


Abstract
Human internal exons have an average size of 147 nt, and most are <300 nt. This small size is thought to facilitate exon definition. A small number of large internal exons have been identified and shown to be alternatively spliced. We identified 1115 internal exons >1000 nt in the human genome; these were found in 5% of all protein-coding genes, and most were expressed and translated. Surprisingly, 40% of these were expressed at levels similar to the flanking exons, suggesting they were constitutively spliced. While all of the large exons had strong splice sites, the constitutively spliced large exons had a higher ratio of splicing enhancers/silencers and were more conserved across mammals than the alternatively spliced large exons. We asked if large exons contain specific sequences that promote splicing and identified 38 sequences enriched in the large exons relative to small exons. The consensus sequence is C-rich with a central invariant CA dinucleotide. Mutation of these sequences in a candidate large exon indicated that these are important for recognition of large exons by the splicing machinery. We propose that these sequences are large exon splicing enhancers (LESEs).
INTRODUCTION
A major point of regulation during eukaryotic gene expression is the splicing of exons to yield a contiguous transcript that can be translated to a functional protein. The splicing machinery has to efficiently recognize small exons flanked by much larger introns many thousands of times during each cell cycle. In different tissues different exons are included, resulting in alternative splicing that yields a greater diversity of proteins (1). In humans, 90% of genes are alternatively spliced (2,3). Splicing regulates both the levels of gene expression and tissue-specific expression (4), with splicing deregulation often leading to disease (5).
Early studies of gene architecture revealed that most internal exons range from 50 to 200 nt, while terminal exons can be much longer (6,7). In humans, the mean size of internal exons is 147 nt, and nearly all appear to be <300 nt (8). The conserved size of internal exons in many species suggests it may be an important factor in splicing. To test this hypothesis, exons were artificially expanded. Expansion of exon size >300 nt diminishes splicing in most but not all cases (9–11).
A necessary step in correct pre-mRNA splicing is the identification of exons and introns by the recognition of splice sites. These recognition events are driven by splice site sequence complementarity to corresponding snRNAs that are part of splicing snRNPs. Although in lower eukaryotes this process is facilitated by the recognition of splice sites across a small intron, it is hypothesized that in higher eukaryotes splicing is facilitated by the recognition of splice sites across an exon (12,10).
Berget and coworkers proposed the exon definition model, which invokes the necessary communication of 3′- and 5′-splice site complexes across an exon for efficient splicing (10,13). This model is based, in part, on observations that a downstream 5′-splice site enhances splicing at the upstream 3′-splice site of the same exon (10,14,15). This model was supported by mutations in either splice site that caused the skipping of the internal exon (16,17). The requirement for communication across an exon and the apparent dearth of large exons in the genome led to the model that splice site recognition is more efficient when exons are small, and exon inclusion decreases with increasing exon size (13). To date, examples of internal exons >500 nt have all been found to be alternatively spliced (18–21).
The communication of snRNP complexes across an exon is influenced by multiple sequences within both exons and introns that either promote or prevent the inclusion of a given exon (22–26). Exonic splicing enhancers (ESE) bind serine/arginine rich-proteins (SR proteins) to recruit the splicing machinery to nearby splice sites, thereby leading to inclusion of the exon (27). Similarly, other enhancer sequences present in adjacent introns called intronic splicing enhancers (ISE) and suppressors in exons and introns called ESS and ISS influence the splicing of a given exon by binding proteins that interact with complexes necessary for proper splicing (28). These sequences, along with splice site strength, local RNA structure, nucleosome density and rate of pre-mRNA synthesis, influence exon recognition (12,29–33).
We found a total of 1115 internal exons >1000 nt in the human genome, and these were present in 1040 different genes (Refseq database). To investigate whether these large exons were frequently skipped, we analyzed 120 RNA-seq datasets deposited in the public sequence read archive (SRA). Surprisingly, we found that 42% of the large exons were expressed at levels similar to their corresponding upstream and downstream exons, indicating large exon inclusion. However, in general large exons were more frequently alternatively spliced than a random set of internal exons <250 nt. Further analysis indicated many of these large exons had strong splice sites and many ESEs. Most of these large exons are evolutionarily conserved in placental mammals, and constitutive large exons are more conserved than alternative large exons. Furthermore, we identified 38 sequences enriched in large exons that promote their recognition and splicing and propose that they are large exon splicing enhancers (LESEs).
MATERIALS AND METHODS
Genomic data
All the annotated genes from the human genome (hg19, February 2009) were extracted from the knownGene table in the UCSC genome browser. Exon and intron lengths were calculated using the exon start and exon end coordinates for each gene. Any redundant entries, pseudogenes and single exon genes were removed from the dataset. This yielded a total of 22 539 genes with more than one exon. These 22 539 genes were used in the following analysis. The entire sequence of each of these large exon-containing genes was extracted from the human genome. These sequences were used to build a Bowtie index, and this index was used to align all the RNA-seq datasets and ribosome footprint datasets using Bowtie (34). All Bowtie alignments were carried out with the following parameters to decrease false alignments, n = 0, best and l = 30.
RNA-seq analysis
In order to analyze large exon expression, 120 datasets were downloaded from the SRA database at NCBI. All of the datasets had been sequenced on one of the Illumina sequencing platforms. Each RNA-seq dataset was aligned to the large exon Bowtie index (34). The Bowtie output was then analyzed using custom scripts to calculate reads per exon of every exon in a large exon-containing gene. The reads per exon were then converted to reads per kilobase per million reads (RPKM) values by dividing the number of reads by the size of the exon in kb.
To investigate the distribution of RNA-seq reads across a large exon, the alignment position output of Bowtie was used to calculate the number of times each nucleotide is represented by an RNA-seq read. This value was then plotted against position in the large exon.
In order to analyze the presence of wild-type and alternate splice junctions, all possible splice junctions sequences were generated using the annotated splice sites. Fifty nucleotides from the end of an upstream exon were fused to 50 nt from the beginning of the downstream exon. These sequences were used to create a splice junction Bowtie index, which was then used to align all RNA-seq datasets.
Ribosome footprint analysis
In order to investigate whether a given large exon was being translated, we aligned ribosome footprints from HeLa cells (35) to the large exon Bowtie index. Similar to the RNA-seq analysis, the alignment position output of Bowtie was used to calculate the number of times each nucleotide was translated.
Average exon inclusion index calculations
To classify whether a large exon was under expressed relative to upstream or downstream exons, we calculated fold change of large exon expression relative to an upstream or downstream exon, whichever was greater. This fold change was then binned to five different exon inclusion indexes as follows, 0: gene not expressed, 1: 0 < large exon < 0.25, 2: 0.25 < large exon < 0.50, 3: 0.50 < large exon < 0.75, 4: 0.75 < large exon < 1.25, 5: large exon >1.25. The average exon inclusion index was computed for all the datasets in which a particular large exon-containing gene was expressed.
Splice site calculations
Splice sites for all exons were extracted using custom scripts, and splice strengths were calculated using the maximum entropy model of MaxEntScan (36). The maximum entropy score for both 5′- and 3′-splice sites were used for further analysis.
In order to investigate the presence of internal splice sites in a large exon, all 9-mers starting at every nucleotide were generated for 5′-splice site analysis; 23-mers starting at all nucleotides were generated for 3′-splice site analysis. The strength of all of these sequences was calculated using MaxEntScan (36).
ESE and ESS calculations
RESCUE–ESE sequences were obtained from Fairbrother et al. (24) and ESS sequences were obtained from Wang et al. (37). The number of ESE and ESS sequences were calculated and then scaled by the size of the exon in kb.
Conservation analysis
Conservation scores generated by phastCons (38) for primates and placental mammals were downloaded from the UCSC genome browser. Conservation scores for each nucleotide for the entire large exon gene were extracted for further analysis. We then extracted the conservation score for 500 nt in the upstream intron, the first 500 nt in the large exon, the last 500 nt in the large exon, and 500 nt in the downstream intron for every constitutive and alternative large exon dataset. These data were then averaged for the constitutive and alternative large exons and plotted against nucleotide position.
Identification of large exon-specific hexamers
In order to identify large exon-enriched sequence motifs, we identified hexamers that were enriched in large exons compared with small exons. As in previous studies determining ESEs and ESSs (24), we focused on counting the number of times any given hexamer appeared in a dataset. We calculated the frequency of occurrence for all 4096 hexamers in all large exons, all internal small exons (<300 nt) and all introns of multi-exonic genes. We then computed for each hexamer the difference between the frequency of a hexamer in large exons (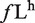 ) and small exons (
) and small exons (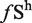 ). The mean and standard deviations for this distribution (ΔLS =
). The mean and standard deviations for this distribution (ΔLS = 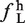 −
− 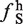 ) was computed. Large-exon enriched hexamers were determined using false discovery rate statistics (39). All hexamers were ranked based on Z-scores (number of standard deviations from mean). The probability that a given hexamer could be enriched by chance was calculated using inverse cumulative normal distribution. All hexamers that had a false discovery rate of <5% were deemed large exon-enriched hexamers.
) was computed. Large-exon enriched hexamers were determined using false discovery rate statistics (39). All hexamers were ranked based on Z-scores (number of standard deviations from mean). The probability that a given hexamer could be enriched by chance was calculated using inverse cumulative normal distribution. All hexamers that had a false discovery rate of <5% were deemed large exon-enriched hexamers.
Hierarchical clustering to determine similar hexamers
In order to identify specific motifs in the large exon-enriched sequences, a dissimilarity index was calculated for all pairs of hexamers, similar to that of Fairbrother et al. (24). The sequences were then clustered using standard average hierarchical clustering as implemented in Python module SciPy v0.11.dev. Sequences for each cluster were aligned using ClustalW (40), and motif logos were rendered using enoLOGOS (41).
Analysis of function of large exon-specific hexamers
Exons 6–8 of the JARID2 gene along with 500 nt of flanking internal introns were cloned into the pcDNA3.1(+) vector. For all 18 hexamer sequences in the JARID2 large exon 7, the internal 4 nt were mutated to complementary nucleotides, and the mutated exon was synthesized as three gBlock fragments (IDT). The mutant large exon was cloned to replace the wild-type large exon in the mini-gene construct. All the original splice sites were maintained and each construct was verified by sequencing. Four micrograms of the constructs was transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen). Total RNA was isolated 36 h post-transfection using RNA-Bee (Tel-Test), and 1 μg of total RNA was reverse transcribed using MMLV-RT (Promega). A vector-specific primer was used in this reverse transcription reaction to avoid amplification from endogenous JARID2 sequences. Splicing was analyzed by polymerase chain reaction (PCR), using primers to detect splice junctions between exons 6 and 7, 7 and 8 and 6 and 8 (skipping of exon 7). Ten percent of the reverse transcription reaction was used in each PCR reaction, and reactions were stopped after 21 cycles, previously shown to be in the linear range by real-time PCR. The PCR reactions were run on 1.5% agarose gels, and bands quantitated using GeneTools (Syngene, Inc).
RESULTS
Large exons are present in almost 5% of the genes in the human genome
Previous analyses of internal exon size in the human genome reported a mean size of 147 nt and most exons were <300 nt (8). This is consistent with the exon definition model of splicing that proposed an upper limit of 300–500 nt for efficient splicing (9–11). In this analysis we searched the annotated human genome (hg19) for internal exons >1000 nt. We found 1153 annotated internal exons >1000 nt in length with a median size of 1503 nt (Figure 1A). Although we did not analyze intermediate-sized exons, we observed 2226 exons between 500 and 1000 nt in length. The remaining 98.5% of internal exons were <500 nt long. Although these 1153 large (>1000 nt) internal exons are a minor fraction (0.5%) of all internal exons, they are present in 1040 different genes making up 4.7% of all human protein-coding genes.
Figure 1.

There are 1153 internal exons >1000 nt in the human genome and 39% of them are next to terminal exons (A) All internal exons <5000 nt were binned into 50-nt bins and plotted as a histogram. The inset histogram is a distribution of all internal exons >1000 nt binned into 200 nt bins. (B) The position of the large exon within a gene is plotted.
There are a small number of human genes with very large internal exons (>10 000 nt); all but one of these genes are in the mucin family of proteins that play a role in protecting the epithelium from the harsh external environment (42). The largest internal exon is exon 3 of human mucin 16 (MUC16), which is >21 000 nt. Other genes in the mucin family with internal exons >10 000 nt include MUC4 (12 708 nt), MUC12 (14 889 nt), MUC17 (12 219 nt) and MUC5B (10 893 nt). The gene with the largest number of internal large exons is titin (TTN) which has five large exons; its largest exon is >17 000 nt in length.
Large exons may have evolved from terminal exons
Although the majority of internal exons are small, the terminal exons of genes do not have any size restrictions (6,7). In fact the biggest terminal exon in the human genome seen in our analysis is 22 000 nt, and the average size of all terminal exons in the human genome is ∼1000 nt. Since terminal exons are so large, we investigated whether large internal exons could have once been terminal exons during an earlier time in evolution, although it is also possible that larger exons in internal positions other than second/penultimate position are more detrimental to organism fitness. If so, we hypothesized that these large exons would be enriched as either the second exon or the penultimate exon in the gene. Consistent with this, we found that 447 (38.7%) of the large exons are either the second exon or the penultimate exon, suggesting these genes may have picked up an additional exon during the course of evolution (Figure 1B). Since there are an average of 13 exons in a human gene, a random distribution of internal large exon positions would result in 15% at exon 2 or the penultimate exon. Of these 447 large exons, 288 are the second exon and the remaining 219 are the penultimate exon of their corresponding genes. The remaining number of large exons are spread across various exon positions.
Interestingly, only 12.5% (36/288) of genes with large exons in the second position had a start codon within the first exons (data not shown), which suggested that many of these new terminal exons are non-coding regulatory elements. Of the remaining genes with large exons in the second position, 52% of the start codons were in the large exon, while another 27% of the start codons were in the third exon (data not shown).
Gene ontology analysis
We asked whether the genes with large exons expressed proteins with particular functions. We found a strong enrichment of cytoskeleton and microtubule-associated proteins (21%), in comparison to 10% of the genes without large exons. Another large subset encoded nuclear proteins (27.8%) in comparison to 21% of genes without large exons. In addition, 35% of the genes with large exons had unknown functions, compared with 25% of genes without large exons. Thus, the genes with large exons represented a subset of all cellular functions.
Expression of large exons
We next wanted to look at the expression of large exons. A Bowtie index (34) was constructed using large exon gene sequences from RNA-seq data obtained from 120 datasets downloaded from the SRA database at NCBI. Analysis of the 1153 large exons showed that 1040 (out of 1078 expressed exons) were fairly evenly transcribed across the large exon, like the GT3C4 exon shown in Figure 2A (RNA-seq reads shown in green). This exon was also translated in HeLa cells, as shown by the ribosome footprints in Figure 2A (bottom, red). We also looked for the presence of internal splice sites and found several, but these were weaker than the splice sites flanking this exon (Figure 2A).
Figure 2.

Analysis of RNA-seq reads and ribosome footprints shows the majority of large exons are transcribed and translated. (A) GT3C4 exon 2 is representative of a true large exon. (B) EFHC1 exon 4 is representative of a large exon that occasionally retains the internal intron. (C) FAM14C exon 2 is representative of a large exon that is likely misannotated in the human genome. (A,B,C) Top panel in green is a plot of the cumulative RNA-seq reads across the large exon from all datasets. The second panel and third panel are plots of the strength of all 5′- and 3′-splice sites in the large exon. The WT splice sites are red in color, while any internal pseudo splice site stronger than the WT splice site is represented in orange. The bottom panel is a plot of ribosome footprints from HeLa cells, where 70% of large exons were translated as in panel A.
In contrast, three of the annotated large exons resembled the EFHC1 exon shown in Figure 2B. The RNA-seq reads were much more abundant at both ends of the exon (∼200 nt regions) than in the center; however, the entire exon was expressed in a fraction of cell types. There were strong splice sites flanking these higher RNA-seq reads, suggesting that they are two independent exons in some cell types. We think this is an example of an intron that is retained in some cells but not in most. These were removed from our dataset.
Lastly, we observed 38 annotated large exons that appeared to have transcripts and ribosome footprints representing only a portion of the exon. An example of this is shown in Figure 2C. These exons usually had a splice site flanking the RNA-seq reads, leading us to believe that these are misannotated large exons that are really small exons; they were removed from our dataset before further analysis.
Large exons are ubiquitously expressed at levels similar to upstream and downstream exons
In order to further assess the expression of large exons and large exon-containing genes, the sequences in 120 RNA-seq datasets were aligned to our Bowtie index of large exon-containing genes. The absolute number of reads aligning to each exon was extracted, and RPKM for each exon was calculated with respect to the total number of reads mapping to the entire gene. The ratio of the RPKM for the large exon relative to the RPKM of the upstream or downstream exon (whichever was larger) was calculated and binned in our large exon inclusion index. (0: gene not expressed, 1: 0 < large exon < 0.25, 2: 0.25 < large exon < 0.50, 3: 0.50 < large exon < 0.75, 4: 0.75 < large exon < 1.25, 5: large exon > 1.25). The average exon inclusion index for each large exon was calculated over the 120 datasets. The internal large exons were only analyzed when both upstream and downstream exons were expressed.
Surprisingly, 42% of the internal large exons had an average exon inclusion index >4 (Figure 3). This means these exons were retained at around the same levels as the flanking exons, so they are considered constitutive exons. Another 37.5% of large exons had an average exon inclusion index between 3 and 4, which means they were present at levels between 50% and 75% relative to the flanking exons. About 15% of the large exons were expressed <50% of the time (bins 1 and 2). Around 5% of large exon-containing genes were not expressed in all 120 datasets at levels required for this analysis (Figure 3). Based on the exon definition hypothesis and previous data, the majority of these large internal exons would be expected to be excluded from transcripts. However, only 166 of the 1040 large exons have an average bin <3, meaning they were excluded more often than included in the mature transcript.
Figure 3.

Forty-five percent of large exons constitutively spliced large exons. The Average exon inclusion index for all 1115 large exons was calculated as described in methods, and the percentage of exons in each index was calculated. Bins 4 and 5 are considered constitutively spliced (75–125%), and bins 1–3 are alternatively spliced (0–75%).
In contrast, an analysis of 2200 random internal exons <250 nt showed a different distribution. Seventy percent of these small internal exons had an average exon inclusion index >4; that is they are constitutively spliced with respect to upstream and downstream exons. In addition, 25% of these had an average inclusion index between 3 and 4 and therefore were present 50–75% of the time in the mature transcript (Figure 3). This indicates that large exons are skipped more often than small internal exons.
The majority of large exons were expressed over a diverse range in different datasets and cell types. Although most of the genes had a maximum exon inclusion index of 5.0, many of them also had a minimum exon inclusion index of 1.0 (Supplementary Figure S1). This means that many of these large exons were expressed at the same levels as flanking exons in some datasets, while they were highly underrepresented in other datasets, indicative of exon skipping.
In order to make sure that these large exons were actually part of contiguous transcripts, we analyzed the presence of splice junctions with upstream and downstream exons. We constructed in silico all possible splice junctions between exons based on annotated splice sites. This was used to make a Bowtie index for alignments. For constitutive large exons, the wild-type splice junction with the upstream and downstream exon was the predominant junction observed (data not shown). Similarly, when the large exon was alternatively spliced, and the exon inclusion index was low, there were more alternative splice junctions fusing the upstream exon with other downstream exons or there was a complete absence of the wild-type splice junction (data not shown). There was very little correlation (Spearman rho = 0.003) between the size of a large exon and the degree of inclusion of the exon in the transcript (Supplementary Figure S2). This analysis indicates that the splicing machinery is capable of splicing large exons, and exclusion or inclusion of large exons is probably determined by other cis- and trans-acting factors. We did not find any correlation between large exon inclusion and the total length of the mRNA (data not shown).
Constitutive large exons are more conserved than alternative large exons
To determine whether large human exons or the flanking introns were conserved in mammals and if constitutive large exons (average exon inclusion index > 4) were more conserved than alternative large exons (average exon inclusion index < 4), we downloaded the UCSC genome browser phastCons (38) conservation scores for primates and all mammals and extracted the conservation score for every nucleotide in large exon-containing genes. We then extracted the conservation score for 500 nt in the upstream intron, the first 500 nt in the large exon, the last 500 nt in the large exon and 500 nt in the downstream intron for every large exon in our dataset. These data were then averaged for the constitutive and alternative large exons and plotted against nucleotide position.
As illustrated in Figure 4, the conservation score across the large exons (average phastCons score = 0.6) was much higher than that in the flanking introns (average phastCons score = 0.2), supporting the idea that these sequences are indeed exons. Also, constitutive large exons were more conserved than alternative large exons. The largest difference in conservation between these datasets was observed at the 3′-end (5′-splice site) of the large exon, although the constitutive exons were more conserved throughout.
Figure 4.

Constitutive large exons are more conserved than alternative large exons. The phastCons score for eight placental mammals was averaged over constitutive or alternative large exons and plotted against position in the flanking intron or within the first 500 nt or last 500 nt of the large exon. The introns were much less conserved than the exons and were similar for the constitutive and alternative large exons.
Large exons flanked by small upstream and downstream introns are efficiently spliced
One factor that could explain the inclusion of large exons is the size of the flanking intron. Previous work indicated that decreasing the size of an upstream intron to around 250 nt led to inclusion of a large exon that would otherwise be excluded (10). Of our 1040 large exons, 66 exons were flanked by upstream and downstream introns <500 nt. Another 220 large exons had either an upstream or downstream intron <500 nt. This corresponds to around 27.5% (286 of 1040 exons) of large exons having at least one flanking intron <500 nt. Interestingly, 27.6% of all internal exons were also flanked by at least one intron <500 nt. These data suggest that there was no enrichment for small introns in the large exon dataset, indicating that intron size probably does not play a big role in inclusion of large exons.
We next investigated whether both upstream and downstream small introns could influence large exon inclusion. A total of 31 large exons had both flanking introns <250 nt, and 28 of these large exons had an average large exon inclusion index of >4.5, demonstrating they are constitutively spliced (Supplementary Figure S3). In contrast to previous data where a small upstream intron was sufficient to promote inclusion of an artificial large exon (10), our analysis indicated that both flanking introns needed to be <250 nt to promote inclusion. We did not see an effect on inclusion if only one of the flanking introns was <250 nt. Furthermore, the remaining large exons with flanking introns <1000 nt did not seem to show enrichment for any particular exon inclusion index.
Many large exons have strong 5′- and 3′-splice sites and are similar to small exons
Since the ends of the constitutive large exons were more conserved than alternative large exons, we asked if constitutive large exons have stronger splice sites. Splice sites are unique sequences that bind splicesomal snRNPs by sequence complementarity during splicing. Previous data have shown that a weak 5′- or 3′-splice site leads to exclusion of the exon from the transcript, when all other factors are controlled. To determine whether large exon inclusion is determined by splice site strength, we extracted all 5′- and 3′-splice sites of large exons and calculated strength using the maximum entropy model of MaxEntScan (36).
The distribution of splice site strength of large exons was similar to that of all internal exons, indicating that there was no selection to make large exon splice sites stronger to compensate for size (Supplementary Figure S4). In Figure 5, the average exon inclusion index is depicted in terms of a heat map with darker colors indicative of a higher index and lighter colors indicative of a lower index. As illustrated in the plot, some large exons with weak splice sites (maximum entropy score of <2.5) have a high average exon inclusion index, and conversely, some exons with very good splice sites have a low exon inclusion index (Figure 5).This indicates that strong splice sites are not the sole determinant of exon inclusion.
Figure 5.

Most large exons have very good 5′- and 3′-splice sites. Large exon 5′- and 3′-splice sites were extracted for each exon, and splice site strength was calculated as described in methods. The 3′-splice site strength was plotted against 5′-splice site strength, and the average exon inclusion index is represented by a color gradient with darker colors indicating a higher average exon inclusion index and lighter colors indicate a lower average exon inclusion index.
The ratio of enhancers to silencers is higher in included large exons
Since constitutive large exons were slightly more conserved than alternative large exons, we asked if there were specific sequences in the exons that might influence their inclusion. ESEs are short sequences that have been shown to increase splicing efficiency and exon retention. It is hypothesized that ESEs are bound by splicing enhancer proteins that help bridge the two splicing complexes at either end of the exon. Previous analysis has identified 238 6-nt RESCUE-ESEs (24) that were enriched in constitutively spliced exons with weak splice sites and absent in introns. We investigated whether there is an increased number of ESEs in large exons to promote their inclusion.
The absolute number of ESEs in all internal exons and all large exons was calculated and then normalized for size (Figure 6A and C). All the large exons had at least 1 ESE/kb; however, 10% of small exons did not have any ESEs. The distribution of ESEs in large exons (Figure 6C) was not significantly different than the distribution of ESEs in all internal exons (Figure 6A), indicating that there was no enrichment of ESEs in large exons to compensate for size. Furthermore, we saw no enrichment of ESEs in large exons with weak splice sites that were spliced efficiently (data not shown). We also did not find any enrichment of ISEs in the introns within 200 nt of either splice site of these large exons (data not shown).
Figure 6.

Ratio of ESE to silencers may contribute to large exon inclusion. ESEs (A) and ESSs (B) in all internal exons and ESEs (C) and ESSs (D) in large exons were calculated and scaled for size and binned. (E) The ratio of ESEs/ESSs was plotted against the average large exon inclusion index.
We wanted to investigate why some large exons with strong 5′- and 3′-splice sites had a very low average exon inclusion index. Apart from having positive sequence elements, exons also contain negative sequence elements called ESSs. These are sequences that are bound by proteins including hnRNPs that inhibit the splicing of the exon (43). First, we investigated whether large exons had a different distribution of ESSs compared with all internal exons. The distribution of ESSs in large exons is not very different than ESSs in all internal exons except at the lower end of the distribution (Figure 6B and D). Surprisingly, there are fewer large exons with very low numbers of ESSs. Next, we calculated the standard deviation from the mean of all large exons with good splice sites to see if those with low splicing were enriched for ESSs. However, there was no enrichment of ESSs in these large exons compared with all other large exons (data not shown).
The inclusion of internal exons is probably influenced by the ratio of ESEs to ESSs. We wanted to see if the ratio of ESEs to ESSs could determine the inclusion of a large exon. Similar to internal small exons, large exons have more ESEs than ESSs. On average there are four times more ESEs than ESSs in large exons (avg. ESEs: 108/kb in large exon, avg. ESSs: 28/kb in large exon) and only 45 of the large exons had more ESSs than ESEs. Interestingly this difference is almost identical to what is observed in small exons. Small exons on average have 110 ESEs/kb and 28 ESS/kb once again indicating that these large exons are indeed exons.
We next investigated whether the ratio of ESEs to ESSs may explain the inclusion of large exons. As illustrated in Figure 6E, some constitutive large exons have a much higher ratio (>5) of ESEs to ESSs in comparison to alternative large exons. Constitutive large exons (inclusion index > 4) have an average of 4.25 times more ESEs than ESSs. Alternative large exons that have an inclusion index of >3 have 3.7 times more ESEs than ESSs and the rest of the alternative large exons have 3.2 times more ESEs than ESSs. This suggests that the ratio of ESEs to ESSs may be an important determining factor in large exon inclusion (Spearman rho = 0.15).
Large exon sequence analysis identifies 38 sequences enriched in large exons
Although there is a modest correlation between the ratio of ESEs/ESSs and the splicing of large exons, we wondered whether there were specific sequences or motifs that identified large exons and promoted their splicing. Since previous analysis to identify splicing enhancers and silencers used hexamers as their basic unit, we decided to use the same approach to try to identify hexamers that were enriched in the large exon dataset. We calculated the frequency of occurrence of all 4096 possible hexamers in the large exon dataset (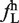 ) and, in all internal small exons of <300 nt (
) and, in all internal small exons of <300 nt (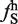 ) (Figure 7A). We then calculated the difference between the large exon frequency and small exon frequency, ΔLS. Using a false discovery rate of 5%, we identified 41 (1.0%) hexamers that were enriched in the large exon dataset (Supplementary Table S1). This represented all hexamers that were >3.2 SD from the mean of the ΔLS distribution and are plotted in red (Figure 7A). Three of these hexamers were previously identified as ESEs (24). We then asked whether any of the remaining 38 large-exon enriched hexamers were representative of intron enriched hexamers. None of the 38 large-exon-enriched hexamers were enriched in introns (Supplementary Figure S5), which means the probability of identifying any of these 38 large exon-enriched hexamers in introns is no greater than random. We also identified 12 sequences that were underrepresented in large exons in comparison to small exons (data not shown).
) (Figure 7A). We then calculated the difference between the large exon frequency and small exon frequency, ΔLS. Using a false discovery rate of 5%, we identified 41 (1.0%) hexamers that were enriched in the large exon dataset (Supplementary Table S1). This represented all hexamers that were >3.2 SD from the mean of the ΔLS distribution and are plotted in red (Figure 7A). Three of these hexamers were previously identified as ESEs (24). We then asked whether any of the remaining 38 large-exon enriched hexamers were representative of intron enriched hexamers. None of the 38 large-exon-enriched hexamers were enriched in introns (Supplementary Figure S5), which means the probability of identifying any of these 38 large exon-enriched hexamers in introns is no greater than random. We also identified 12 sequences that were underrepresented in large exons in comparison to small exons (data not shown).
Figure 7.

Thirty-eight hexamers are enriched in large exons relative to small exons and introns. (A) The values of frequency of hexamer in large exon (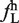 ) and frequency of hexamer in small exon (
) and frequency of hexamer in small exon (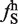 ) were plotted against each other. The black line represents +3.2 SD from the mean of ΔLS (
) were plotted against each other. The black line represents +3.2 SD from the mean of ΔLS (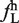 −
− 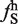 ) distribution. The dots in red represent large exon-enriched hexamers. (B) For each large exon the number of large exon-enriched hexamers were counted, normalized for size and plotted against average exon inclusion index. (C) The 38 sequences were clustered based on a dissimilarity matrix in the dendrogram. The sequences were aligned and motifs calculated based on the frequency of each nucleotide in the alignment.
) distribution. The dots in red represent large exon-enriched hexamers. (B) For each large exon the number of large exon-enriched hexamers were counted, normalized for size and plotted against average exon inclusion index. (C) The 38 sequences were clustered based on a dissimilarity matrix in the dendrogram. The sequences were aligned and motifs calculated based on the frequency of each nucleotide in the alignment.
These 38 enriched sequences were present in all large exons, although constitutive large exons had, on average, a greater number of them (Spearman rho = 0.3, Figure 7B). In order to identify specific motifs that were present in these 38 hexamers, we used standard average hierarchical clustering based on a dissimilarity index to cluster these sequences (Figure 7C). This analysis showed that all 38 hexamers were related to each other with a dissimilarity index of <3. All 38 hexamers were then aligned by ClustalW to calculate a frequency matrix, which was then used to calculate a motif (Figure 7C). The motif is C-rich and there is a central CA dinucleotide that is nearly invariant in all the hexamers. We propose that these sequences may play a role in enhancing large exon splicing.
We then analyzed the presence of these 38 large exon-enriched sequences in the constitutive dataset (average inclusion index > 4) and the alternative dataset (average inclusion index < 3) to try to identify constitutive splicing-enriched hexamers. Similar to previous analysis, the frequency of the 38 hexamers was calculated for both the constitutive large exon dataset and the alternative large exon dataset (data not shown). Constitutive large exon-enriched hexamers were determined based on false discovery rate statistics. This analysis indicated that there were no significantly enriched hexamers in the constitutive large exon dataset compared with the alternative dataset. This may indicate that large exon-enriched hexamers only identify and enhance the splicing of all large exons and do not play a role in alternative splicing of the large exon. The constitutive and alternative nature of a large exon may be determined by previously analyzed features.
Large exon-specific hexamers may define and identify large exons in vivo
To investigate whether this large exon-specific motif plays a role in large exon definition, we mutated the large exon-enriched hexamers in a candidate exon, the 1039-nt exon 7 of the JARID2 gene. This exon is a constitutively spliced in 293 cells. We constructed a mini-gene containing JARID2 exons 6–8 and truncated (to 1 kb) flanking introns (Figure 8A). The JARID2 large exon had a total of 18 different large exon-enriched hexamers. We synthesized a mutant large exon in which the internal 4 nt in each hexamer were mutated. This construct was cloned into the mini-gene to create a JARID2-Mutant mini-gene. Both wild-type and mutant constructs were transfected into HEK293T cells. Total RNA was extracted 36 h later, and each exon–exon junction was assayed using PCR. We assayed the WT (6–7, 7–8) exon–exon junctions as well as the alternative (6–8) exon–exon junction (Figure 8B).
Figure 8.

LESEs identify and promote JARID2 large exon splicing. (A) The gene structure of the JARID2 large exon and flanking exons and introns. Exons 6–8, along with 500 nt of the flanking introns, were cloned and expressed in 293T cells. A representative image of the PCR of splice junctions in the WT and Mutant constructs. (B) Quantitation of splice junctions indicates that large exon hexamers promote large exon splicing.
The JARID2-WT construct spliced as expected with 98% of expressed mRNA demonstrating wild-type splice junctions and 2% showing skipping of exon 7 (6–8 splice junction). This is consistent with this large exon being constitutive in 293 cells and demonstrated that the JARID2-WT mini-gene splices like the endogenous JARID2 pre-mRNA. The ratio between the two wild-type junctions (6–7/7–8) was 0.95 (Figure 8B), which also indicated that all the splice sites were recognized appropriately and spliced with almost equal efficiency.
Next, we asked whether the JARID2-Mutant construct would be spliced properly. The ratio between the two wild-type splice junctions in the JARID-Mutant construct was, 24 to 1, which was very different from that in the JARID2-WT (Figure 8B). While we observed efficient splicing of exon 6 to exon 7, the removal of the downstream intron was reduced (Figure 8B). In addition, we observed a small increase in the relative amount of skipping of exon 7 in the mutant construct. This suggested that the large mutant exon is not recognized as an exon, preventing its 5′-ss from being recognized and spliced properly. The large exon and the retained downstream intron appear to be seen as part of the 3′ terminal exon of this minigene construct (data not shown). This supports the idea that these hexamer sequences promote the identification of large exons and function as LESEs.
DISCUSSION
The majority of human exons are small, and it is thought that the splicing complexes at either end of the exon communicate with each other to define the exon and accurately splice (10). This is consistent with the exon definition model, which predicts that any large exon present in the human genome would be skipped (or alternatively spliced) during the splicing of the pre-mRNA transcript (13). However, we were surprised to find that 5% of all human mRNAs have at least one large internal exon >1000 nt, and 42% of these were constitutively spliced. Our data indicate that the splicing machinery is fairly elastic and is capable of splicing large internal exons efficiently (11). In the cases where the large exon is excluded, this may be a mechanism to control the level of wild-type gene expression.
We observed a high degree of conservation of the large exons across 31 different mammalian species and a much lower degree of conservation of the adjacent introns (Figure 4). Interestingly, the constitutive exons were more conserved than the alternative exons especially at both ends. Because of the increased conservation at both ends of the large exon, we hypothesized that there maybe an interaction between the ends to bring the splice sites closer together. Using UNAfold (44) we analyzed the number of predicted interactions between the first 200 nt and last 200 nt of large exons in the lowest energy structure of the entire large exon (data not shown). We did not find any difference in total folding energy (data not shown) or any increase in interactions between the ends when compared with a sequence that was randomized with the same dinucleotide frequency (data not shown). Although we did not observe any structural elements in these conserved regions, it will be interesting to study this region further to determine whether regulatory proteins might bind in these areas. We also did not observe a difference between the nucleosome occupancy of small exons and large exons (Supplementary Figure S6), which is consistent with a previous analysis that looked at nucleosome density of exons > 500 nt (45). The average number of nucleosome footprints per size was not higher in large exons and the distribution of the nucleosomes with respect to splice sites was also similar.
We asked whether large exon inclusion can be explained by determinants involved in regulating the splicing of smaller exons. Although none of the variables alone seemed to correlate with the distribution of large exon expression, each variable probably contributes to the inclusion of the large exon. For example, many of the large exons with short flanking introns (<250 nt) had a high average exon inclusion index (Supplementary Figure S2). Similarly, many large exons with strong splice sites also had a high average exon inclusion index (Figure 5).
Another variable that directly influences the inclusion and exclusion of any internal exons are the levels of SR and hnRNP proteins in each of the cell types (27). Since constitutive large exons seem to have a higher ratio of ESEs to ESSs (Figure 6E), the competition between these two protein families could determine the inclusion of a large exon. It would be interesting to identify the presence or absence of SR or hnRNP proteins when a particular large exon is included or excluded. A RIP-seq analysis of these proteins could explain more about the inclusion of many of these large exons.
Furthermore, we identified 38 LESEs and one motif that are enriched in large exons and may potentially regulate their inclusion or exclusion in an mRNA (Figures 7 and 8). We have made mini-gene constructs (Figure 8A), which will allow us to test which proteins are involved in large exon splicing. We analyzed these RNA sequences for potential protein-binding sites, using the RNA-binding protein data base (46). This analysis identified RBMX/hRNPG (CCCG,CCAC,CCAG) as a binding protein in 36 of the sequences and SFRS1 (AGGA) and SFRS9 (AGGAG) as a binding protein in two of the hexamers. Interestingly, all three of these proteins have been identified as splicing regulators, influencing many genes as in the case of SFRS1 (47–49). It is possible that these proteins, amongst others, could bind and identify large exons and promote their inclusion.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–6.
FUNDING
Funding for open access charge: National Institutes of Health [R01CA103891 to K.L.B.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Nicholas Ingolia for review of this article and valuable suggestions regarding statistical calculations for hexamers. The authors also thank Dr David Zappulla, Dr Johanna Withers and Dr Thomas O’Hare for suggestions and review of this article.
REFERENCES
- 1.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 3.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matlin AJ, Clark F, Smith CWJ. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell. Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 5.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naora H, Deacon NJ. Relationship between the total size of exons and introns in protein-coding genes of higher eukaryotes. PNAS. 1982;79:6196–6200. doi: 10.1073/pnas.79.20.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawkin JD. A survey on intron and exon lengths. Nucleic Acids Res. 1988;16:9893–9908. doi: 10.1093/nar/16.21.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 9.Zillmann M, Rose SD, Berget SM. U1 small nuclear ribonucleoproteins are required early during spliceosome assembly. Mol. Cell. Biol. 1987;7:2877–2883. doi: 10.1128/mcb.7.8.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robberson BL, Cote GJ, Berget SM. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 1990;10:84–94. doi: 10.1128/mcb.10.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen IT, Chasin LA. Large exon size does not limit splicing in vivo. Mol. Cell. Biol. 1994;14:2140–2146. doi: 10.1128/mcb.14.3.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hertel KJ. Combinatorial control of exon recognition. J. Biol.Chem. 2008;283:1211–1215. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 13.Berget SM. Exon recognition in vertebrate splicing. J. Biol.Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- 14.Nasim FH, Spears PA, Hoffmann HM, Kuo HC, Grabowski PJ. A sequential splicing mechanism promotes selection of an optimal exon by repositioning a downstream 5′ splice site in preprotachykinin pre-mRNA. Genes Dev. 1990;4:1172–1184. doi: 10.1101/gad.4.7.1172. [DOI] [PubMed] [Google Scholar]
- 15.Kreivi JP, Zefrivitz K, Akusjarvi G. A U1 snRNA binding site improves the efficiency of in vitro pre-mRNA splicing. Nucleic Acids Res. 1991;19:6956. doi: 10.1093/nar/19.24.6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 17.Carothers AM, Urlaub G, Grunberger D, Chasin LA. Splicing mutants and their second-site suppressors at the dihydrofolate reductase locus in Chinese hamster ovary cells. Mol. Cell. Biol. 1993;13:5085–5098. doi: 10.1128/mcb.13.8.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barthels D, Santoni M, Vopper G, Boned A, Goridis C, Wille W. Differential exon usage involving an unusual splicing mechanism generates at least eight types of NCAM cDNA in mouse brain. EMBO J. 1989;8:385–392. doi: 10.1002/j.1460-2075.1989.tb03389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruce SR, Dingle RC, Peterson ML. B-cell and plasma-cell splicing differences: a potential role in regulated immunoglobulin RNA processing. RNA. 2003;9:1264–1273. doi: 10.1261/rna.5820103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humphrey M, Bryan J, Cooper T, Berget S. A 32-nucleotide exon-splicing enhancer regulates usage of competing 5′ splice sites in a differential internal exon. Mol. Cell. Biol. 1995;15:3979–3988. doi: 10.1128/mcb.15.8.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miki Y, Swensen J, Shattuckeidens D, Futreal P, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett L, Ding W, et al. A strong candidate for the breast and ovarian-cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 22.Liu H-X, Zhang M, Krainer AR. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998;12:1998–2012. doi: 10.1101/gad.12.13.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian H, Kole R. Selection of novel exon recognition elements from a pool of random sequences. Mol. Cell. Biol. 1995;15:6291–6298. doi: 10.1128/mcb.15.11.6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fairbrother WG, Yeh R-F, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 25.Chasin LA, Blencowe B, Graveley B, editors. Alternative Splicing in the Postgenomic Era. Springer: Landes Bioscience; 2007. [Google Scholar]
- 26.Kan JL, Green MR. Pre-mRNA splicing of IgM exons M1 and M2 is directed by a juxtaposed splicing enhancer and inhibitor. Genes Dev. 1999;13:462–471. doi: 10.1101/gad.13.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mrna directed by competing intronic RNA secondary structures. Cell. 2005;123:65–73. doi: 10.1016/j.cell.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de la Mata M, Kornblihtt AR. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat. Struct. Mol. Biol. 2006;13:973–980. doi: 10.1038/nsmb1155. [DOI] [PubMed] [Google Scholar]
- 32.Tilgner H, Nikolaou C, Althammer S, Sammeth M, Beato M, Valcarcel J, Guigo R. Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 2009;16:996–1001. doi: 10.1038/nsmb.1658. [DOI] [PubMed] [Google Scholar]
- 33.Andersson R, Enroth S, Rada-Iglesias A, Wadelius C, Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009;19:1732–1741. doi: 10.1101/gr.092353.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R.25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Computat. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 38.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. 1995;57:289–300. [Google Scholar]
- 40.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 2005;33:W389–W392. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 2008;70:431–457. doi: 10.1146/annurev.physiol.70.113006.100659. [DOI] [PubMed] [Google Scholar]
- 43.Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Molecular Cell. 2001;8:1351–1361. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
- 44.Markham NR, Zuker M. UNAFold. In: Keith JM, editor. Bioinformatics. Springer: Humana Press; 2008. [Google Scholar]
- 45.Andresson R, Enroth S, Rada-Iglesias A, Wadelius C, Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009;19:1732–1741. doi: 10.1101/gr.092353.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2010;36:D301–D308. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elliott DJ. The role of potential splicing factors including RBMY, RBMX, hnRNPG-T and STAR proteins in spermatogenesis. Int. J. Androl. 2004;27:328–334. doi: 10.1111/j.1365-2605.2004.00496.x. [DOI] [PubMed] [Google Scholar]
- 48.Sanford JR, Wang X, Mort M, VanDuyn N, Cooper DN, Mooney SD, Edenberg HJ, Liu Y. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–394. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simard MJ, Chabot B. SRp30c is a repressor of 3′ splice site utilization. Mol. Cell. Biol. 2002;22:4001–4010. doi: 10.1128/MCB.22.12.4001-4010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.