the full coding sequence of IBM1 cDNA. IBM1 cDNA was generated from total RNA isolated from WT Col-0 plants by RT-PCR and amplified using the primers IBM1-NS and IBM1-CAS (Supplementary Table S10). The product was digested with BamHI-SalI and cloned into the binary vector pCAMBIA1300 containing the CaMV35 promoter and YFP.
For plant transformation, the constructs were introduced into Agrobacterium tumefaciens strain GV3101, and then transformed into plants via the floral dip method (30). T1 transformants were selected using hygromycin; single insertion lines were obtained based on their segregation rates of antibiotic resistance.
Illumina mRNA sequencing and bioinformatic analysis
mRNA samples were purified and subjected to Illumina sequencing as described in the Illumina mRNA sequencing sample preparation guide. Briefly, mRNA was specifically enriched from total RNA using oligo(dT) beads and sheared into small pieces. The fragments were then reverse-transcribed into first-strand cDNA using random hexamer primers, followed by second-strand synthesis using DNA polymerase I. The short cDNA strands were ligated with 3′- and 5′-adapters for amplification and sequencing.
All reads 42 bases in length were mapped to the reference genome of Arabidopsis thaliana (TAIR10). Bowtie (31) was used to map those sequence reads with no more than three mismatches, and only uniquely mapped reads were used in our subsequent analyses. Reads mapped to exonic regions of the annotated gene models were normalized against the length of the transcript and sample size for further analysis. Cutoff values were based on the average number of reads in the introns and intergenic regions. The raw data have been deposited in the NCBI database under GEO number GSE32284.
RT-PCR and real-time qPCR analysis
Total RNA was isolated from 10-day-old seedlings grown on MS plates using RNAiso plus (Takara) according to the manufacturer’s instructions then treated with RNA-free DNase (Promega). Three milligrams of total RNA were reverse-transcribed by a two-step method using M-MuLV Reverse Transcriptase (Fermentas). Quantitative PCR (qPCR) was performed using a 7500 Fast Real-Time PCR system (Applied Biosystems) with SYBR® Premix Ex TaqTM (Takara) in a total volume of 20 μl. The siRNA was quantified by a TaqMan® small RNA assay (ABI) as described previously (32). Three biological replicates were performed to evaluate the relative mRNA abundance and to determine the standard error (S.E.) between the replicates. The primers used for RT-PCR and qPCR are listed in Supplementary Table S10.
ChIP assays
ChIP was performed as described previously (33,34). Briefly, plant tissues were harvested after 10 days of growth on MS medium and cross-linked by vacuum infiltration with 1% formaldehyde prior to being ground in liquid nitrogen. The isolated chromatin was disrupted into ∼500-bp fragments by sonication and precipitated using H3K9me2- (Millipore CS200587), H3K4me3- (Upstate 04-745), YFP- (ABcam ab290) and Pol II-specific antibodies (COVANCE MMS-126R, MMS-129R and MMS-134R). After elution from the beads and reverse cross-linking, the DNA fragments in immunocomplexes were extracted with phenol/chloroform and recovered by ethanol precipitation. The abundance of immunoprecipitated chromatin was determined by qPCR using the primers given in Supplementary Table S10. The results of three biological replicates are shown as the absolute enrichment compared with the input.
DNA methylation analysis
Genomic DNA from the appropriate genotypes was subjected to bisulfite conversion using a Methylamp DNA Modification kit (Epigentek) according to the manufacturer’s instructions. The regions of interest, RDR2 (+2509 to +2702) and DCL3 (+4204 to +4396), were amplified by PCR and cloned into the vector pEASY-Blunt (Transgen). At least 11 clones for each converted sample were sequenced using the primer M13F. Bisulfite sequencing to determine the methylation state of tandem repeats in the SDC promoter was performed as described (18). During our analysis of the bisulfite sequencing data, the percentage of methylated sites in the clones was calculated for various sequence contexts. Further information on the clones and bisulfite data are provided in Supplementary Tables S4, S5, S7 and S8; the primers used for amplification are listed in Supplementary Table S10.
Small RNA sequencing and analysis
The preparation of small RNA samples for Illumina sequencing was done according to the Illumina small RNA sample preparation guide. Using total RNA extracted from 10-day-old plants, small RNAs were enriched by PEG precipitation and separated by 15% TBE-urea PAGE. Following SYBR-gold staining, the small RNA fraction (18–26 nucleotides [nt] in length) was recovered by gel purification and ligated sequentially to 5′- and 3′-adapters. The products of the second ligation were reverse-transcribed and amplified using the corresponding primers. The sequences of the adapters and primers used were as described in the Illumina user manual. Small RNA sequencing was done according to the Illumina 1G sequencing protocol. The raw data have been deposited in the NCBI database under GEO number GSE32284.
The 3′-adapter sequences were removed by a custom Perl script, and the trimmed small RNA reads were mapped to the Arabidopsis genome using Bowtie with perfect matches. After mapping, the positions of those reads matching with multiple loci were randomly selected and counted together with those reads mapping to unique loci in our subsequent analyses. Sequences matching with tRNAs, rRNAs, small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs) that might represent degradation products of these abundant RNA species were discarded. Reads were selected as miRNAs and tasiRNAs by comparing the remaining small RNA reads against miRNA and tasiRNA precursor genes. The programs Tandem Repeat Finder (35) and Inverted Repeat Finder (36) were used to identify tandem repeats and inverted repeats, respectively. Other repeats were identified using the program RepeatMasker with the repeat library from RepBase (37).
Small RNA reads from WT and ibm1-3 were pooled together and clustered by merging the regions with at least four small RNA reads, each separated from its nearest neighbor by no more than 200 nt.
Analysis of differentially expressed genes
In all comparisons, the read counts for a gene or genomic region from each library were normalized to the Reads Per Kilobase of exon model per Million mapped reads (RPKM); the read number for each gene or region was divided by the total read number in each library and multiplied by 109. Significant differences in gene expression and small RNA generation between the compared lines were identified by the chi-squared test. Next, the P-values were adjusted to q-values for multiple testing corrections (38).
RESULTS
IBM1 regulates RDR2 and DCL3 expression
To identify novel targets of IBM1, we performed a genome-wide analysis of gene expression using Illumina mRNA-Seq, which sequences cDNAs synthesized from pre-sheared and poly(A)-enriched RNA directly (39,40). This approach keeps the proportion of sequence reads from rRNA low, and avoids the bias against mRNAs with a short poly(A) tail (41). We obtained >17 million reads of 42 bp, representing >6 times coverage of the Arabidopsis genome, from whole 10-day-old WT or ibm1-3 plants. By mapping the sequence reads to the reference genome (TAIR10), ∼71% of the WT reads and 62% of the ibm1-3 reads mapped to unique genomic locations; of these, ∼98% mapped to annotated exons (Supplementary Table S1). Furthermore, 24 013 (71.5%) of all annotated genes in the TAIR10 database were mapped to by at least one read (Figure 1a). Thus, mRNA-Seq provides quantitative data for studies of transcription (39,42).
Figure 1.
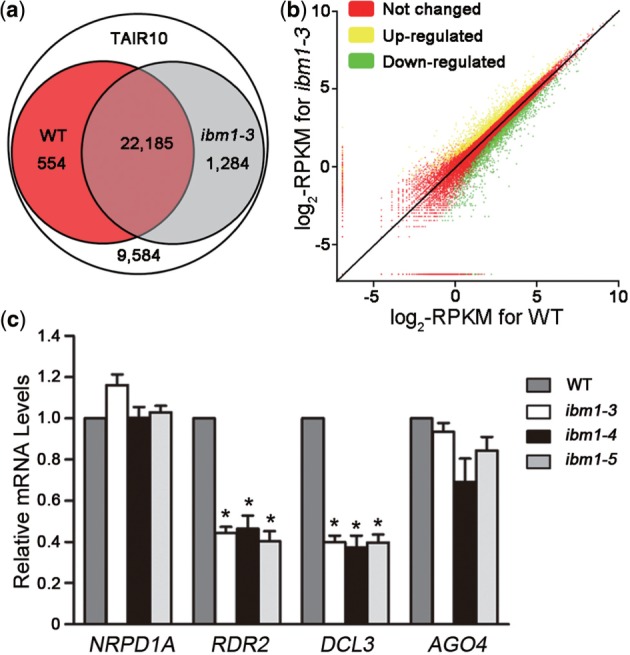
Detection of IBM1-regulated genes by mRNA-Seq and qPCR. (a) Overlap between the libraries produced from WT and ibm1-3 plants based on mRNA-Seq and the TAIR10 database. Those loci with at least one matched read were counted in the mRNA-Seq libraries. (b) Comparison of the level of transcription of individual loci between WT and ibm1-3 plants. To evaluate the significance of the transcriptional level at each locus, the absolute number of matched reads was normalized to the scale of the libraries and to the length of the gene-coding sequence, respectively. (c) mRNA levels of siRNA biosynthesis pathway genes in the ibm1 mutants. Plants of each genotype were grown under LD conditions for 10 days then harvested for qPCR analysis. The results from three independent biological replicates are shown; error bars represent the S.E. Asterisks indicate a significant difference using Student’s t-test (P < 0.05).
At P ≤ 0.05 and Q ≤ 0.01, 4912 genes were identified as differentially expressed between ibm1-3 and WT, with an estimated absolute log2-fold change >0.5, and 40.9% (2007 genes) of these exhibited a log2-fold change >1 (Figure 1b and Supplementary Table S2). Unlike the variations in DNA methylation seen previously in ibm1 (i.e. elevated DNA methylation levels in more than 1000 genes but reduced levels in just a few) (29), almost equal numbers of genes were up- or downregulated at the level of transcription in our mRNA-Seq analysis (2869 up- to 2043 downregulated) (Figure 1b and Supplementary Table S2). Since IBM1 is a histone demethylase with the ability to remove methyl group(s) from H3K9me2, a marker of transcriptional repression, the loss of IBM1 may lead to the reduced expression or silencing of its targets. Hence, the enhanced expression of multiple genes seen in this study suggests the indirect regulation of gene expression by IBM1.
From the list of modulated genes, we found that RDR2 and DCL3, which encode necessary factors in siRNA biogenesis and the RdDM pathway, were obviously decreased in ibm1; in contrast, RDR6 and DCL4, homologs of RDR2 and DCL3 with divergent functions, remained unchanged in ibm1 compared with WT plants (Supplementary Table S3). Further examination by qPCR verified the downregulation of RDR2 and DCL3 in ibm1. However, there was no significant difference in the expression of NRPD1A and AGO4, two other key members of the RdDM pathway (Figure 1c). These results were reproducible in three ibm1 alleles (Figure 1c), suggesting that IBM1 is required to maintain normal transcriptional levels of RDR2 and DCL3 in Arabidopsis.
IBM1 associates directly with and demethylates RDR2 and DCL3 chromatin
In a previous study, IBM1 was clustered with the human JHDM2 subfamily, which is known to specifically demethylate H3K9me2 and H3K9me1 (43–45); IBM1 was also shown to catalyze H3K9me2 and H3K9me1 demethylation in Arabidopsis (26). To address whether IBM1 regulates the expression of RDR2 and DCL3 by affecting the histone methylation status of the chromatin at these loci, we performed a ChIP assay using a series of primers covering different regions across RDR2 and DCL3 (Figure 2a).
Figure 2.

IBM1 controls the chromatin modification state at RDR2 and DCL3. (a) Schematic genetic structures of RDR2 and DCL3 and of the regions used for RT-PCR and real-time qPCR after ChIP. The black boxes represent exons; the gray lines represent introns or 3′-UTRs. (b) Detection of the H3K9me2 and H3K4me3 levels in RDR2 and DCL3 chromatin by RT-PCR following ChIP. ChIP was conducted using antibodies against H3K9me2 and H3K4me3. Plants of each genotype were grown under LD conditions for 10 days before harvest for ChIP. Input, chromatin before IP; NoAB, control samples without antibodies in the IP steps. (c–f) qPCR analysis of the H3K9me2 ([c] and [e]) and H3K4me3 levels ([d] and [f]) at RDR2 and DCL3, respectively. Our results for WT and ibm1-4 are normalized to the input. The results from three biological replicates are shown; error bars represent the S.E. Asterisks indicate a significant difference using Student’s t-test (P < 0.05).
In WT plants, the H3K9me2 level was extremely low across RDR2 and DCL3 (Figure 2b, c and e), which is consistent with the active function of these genes in growth and development. The H3K9me2 levels in ibm1 were distinctly increased in regions P2 and P3, which represent the gene body of RDR2 (Figure 2b and c). In addition, the level of H3K4me3, which is a gene activation marker and antagonist to H3K9me2, was obviously reduced in ibm1 in the intragenic, but not in the upstream or downstream region, of RDR2 (Figure 2b and d).
Nearly the same results were obtained for DCL3 by ChIP: the H3K9me2 level was distinctly increased in regions P2–P7 of the DCL gene body, whereas the H3K4me3 level in DCL3 chromatin was regulated in an antagonistic manner to H3K9me2 in ibm1 (Figure 2b, e and f). These results indicate that IBM1 induces RDR2 and DCL3 expression by demethylating H3K9me2 within their loci in vivo.
To determine how K3K9 methylation regulates the transcription of RDR2 and DCL3, we examined the recruitment of Pol II to RDR2 and DCL3 chromatin in WT and ibm1 plants. Pol II accumulation was decreased at RDR2 and DCL3 chromatin when IBM1 was absent (Supplementary Figure S2a and b), which is consistent with the downregulation of their expression in ibm1. However, accumulation of the initiation (Ser5 phosphorylated) and elongation (Ser2 phosphorylated) forms of Pol II at RDR2 and DCL3 chromatin was unchanged in ibm1 (Supplementary Figure S2c–f), suggesting that H3K9 methylation is not involved in the transcriptional initiation or the elongation of RDR2 and DCL3.
The above results suggest that IBM1 binds directly to RDR2 and DCL3 chromatin to regulate their H3K9 methylation status locally. To investigate the direct association of IBM1 with RDR2 and DCL3 chromatin, we performed a ChIP assay using transgenic plants expressing 35S::YFP::IBM1 in an ibm1-3 background. The integrated IBM1 in the transgenic plants was expressed (Figure 3d) and was exclusively nuclear-localized in protoplasts (Figure 3a). Also, a functional YFP-IBM1 fusion protein was produced in vivo, as demonstrated by its ability to complement all of the mutant phenotypes of ibm1-3, including its embryonic defect and abnormally sharp rosette leaves (Figure 3b and c).
Figure 3.
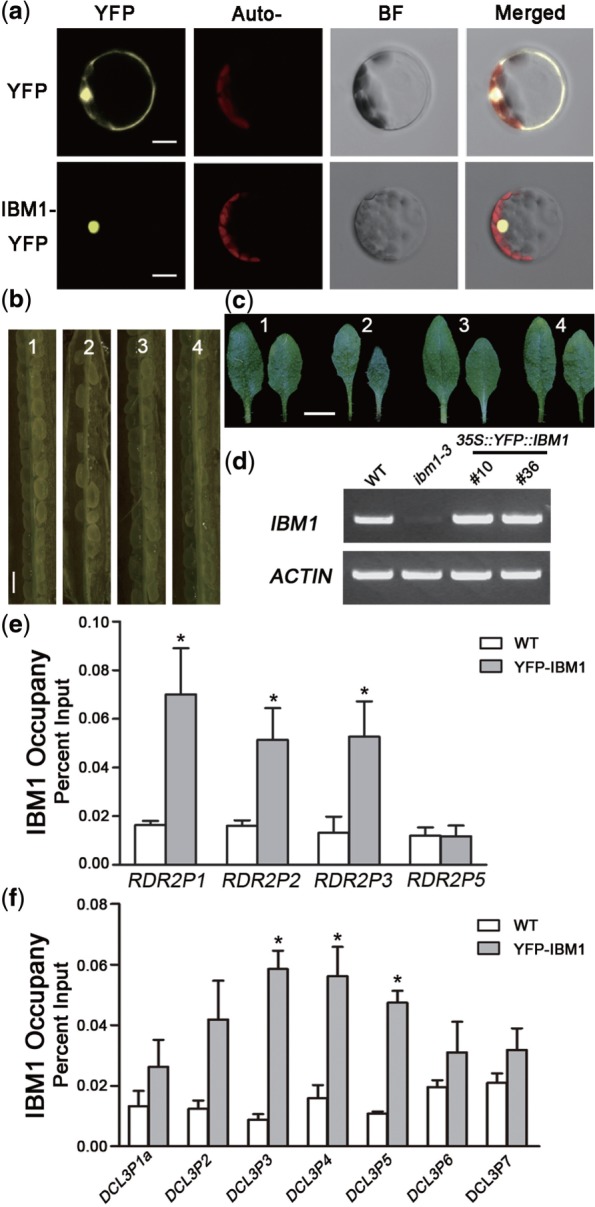
IBM1 associates directly with RDR2 and DCL3 chromatin. (a) YFP-IBM1 was nuclear-localized in leaf mesophyll protoplasts. Bar = 10 μm. The defects in embryonic (b) and rosette leaf development (c) in ibm1 were rescued by the transformation of 35 S::YFP::IBM1 into ibm1-4. The number 1-4 refers to WT, ibm1-4, and two independent T1 lines (#10 and #36) of 35 S::YFP::IBM1/ibm1-4, respectively. Siliques from 40-day-old plants were stripped and photographed. Bar = 0.5 mm. The 6th and 7th rosette leaves from 25-day-old plants of each genotype were photographed. Bar = 1 cm. (d) RT-PCR analysis of the transcriptional levels of IBM1 in the complemented plants. The ACTIN mRNA level was used as an endogenous control. (e) and (f) ChIP analysis of the association of IBM1 with RDR2 and DCL3, respectively. 10-day-old plants were used for ChIP, and the enrichments were normalized to the input. All results are shown as the mean value ± S.E. of three biological replicates. Auto: auto-fluorescence; BF: bright field. Asterisks indicate a significant difference using Student’s t-test (P < 0.05).
Using YFP-specific antibodies, the chromatin associated with IBM1 was immunoprecipitated and measured by qPCR. Our results indicate that DNA fragments from the upstream and intragenic regions of RDR2, and from the intragenic region of DCL3, were obviously enriched by IBM1 in the transgenic plants (Figure 3e and f). The occupancy of IBM1 was detected at a higher density in regions P3, P4 and P5 of DCL3, and in the P1, P2 and P3 regions of RDR2 than in any other region within that locus, in agreement with the increase in H3K9me2 induced by a defect in IBM1 (Figure 2b, c and e). In summary, these data suggest that IBM1 associates directly with the gene body of DCL3, and with both the promoter and gene body of RDR2 in vivo, and that it regulates the H3K9me2 status at these loci to control their expression.
IBM1 mediates the non-CG DNA methylation of RDR2 and DCL3
Previous genetic and molecular studies have demonstrated a close relationship between H3K9me2 and non-CG DNA methylation (20,25). Given the function of IBM1 in the regulation of H3K9 methylation, we examined the cytosine methylation state of target loci of IBM1. Using a bisulfite assay, the intragenic regions of RDR2 (+2509 to +2702) and DCL3 (+4204 to +4396) were amplified, cloned and sequenced. The regions in RDR2 and DCL3 selected for bisulfite sequencing had the highest IBM1 occupancy and greatest increase in H3K9me2 in ibm1 (Figure 4).
Figure 4.
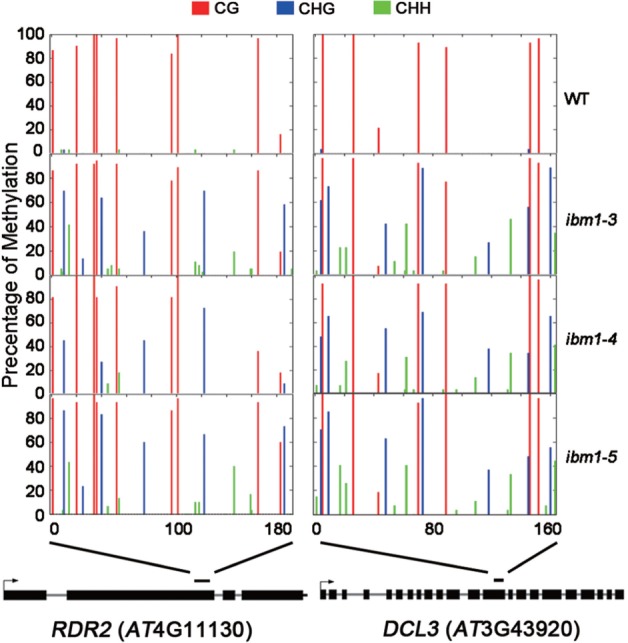
IBM1 is required to protect RDR2 and DCL3 from non-CG DNA methylation. The DNA methylation states of RDR2 and DCL3 were analyzed by bisulfite sequencing in both WT plants and three ibm1 alleles. The average percentage of methylation at each cytosine was determined for at least 11 clones (indicated by vertical bars: red, CG; blue, CNG; and green, asymmetric cytosine). The schematic structures of RDR2 and DCL3 at the bottom of the figure show the regions included in our DNA methylation analysis.
Considerable CG methylation was detected in the WT plants, but almost no methylated cytosines were found in the context of CHG or asymmetric cytosines in RDR2 and DCL3 (Figure 4 and Supplementary Tables S4 and S5). It has been reported that a large number of genes carry CG methylation within their coding sequences (3). Unlike non-CG methylation in the promoter or 5′ region, intragenic CG methylation clearly does not function to silence gene expression (4,46). In all three ibm1 alleles, the non-CG methylation levels at RDR2 and DCL3 were remarkably elevated (Figure 4 and Supplementary Tables S4 and S5), in agreement with previous data showing a correlation between H3K9 and DNA cytosine methylation. The non-CG hypermethylation of RDR2 and DCL3 in ibm1 indicates that the increase in expression of RDR2 and DCL3 caused by IBM1 is a consequence of integrated regulation by different epigenetic modifications.
The mutation of IBM1 causes a decrease in gene-specific siRNAs
In Arabidopsis, a whole-genome tiling array analysis revealed dozens of upregulated transcripts in rdr2, some of which have predicted siRNA loci (47). Previous studies revealed the dramatic enrichment of 21-nt miRNAs and tasiRNAs, and a corresponding reduction in 24-nt siRNAs, in rdr2 (48–51). Also, dcl3 shows a similar pattern to rdr2 in terms of its small RNA population (49).
To determine whether the reduction in RDR2 and DCL3 expression in ibm1 affects the global profile of siRNA biogenesis, we performed unbiased Illumina small RNA sequencing using WT and ibm1-3 plants. A total of 24 911 693 reads in WT and 24 845 025 reads in ibm1-3 were obtained. After eliminating unmatched nucleotides, 21 461 549 sequences (86.2% of the total reads) in WT and 22 014 858 sequences (88.6% of the total reads) in ibm1-3 were found to have at least one perfect match in the Arabidopsis nuclear genome, whereas 5 088 056 reads in WT and 6 562 528 reads in ibm1-3 were considered to be small RNAs after eliminating those reads corresponding to tRNAs, rRNAs, snRNAs and snoRNAs. A total of 45.4% of the small RNAs in WT (2 310 925 reads) and 48.7% of those in ibm1-3 (3 195 421 reads) were mapped to unique genomic loci. Those small RNAs that mapped to the nuclear genome represented 1 836 926 unique sequences in WT, whereas those from ibm1-3 represented 2 140 106 unique sequences (Supplementary Table S6).
Size profiling revealed that although the population of 21-nt small RNAs was increased in ibm1-3, similar to rdr2 (50,52), the population of 24-nt siRNAs was not dramatically reduced (Figure 5b and Table 1). These data suggest that 24-nt heterochromatin siRNAs remained in ibm1-3 even though the transcript levels of RDR2 and DCL3 were obviously decreased (Figure 1c), and they indicate a distinct requirement for RDR2 and DCL3 in the biogenesis of different 24-nt siRNAs.
Figure 5.

Small RNA sequencing in WT and ibm1 plants. (a) Scrolling-window analysis of small RNAs in ibm1-3 and WT. The reads produced by Illumina sequencing were mapped to the Arabidopsis genome, and the small RNA density distributions in 100-kb sliding windows with a 50-kb slide were counted along the chromosomes and are shown as the read number per bp. Bars with >10 reads/bp are not shown in this figure. (b) Comparison of the size distribution of small RNAs in WT and ibm1-3. The percentage of reads of a given length in all small RNA reads was calculated. (c) Scatterplot comparing the amounts of endogenous siRNAs generated from each locus between ibm1-3 and WT. The number of siRNA reads for each locus was counted and normalized to the scale of the libraries. RPM: reads per million.
Table 1.
Global view of the small RNA clusters in ibm1 and WT plants
| Categories | WT reads of small RNA | Categories in WT (%) | WT signatures | ibm1-3 reads of small RNA | Categories in ibm1-3 (%) | ibm1-3 signatures | |
|---|---|---|---|---|---|---|---|
| Total | 5 088 056 | 100 | 1 836 926 | 6 562 528 | 100 | 2 140 106 | |
| miRNA | 481 980 | 9.47 | 3 032 | 1 050 165 | 16.00 | 3 373 | |
| tasiRNA | 142 722 | 2.81 | 6 622 | 254 005 | 3.87 | 7 697 | |
| Tandem repeats | 137 206 | 2.70 | 45 375 | 144 885 | 2.21 | 52 675 | |
| Transposon | 253 933 | 5.00 | 162 311 | 301 960 | 4.60 | 178 678 | |
| LTR | 373 619 | 7.34 | 56 725 | 408 169 | 6.22 | 70 966 | |
| Non-LTR | 103 559 | 2.04 | 67 613 | 114 238 | 1.74 | 74 702 | |
| Inverted repeats | 75 791 | 1.49 | 28 293 | 100 064 | 1.52 | 31 455 | |
| Protein-coding genes | 888 374 | 17.46 | 796 989 | 1 084 449 | 16.52 | 953 938 | |
| Promoter regions | 1 194 633 | 23.48 | 230 657 | 1 418 967 | 21.62 | 246 057 | |
| Downstream regions | 157 753 | 3.10 | 58 755 | 187 536 | 2.86 | 65 015 | |
| Others | 1 278 486 | 25.13 | 380 554 | 1 498 090 | 22.83 | 455 550 | |
To analyze the distribution of loci generating small RNAs in the genome, we next clustered the small RNAs in both WT and ibm1-3 using a proximity-based algorithm to compare their similarities and differences (52). Almost all of the clusters (99.2%) detected in WT also existed in ibm1-3, and vice versa (95.3%) (Supplementary Figure S2). In addition, the small RNAs in both WT and ibm1-3 displayed pericentromeric concentrations, which were missing in rdr2 because of the loss of heterochromatic siRNA (Supplementary Figure S2). Also, there was no detectable change in a scrolling-window analysis (53) of small RNA-homologous sequences across each chromosome (Figure 5a). Our data demonstrate that the downregulation of RDR2 and DCL3 in ibm1 did not affect the generation and distribution of small RNAs on a genome-wide scale.
To further investigate the regulation of small RNA biogenesis by IBM1, the small RNAs mapped to the Arabidopsis genome were categorized based on their sizes, species, functions and genomic locations (54,55). The locations included intergenic regions, annotated protein-coding genes, transposable elements, repetitive sequences and the promoter or 3′ region of coding genes (Table 1). Based on the known proportions in the miRNA library, the overall enrichment of miRNA in ibm1-3 was 1.7-fold, which is similar to the previously reported 1.8-fold increase in rdr2 (48). In comparison, the overall enrichment of tasiRNAs in ibm1-3 was 1.4-fold (Table 1 and Supplementary Figure S3). Although the mechanism of miRNA and tasiRNA enrichment by the knockout or downregulation of RDR2 and DCL3 is unclear, it is most likely an indirect effect. In comparison, other fractions of small RNAs, like the 24-nt siRNAs associated with genic, intergenic and transposable sequences, showed no obvious differences between WT and ibm1-3 (Table 1 and Supplementary Figure S3).
Although the disruption of RDR2 and DCL3 expression in ibm1 did not affect global siRNA biogenesis in Arabidopsis, it was expected that RDR2- and DCL3-dependent siRNAs homologous to some loci should be decreased in ibm1. To identify candidate loci affected indirectly by IBM1 through the RdDM pathway, 24-nt siRNAs homologous to the coding regions of genes, as well as their 1000-bp upstream promoters and 500-bp downstream regions were selected and counted. Using this approach, we identified 2926 loci (8.78%, P ≤ 0.01) with a significant reduction in local siRNAs in ibm1, as well as 1144 loci (3.43%, P ≤ 0.01) that were enriched (Figure 5c). This suggests that, unlike rdr2 and dcl3, in which siRNA biosynthesis was completely blocked, the ∼50% decrease in RDR2 and DCL3 in ibm1 only affected a small portion of siRNAs.
Further, we found the distinct accumulation of siRNAs mapping to the genic regions of RDR2 in ibm1-3, which is in agreement with the increase in non-CG methylation in the gene body and repression of mRNA transcription (Figures 1c and 4 and Supplementary Table S3). In summary, the defect in siRNA biogenesis detected in ibm1 is specific to a group of genes, instead of being a genome-wide effect.
Genes regulated by IBM1 through RdDM
SDC is a protein-coding gene silenced by siRNA-mediated DNA methylation in WT plants. The 24-nt siRNAs homologous to tandem repeats in the promoter of SDC are undetectable in rdr2 and dcl3 (18), suggesting a genetic requirement for RDR2 and DCL3 in establishing and maintaining SDC in a silent state. To test whether the reduction in RDR2 and DCL3 expression in ibm1 resulted in defective siRNA biogenesis and RdDM, we used SDC expression, which indicates the functional activity of the RdDM pathway in Arabidopsis, as a molecular marker.
Using mRNA-Seq, the reads that mapped to exons of SDC were distinctly enriched in ibm1-3 (Supplementary Figure S4a and Supplementary Table S3). In addition, qPCR confirmed the ≥20-fold accumulation of SDC mRNA in all three ibm1 alleles (Supplementary Figure S4b). Subsequently, we analyzed DNA methylation in the entire tandem repeats and in the regions upstream and downstream of the SDC repeats by bisulfite sequencing. In WT plants, all of the sequence contexts were densely methylated (Supplementary Figure S4c and Supplementary Table S7). Consistent with this, DNA methylation at CHG and CHH sites in the SDC tandem repeats was slightly reduced in the ibm1 alleles, whereas that in the regions flanking the repeats was not obviously changed (Supplementary Figure S4c and Supplementary Table S7). These data show that the defect in IBM1 reduced RDR2 and DCL3 transcription and partially affected the small RNA-mediated DNA methylation of the SDC locus.
To investigate whether the reduction in local siRNAs could influence the expression of other genes, we combined our mRNA-Seq and small RNA sequencing data. Since RdDM exerts a repressive effect at the level of transcription, the downregulation of RDR2 and DCL3, and the resulting decrease in siRNAs in ibm1, should release the repressed genes. Thus, we expected to find genes with upregulated expression and downregulated local siRNA production caused by the mutation of IBM1.
Screening based on these criteria revealed 246 loci (7.41%) whose local siRNA levels were reduced in ibm1-3, and which were upregulated by a log2-fold change >0.5 in terms of mRNA accumulation; among them, 127 (3.83%) loci were upregulated by a log2-fold change >1. Our real-time PCR results confirmed that the mRNA levels of these randomly selected genes were upregulated in ibm1-3 (Figure 6a). In addition, all of them were significantly upregulated in both rdr2 and dcl3 (Figure 6a).
Figure 6.

IBM1 represses gene expression indirectly through the RdDM pathway. (a) Increases in the expression of representative indirect IBM1 targets were validated by qPCR in ibm1-3. The expression of these representative genes were also increased in rdr2 and dcl3. RNA from 10-day-old plants was used for qPCR analysis. The level of expression was normalized to that of ACTIN. Error bars indicate the S.E. from three biological replicates. (b) The DNA methylation states of these genes were analyzed by bisulfite sequencing in both ibm1-3 and WT. The average percentage of methylation at each cytosine was determined from all clones sequenced. The bisulfite sequencing data are included in Supplementary Table S8. (c) The levels of three representative siRNAs in ibm1 and WT as detected by TaqMan® small RNA assays. The gene codes corresponding to the siRNAs are indicated. Asterisks indicate a significant difference using Student’s t-test (P < 0.05).
To detect the reduction in siRNAs in ibm1, we conducted northern blotting to analyze the corresponding siRNA levels of several randomly selected genes from the above-mentioned 246 loci in WT and ibm1 plants. Due to the low levels of those siRNAs, we could not detect the siRNA signals, even in WT plants, by northern blotting. However, we detected a reduction in the siRNA levels of two RDR2- and DCL3-dependent marker transposable elements, SIMPLEHAT2 and AtREP2, by northern blotting (Supplementary Figure S5), which is consistent with the downregulation of RDR2 and DCL3 in ibm1. Thus, we employed a more sensitive approach to detect the siRNA levels: the TaqMan® small RNA assay. Among the four loci we selected, the target siRNAs of three were specifically detected, and all three showed an obvious decrease in ibm1 (Figure 6c).
To determine whether these loci with increased expression and decreased siRNA levels are regulated by the RdDM pathway, we examined their DNA methylation status by bisulfite sequencing. For all eight randomly selected loci, the methylation levels in ibm1 were decreased (Figure 6b and Supplementary Table S8), consistent with the observed reduction in siRNAs and transcriptional release. These results indicate that the indirect repression of these novel targets by IBM1 is dependent on RdDM-directed DNA hypermethylation.
In summary, by analyzing the transcriptome and small RNA profiles of WT and ibm1-3 plants, more than 100 candidate genes were identified that exhibited an increased mRNA level and decreased siRNA accumulation in ibm1. These data suggest a mechanism whereby IBM1 regulates a series of protein-coding genes through the RdDM-dependent control of siRNA biogenesis.
DISCUSSION
The expression level of a gene is controlled by its histone modification status. For example, H3K4me3 is an activation marker, whereas H3K9me2 is a repression marker (56,57). IBM1 is a conserved JmjC family histone demethylase in Arabidopsis that specifically removes methyl group(s) from H3K9me2 and H3K9me1; thus, it works as a gene activator (26,29). Therefore, we expected that the expression of multiple genes would be repressed in ibm1. However, the results of our mRNA-Seq analysis revealed that in addition to the repression of more than 2000 genes, a similar number of genes were de-repressed (Figure 1b and Supplementary Table S2). This suggests that IBM1 directly represses gene expression by demethylating H3K9me2 chromatin, and that it indirectly increases gene expression by an unknown mechanism.
We compared the genes with a hypermethylated cytosine in ibm1 from Miura et al. (29) with the genes that exhibited transcriptional downregulation in ibm1-3 based on our mRNA-Seq data, and found that among 2043 genes, 309 (15.1%) were also hypermethylated at cytosines in ibm1 by at least 1.41-fold (Supplementary Figure S6a). We also observed the accumulation of 24-nt small RNAs in the gene body regions of these hypermethylated genes (Supplementary Table S9). These results indicate that the genes exhibiting both DNA hypermethylation and downregulated expression in ibm1 may be direct targets of IBM1.
In our high-throughput mRNA-Seq analysis, we found that the expression of two essential factors required for siRNA biogenesis and the RdDM pathway, RDR2 and DCL3, was downregulated in ibm1, and this observation was confirmed by qPCR (Figure 1c and Supplementary Table S3). Consistent with this observation, we found that among the 497 genes upregulated significantly in rdr2 and dcl3 (49), 176 (35.4%) were also upregulated in ibm1 (Supplementary Figure S6b). Furthermore, among the eight randomly selected genes that were upregulated in ibm1, all of them were upregulated in both rdr2 and dcl3 (Figure 6a). We also detected a reduction in the siRNA levels of two RDR2- and DCL3-dependent marker transposable elements, SIMPLEHAT2 and AtREP2, in ibm1 (Supplementary Figure S5).
In addition, our ChIP results showed that IBM1 could directly bind (Figure 3e and f) and remove the methyl group(s) from H3K9me2 (Figure 2) at RDR2 and DCL3 chromatin. Furthermore, the loss of IBM1 increased the non-CG DNA methylation of RDR2 and DCL3 (Figure 4); thus, the H3K9 methylation and DNA methylation of these loci are coupled (Figure 4 and Supplementary Tables S4 and S5). Therefore, our results not only indicate that RDR2 and DCL3 are direct targets of IBM1, they also show that IBM1 controls its targets epigenetically through histone modification and DNA methylation. In addition, we found that the levels of RDR2 and DCL3 were downregulated by only ∼50% in ibm1 (Figure 1c), suggesting that other regulators besides IBM1 are necessary for the regulation of RDR2 and DCL3 expression and the RdDM pathway. Further, besides RDR2 and DCL3, there are other direct targets of IBM1 in the genome, and a number of them were downregulated and hypermethylated in ibm1 (Supplementary Figure S6a).
RDR2 and DCL3 play essential roles in siRNA biogenesis in cells, and especially in the production of 24-nt siRNAs (10). A genome-wide expression profile analysis of the small RNAs in ibm1 revealed a reduction in siRNAs specific for a group of loci (Figure 5c). The reduction in these siRNAs was confirmed by a TaqMan® small RNA assay (Figure 6c). Subsequent experiments showed that the locus-specific regulation of siRNAs by IBM1 regulated the DNA methylation of their target genes, and ultimately mediated the expression of these genes (Figure 6), demonstrating the indirect function of IBM1 in the regulation of gene expression via the RdDM pathway. We also observed that there were no global changes in 24-nt siRNAs between WT and ibm1 (Figure 5 and Supplementary Figure S2). This may be due to functional redundancy among RDR and DCL proteins, or to the fact that RDR2 and DCL3 expression was only reduced by 50% in ibm1 (Figure 1c).
Besides its direct function in protecting genes from silencing by preventing the coupling of histone and DNA methylation, IBM1 also works as a precise regulator that indirectly represses gene expression by mediating siRNA biogenesis and specifically controlling a set of siRNA-targeted genes through the RdDM pathway. These observations suggest that IBM1 regulates gene expression through two distinct pathways in plants to coordinate the response to environmental signals and endogenous cues: the direct inducement of gene expression and indirect repression.
During the processes whereby IBM1 achieves its function in gene regulation, RDR2 and DCL3, two components of the RdDM pathway, play an important role in the regulation of gene expression by IBM1, as they link the two pathways. In rdr2 and dcl3, 24-nt siRNAs are distinctly underrepresented (48,49). However, in ibm1, in which the expression of RDR2 and DCL3 was downregulated (Figure 1c), there was no detectable difference in the abundance or distribution of 24-nt siRNAs by cluster or scrolling-window analysis (Figure 5a and Supplementary Figure S1). In addition, a pericentromeric concentration of heterochromatic siRNA was detected in ibm1 (Supplementary Figure S1), and the reduction in siRNAs in ibm1 was limited to a specific group of loci, rather than being on a genome-wide scale (Figure 5c). These results indicate distinct dosage requirements for RDR2 and DCL3 in the biogenesis of different 24-nt siRNAs. However, the biochemical mechanism underlying the differential requirement for these enzymes in the biosynthesis of siRNAs remains unclear.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–10 and Supplementary Figures 1–6.
FUNDING
Funding for open access charge: National Basic Research Program of China (973 Program) [2012CB910900 and 2012CB114200]; Hebei Province key laboratory program [109960121D]; and National Natural Science Foundation of China [31071124].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Jessica Habashi for critical reading of the manuscript, Jianguang Zhang and Dr. Tao Zhao for their help and suggestions on small RNA preparation and high-throughput sequencing, and the ABRC for the T-DNA insertion line.
REFERENCES
- 1.Chan SW, Henderson IR, Jacobsen SE. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005;6:351–360. doi: 10.1038/nrg1601. [DOI] [PubMed] [Google Scholar]
- 2.Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 5.Saze H, Scheid O, Paszkowski J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003;34:65–69. doi: 10.1038/ng1138. [DOI] [PubMed] [Google Scholar]
- 6.Cao X, Jacobsen SE. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl Acad. Sci. USA. 2002;99:16491–16498. doi: 10.1073/pnas.162371599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002;12:1138–1144. doi: 10.1016/s0960-9822(02)00925-9. [DOI] [PubMed] [Google Scholar]
- 8.Jones L, Hamilton AJ, Voinnet O, Thomas CL, Maule AJ, Baulcombe DC. RNA-DNA interactions and DNA methylation in post-transcriptional gene silencing. Plant Cell. 1999;11:2291–2301. doi: 10.1105/tpc.11.12.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 10.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature. 2007;447:418–424. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- 12.Onodera Y, Haag JR, Ream T, Nunes PC, Pontes O, Pikaard CS. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell. 2005;120:613–622. doi: 10.1016/j.cell.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Herr AJ, Jensen MB, Dalmay T, Baulcombe DC. RNA polymerase IV directs silencing of endogenous DNA. Science. 2005;308:118–120. doi: 10.1126/science.1106910. [DOI] [PubMed] [Google Scholar]
- 14.Li CF, Pontes O, El-Shami M, Henderson IR, Bernatavichute YV, Chan SW, Lagrange T, Pikaard CS, Jacobsen SE. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell. 2006;126:93–106. doi: 10.1016/j.cell.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 15.Pontes O, Li CF, Nunes PC, Haag J, Ream T, Vitins A, Jacobsen SE, Pikaard CS. The Arabidopsis chromatin-modifying nuclear siRNA pathway involves a nucleolar RNA processing center. Cell. 2006;126:79–92. doi: 10.1016/j.cell.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 16.Qi Y, He X, Wang XJ, Kohany O, Jurka J, Hannon GJ. Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature. 2006;443:1008–1012. doi: 10.1038/nature05198. [DOI] [PubMed] [Google Scholar]
- 17.Mosher RA, Schwach F, Studholme D, Baulcombe DC. PolIVb influences RNA-directed DNA methylation independently of its role in siRNA biogenesis. Proc. Natl Acad. Sci. USA. 2008;105:3145–3150. doi: 10.1073/pnas.0709632105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henderson IR, Jacobsen SE. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev. 2008;22:1597–1606. doi: 10.1101/gad.1667808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 20.Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 21.Ebbs ML, Bartee L, Bender J. H3 lysine 9 methylation is maintained on a transcribed inverted repeat by combined action of SUVH6 and SUVH4 methyltransferases. Mol. Cell Biol. 2005;25:10507–10515. doi: 10.1128/MCB.25.23.10507-10515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebbs ML, Bender J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell. 2006;18:1166–1176. doi: 10.1105/tpc.106.041400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernatavichute YV, Zhang X, Cokus S, Pellegrini M, Jacobsen SE. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS ONE. 2008;3:e3156. doi: 10.1371/journal.pone.0003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Callis J, Jacobsen SE. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 2007;17:379–384. doi: 10.1016/j.cub.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindroth AM, Shultis D, Jasencakova Z, Fuchs J, Johnson L, Schubert D, Patnaik D, Pradhan S, Goodrich J, Schubert I, et al. Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J. 2011;30:1874. doi: 10.1038/sj.emboj.7600430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inagaki S, Miura-Kamio A, Nakamura Y, Lu F, Cui X, Cao X, Kimura H, Saze H, Kakutani T. Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J. 2010;29:3496–3506. doi: 10.1038/emboj.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saze H, Shiraishi A, Miura A, Kakutani T. Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science. 2008;319:462–465. doi: 10.1126/science.1150987. [DOI] [PubMed] [Google Scholar]
- 28.Sun Q, Zhou DX. Rice jmjC domain-containing gene JMJ706 encodes H3K9 demethylase required for floral organ development. Proc. Natl Acad. Sci. USA. 2008;105:13679–13684. doi: 10.1073/pnas.0805901105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miura A, Nakamura M, Inagaki S, Kobayashi A, Saze H, Kakutani T. An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J. 2009;28:1078–1086. doi: 10.1038/emboj.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 31.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowler C, Benvenuto G, Laflamme P, Molino D, Probst AV, Tariq M, Paszkowski J. Chromatin techniques for plant cells. Plant J. 2004;39:776–789. doi: 10.1111/j.1365-313X.2004.02169.x. [DOI] [PubMed] [Google Scholar]
- 34.Saleh A, Alvarez-Venegas R, Avramova Z. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat. Protoc. 2008;3:1018–1025. doi: 10.1038/nprot.2008.66. [DOI] [PubMed] [Google Scholar]
- 35.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warburton PE, Giordano J, Cheung F, Gelfand Y, Benson G. Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 2004;14:1861–1869. doi: 10.1101/gr.2542904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- 38.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bahler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 43.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 44.Lu F, Li G, Cui X, Liu C, Wang XJ, Cao X. Comparative analysis of JmjC domain-containing proteins reveals the potential histone demethylases in Arabidopsis and rice. J. Integr. Plant Biol. 2008;50:886–896. doi: 10.1111/j.1744-7909.2008.00692.x. [DOI] [PubMed] [Google Scholar]
- 45.Hong EH, Jeong YM, Ryu JY, Amasino RM, Noh B, Noh YS. Temporal and spatial expression patterns of nine Arabidopsis genes encoding Jumonji C-domain proteins. Mol. Cells. 2009;27:481–490. doi: 10.1007/s10059-009-0054-7. [DOI] [PubMed] [Google Scholar]
- 46.Vaughn MW, Tanurdzic M, Lippman Z, Jiang H, Carrasquillo R, Rabinowicz PD, Dedhia N, McCombie WR, Agier N, Bulski A, et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007;5:e174. doi: 10.1371/journal.pbio.0050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurihara Y, Matsui A, Kawashima M, Kaminuma E, Ishida J, Morosawa T, Mochizuki Y, Kobayashi N, Toyoda T, Shinozaki K, et al. Identification of the candidate genes regulated by RNA-directed DNA methylation in Arabidopsis. Biochem. Biophys. Res. Commun. 2008;376:553–557. doi: 10.1016/j.bbrc.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 48.Lu C, Kulkarni K, Souret FF, Valliappan R, Tej SS, Poethig RS, Henderson IR, Jacobsen SE, Wang W, Green PJ, et al. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res. 2006;16:1276–1288. doi: 10.1101/gr.5530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Carrington JC. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;5:e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004;2:E104. doi: 10.1371/journal.pbio.0020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia Y, Lisch DR, Ohtsu K, Scanlon MJ, Nettleton D, Schnable PS. Loss of RNA-dependent RNA polymerase 2 (RDR2) function causes widespread and unexpected changes in the expression of transposons, genes, and 24-nt small RNAs. PLoS Genet. 2009;5:e1000737. doi: 10.1371/journal.pgen.1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu C, Tej SS, Luo S, Haudenschild CD, Meyers BC, Green PJ. Elucidation of the small RNA component of the transcriptome. Science. 2005;309:1567–1569. doi: 10.1126/science.1114112. [DOI] [PubMed] [Google Scholar]
- 53.Ha M, Lu J, Tian L, Ramachandran V, Kasschau KD, Chapman EJ, Carrington JC, Chen X, Wang XJ, Chen ZJ. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl Acad. Sci. USA. 2009;106:17835–17840. doi: 10.1073/pnas.0907003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao T, Li G, Mi S, Li S, Hannon GJ, Wang XJ, Qi Y. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 2007;21:1190–1203. doi: 10.1101/gad.1543507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5' terminal nucleotide. Cell. 2008;133:116–127. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 57.Li F, Huarte M, Zaratiegui M, Vaughn MW, Shi Y, Martienssen R, Cande WZ. Lid2 is required for coordinating H3K4 and H3K9 methylation of heterochromatin and euchromatin. Cell. 2008;135:272–283. doi: 10.1016/j.cell.2008.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.