


Abstract
We present One Hand Clapping (OHC), a method for the detection of condition-specific interactions between transcription factors (TFs) from genome-wide gene activity measurements. OHC is based on a mapping between transcription factors and their target genes. Given a single case–control experiment, it uses a linear regression model to assess whether the common targets of two arbitrary TFs behave differently than expected from the genes targeted by only one of the TFs. When applied to osmotic stress data in S. cerevisiae, OHC produces consistent results across three types of expression measurements: gene expression microarray data, RNA Polymerase II ChIP-chip binding data and messenger RNA synthesis rates. Among the eight novel, condition-specific TF pairs, we validate the interaction between Gcn4p and Arr1p experimentally. We apply OHC to a large gene activity dataset in S. cerevisiae and provide a compendium of condition-specific TF interactions.
INTRODUCTION
Homeostasis, the ability to respond to a plethora of environmental challenges, is vital to the cell. This adaptation is achieved by an orchestrated regulation of gene expression. It was discovered that some transcription factors (TFs) act as master regulators in many different conditions, and that the specificity of the regulatory response is obtained through dispatching the signal from the master regulators to downstream TFs (1). It is quite clear that direct TF interactions (TFIs), both physical and genetic, are the prevalent mechanisms of this dispatching (2–4). A method for the detection of functionally relevant, condition-specific TFIs would therefore greatly contribute to our understanding of gene regulation.
A necessary first step toward the detection of TFIs is the quantification of individual TF activity. It is difficult to deduce the activity of a TF by its expression alone [only a small fraction of TFs show expression levels that correlate with those of their target genes (5)], as there are many alternative mechanisms to activate TFs. A complementary approach is the quantification of TF-DNA binding with chromatin immunoprecipitation (ChIP) assays (6). Computational approaches rely on a known TF-target interaction graph (6,7). A linear model that describes gene expression as the product of a position-specific activity matrix derived from binding data, and the unknown TF activities are presented in (8). The experimental detection of TFIs is based on techniques such as co-immunoprecipitation and protein binding arrays (6,9), which are costly and time-consuming. A statistical framework to deduce TF cooperativity from overrepresentation of common TF motifs at the promoter region of target genes is presented in (10,11). However, these approaches do not make direct use of gene expression profiles, nor are their predictions condition-specific. The most promising approaches integrate multiple sources of information, e.g. expression data with binding sites from ChIP. The idea is that if two TFs act cooperatively then there should exist a sufficiently large target gene set to which both TFs bind, and the expression profiles of these target genes should be similar across a series of experiments (12). This concept is used to rigorously assess cooperativity among TFs in the yeast cell cycle (13). Bar-Joseph et al. (14) construct regulatory gene modules by requiring co-regulation and the co-occurrence of binding sites for a pair of interacting TFs. Beer et al. (15) cluster gene expression profiles in a preliminary step and apply a Bayesian classifier to predict TF modules, i.e. groups of TFs that act together in regulating a set of targets. Advanced statistical models for the integration of binding data and expression data are used in (16). Single TFs and TF sets are modeled as hidden variables in a sparse regression model. In this way, the authors can assign a significance value for the combinatorial activity of each TF set. Wang et al. (17) view the problem of TFI identification as a learning task and use Bayesian networks for the integration of multiple sources of evidence to predict cooperatively binding TFs.
Although there are only few studies that focus on TFIs, genetic interactions in general have been investigated extensively. Classically, the biological concept of genetic interaction (e.g. epistasis) between two components relies on the simultaneous perturbation of two components that yields an effect which is different from what one would expect from the perturbation of the individual components. This was applied at large scale in synthetic lethality/growth defect screens like (18–20), to name a few of them. Typically, as many genes as possible are screened for interaction in an automated way by measuring the fitness of single and double gene deletions. Both fitness measures (growth and lethality) are one dimensional. It is still under debate how the deviation of the double deletion fitness from the fitness of the single deletions can be appropriately measured and tested in a rigid mathematical framework (21,22). While this direct interaction measure proved to be rather fragile, the comparison of interaction profiles (the vector of all interaction scores of one gene with all others) yielded surprisingly robust and good results (22). Furthermore, it became evident that the experimental effort can be reduced considerably if not all pairwise combinations of the genes of interest [∼5.4 million combinations tested in (18)] are screened, and that even more information can be gained from measurements under different conditions. This insight is reflected in the work of Bandyopadhyay et al. (23) which identified genes interacting with DNA damage-specific partners, screening a comparably low number of 80 000 double mutants.
In the present work, we extend the concept of genetic interaction to high-dimensional phenotypes (e.g. genome-wide messenger RNA (mRNA) measurements, RNA-seq) as these become increasingly available. We formulate a mathematical concept of TFI which relies on the assumption that the common targets of interacting TFs should behave significantly different than the genes targeted by only one TF alone. So far, each pairwise genetic interaction had to be tested in an individual experiment, requiring a huge number of combinatorial perturbations. Our method instead needs only one global intervention to the system [the fact that led to the name One Hand Clapping (OHC)] in the form of an environmental stimulus, and a high-dimensional gene activity readout in order to score all pairwise TFIs. As in the case of synthetic genetic arrays, we compare the obtained interaction profiles between TFs to obtain reliable and stable predictions. A first proof of concept of this method was given in (24), where we applied OHC to transcriptional activity data obtained under osmotic stress. Here, we establish a solid methodological basis and provide a proof of its universal applicability. After benchmarking the performance of OHC, we construct a compendium of high confidence, condition-specific TFIs based on a large gene expression screen (25). Finally, we validate two of the novel TFI predictions under osmotic stress, one of them in silico, the other one in vivo. OHC is available as an open source, user-friendly R package (see Supplementary data). The current best practice in the study of gene regulation, consisting of quantification of differential expression and gene set enrichment analysis, can now be extended by the screening for combinatorial TF activity.
MATERIALS AND METHODS
TFI model
Let there be gene activity measurements eg for all genes g ∈ G. G is the set of all genes of the organism. In our case, the values eg will be the log folds of the activity in a perturbation experiment versus a wild-type control. Suppose, we knew all TF-target relations (for a discussion how to obtain such a TF-target annotation see the next subsection). For each TF T, we then had a binary indicator function I(g ∈ T) taking on value 1 if gene g belongs to the target set of T and 0 otherwise. The main idea of our method is to divide the set of all genes into four subsets (Figure 1): those genes that are targeted by none of the two TFs those that are targeted by only one of the TFs and those that are targeted by both TFs. Apart from a possible baseline shift β0 in gene activity, TF Tj alone is assumed to have an effect βj on its targets (j = 1, 2). Disregarding the baseline shift, the common targets of T1 and T2 are expected to show a change in activity that amounts to β1 + β2, if the two TFs do not interact. The deviation from this expectation is quantified by the interaction term β12, which presents the most interest. Formally, this can be cast as a second-order linear regression of eg versus the covariates I(g ∈ T1) and I(g ∈ T2),
with g ∈ G. The regression is performed for each TF pair separately, since including more TFs and their interaction terms would lead to overfitting. This cannot be alleviated by using regularization methods such as ridge regression or lasso regression (data not shown). Running the regression in an all-against-all fashion for a set of TFs T results in a symmetric |T| × |T| interaction matrix M containing all interaction terms β12 (see Supplementary Figures S3–S6). We noticed that the interaction terms alone are not strong predictors of interaction (data not shown). The possible explanations for this are 3-fold: the definition of the target sets T1 and T2 is imperfect, the expression measurements are prone to unsystematic variation or the model of TF activity might be too simplistic in some cases.
Figure 1.
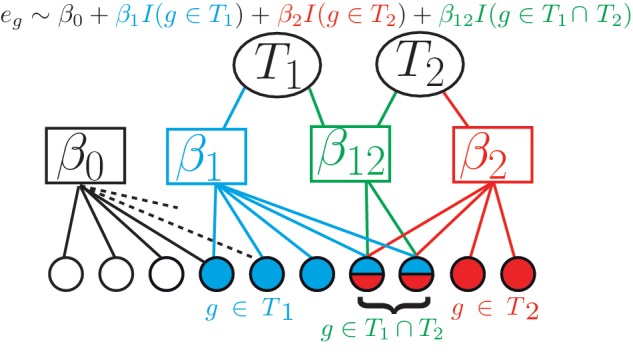
Schematic description of the linear regression model: for two TFs T1 and T2, expression of all genes that are targets of T1 is described by coefficient β1 (cyan), expression of genes that are targets of T2 is described by coefficient β2 (red) and expression of genes that are targets of both TFs is described by coefficient β12 (green). β0 (white) is the coefficient for the baseline activity. It is connected to all genes including those that are targets of neither T1nor T2 (white circles). Remaining connections are symbolized by dotted lines. Circles at the bottom symbolize genes and are colored according to the TF that targets them. The whole formula of the logistic linear regression is shown at the top, with the relevant parts highlighted at the bottom. For a detailed description of the model, refer to ‘Materials and Methods’ section.
TF annotation
One cornerstone for finding TFIs by looking at commonly regulated target genes is the availability of a sufficiently accurate TF-target gene mapping. Such a mapping is rarely available, especially for different growth conditions. This is a limitation of the method that will hopefully be alleviated with the advent of ChIP-seq data of TFs in many organisms, as they are being generated by the ENCODE and modENCODE consortia (26–28).
For Saccharomyces cerevisiae, there are fortunately several several high-quality TF-target mappings available. TF-target relations mined from a manually curated literature repository can be found in the YEASTRACT database (29) which is used in this work. We filter this annotation removing TFs with <10 annotated target genes. This leaves 165 TFs with a median of 167 annotated genes per TF. The size distribution of the annotated gene groups is shown as Supplementary Figure S1.
Supplementary Figure S2A shows a box plot of expression folds (total fraction) of the TFs from YEASTRACT. Prominent differentially expressed TFs are explicitly shown (XBP1, MAG1, SIP4, CIN5, NRG1, CUP2, TEC1, ASH1 and BAS1). Most of these outliers are not directly involved in the salt stress or general stress response pathways, confirming that TF activity is not regulated at the transcriptional level.
When looking at the coefficients β1, β2 and β12 from the regression model of all TF pairs in the YEASTRACT database, there is no apparent structure (Supplementary Figure S2B). Closer investigation reveals extreme values that are due to pairwise interactions between a small set of four TFs (Hot1p, Sps18p, Gis1p and Gat4p, see Supplementary Figure S2C). Indeed these TFs have target genes that are strongly differentially expressed, thus giving rise to a high β12 coefficient to every TF having a considerable overlap with one of these four TFs. The mean expression of all target genes is above that of all other TFs (Supplementary Figure S2C). A Gene Ontology analysis revealed that they are stress responder genes involved in response to various stimuli and to heat shock (Supplementary Table S1). We removed these four outlier TFs from the TF-target graph, leaving us with a final annotation containing 161 TFs.
TFI prediction
To arrive at robust TFI predictions, we use a ‘guilt-by-association’ principle that has been commonly applied in genetic interaction screens (18). Instead of comparing single interaction values, we compare the interaction profiles of each TF (the rows of the interaction matrix M) by means of their correlation. More specifically, we use 1−Pearson correlation as a distance measure. We apply hierarchical average linkage clustering to the rows of M using this distance measure. The resulting clustering dendrogram is shown in Supplementary Figures S7–S10. The two descendants of the terminal branches of this dendrogram define our TFI predictions (Supplementary Algorithm 1). The reasoning behind this is that we expect many TFs to have at least one interaction partner in a given condition, and the most likely partner is the one with the most similar interaction profile. Alternatively, we tried to predict TFIs based on P-values derived from a null distribution of the correlation distances. Such null distributions can be either derived from Pearson's Product Moment Coefficient (30) or, more conservatively, from resampling procedures (shuffling target genes). Still our simple clustering procedure works best in terms of area under the curve (AUC) (data not shown; for a definition of AUC see section ‘Results’).
Gene activity data
In this article, we use several datasets as input to our OHC method. First, we use mRNA expression data from a time course experiment exposing a wild-type yeast strain to osmotic stress by adding 0.8 M NaCl (see (24) for more details). The article provides standard total mRNA expression data after 36 min of osmotic stress (dataset D1), as well as the corresponding measurements of ‘newly synthesized’ mRNA (dataset D2), which are roughly proportional to the mRNA synthesis rates at the time of measurement. Throughout this article, we always mean log expression folds (log quotient of expression under the experimental condition against expression in the control experiment) when referring to expression data. To test the reliability of our method, we included an unrelated gene expression dataset generated by Mitchell et al. (31) obtained from S. cerevisiae under osmotic stress. The total mRNA expression level (corresponding to the total fraction of Miller et al.) 30 min after addition of 0.8 M NaCl was measured (dataset D3). The gene expression datasets from Miller et al. and Mitchell et al. should be highly comparable, since the same yeast strain, the same microarray platform and a similar protocol were used. Microarray data were downloaded as raw files from GEO (32) (accession number: GSE15936) for Mitchell et al. data and from ArrayExpress (accession number: E-MTAB-439) for Miller et al. Normalization was performed using gcrma (33) (as implemented in R/Bioconductor (34)) without quantile normalization, since we expect global effects of the perturbation on mRNA expression. As a completely different way of assessing gene activity, Miller et al. (24) also provide RNA polymerase II (Pol II) occupancies from ChIP-chip experiments 24 min after addition of salt. We use their Pol II mean occupancy on each gene (between transcription start site and polyadenylation site) as another proxy for gene activity (dataset D4).
Yeast strains and growth assays
The S. cerevisiae deletion strains hog1Δ, arr1Δ and gcn4Δ, as well as the wild-type strain BY4741 were obtained from Open Biosystems (Huntsville, USA). The double deletion strain arr1Δ/gcn4Δ was generated by integrating a ClonNat cassette in the ARR1 locus of the gcn4Δ strain. Correct gene disruptions were verified by polymerase chain reaction (PCR). Spot dilutions were done to assess fitness and growth under osmotic stress. Equal amounts of freshly grown yeast cells in YPD were re-suspended in water, 10-fold dilutions were spotted on YPD plates and YPD plates with 1.2 M NaCl. Plates were incubated for 4 days at 30°C. Results are found in Supplementary Figure S11.
Gene expression microarrays
Overnight cultures were diluted in fresh synthetic complete medium with 2% glucose to OD600nm = 0.1 (120 ml cultures, 160 rpm shaking incubator, 30°C). In the early log phase (OD600nm = 0.8), 20 ml of the culture was harvested by centrifugation (no salt stress sample). Afterwards, NaCl was added to the remaining culture to a final concentration of 0.8 M, 30 min after addition, 20 ml of culture was harvested (salt stress sample). Total RNA was prepared after cell lysis using a FastPrep-24 instrument (Millipore) and subsequent purification using the RiboPure-Yeast Kit (Ambion) following the manufacturer's instructions. All following steps were conducted according to the Affymetrix GeneChip 3′IVT Express Kit protocol. Briefly, one-cycle complementary DNA (cDNA) synthesis was performed with 300 ng of total RNA. In vitro reverse transcription labeling was performed for 16 h. The fragmented samples were hybridized for 16 h on ‘Yeast Genome 2.0’ expression arrays (Affymetrix), washed and stained using a Fluidics 450 station and scanned on an Affymetrix GeneArray scanner 3000 7G. Micorarray data have been deposited to the ArrayExpress database (http://www.ebi.ac.uk/microarray) under accession number E-MEXP-3566.
RESULTS
OHC accurately predicts pairwise TFIs
We first applied OHC to mRNA expression data from the total mRNA fraction of Miller et al. (dataset D1) using the filtered YEASTRACT database as TF-target annotation (see ‘Materials and Methods’ section). The resulting interaction matrix is shown as a heatmap (Supplementary Figure S3). The rows of the matrix were clustered and TFI predictions were made as described in ‘Materials and Methods’ section. We predict 59 mutually disjoint TFI pairs, while for 43 single TFs no interaction partners were predicted. Validation of the predictions was done through the BioGRID database [(35), version 3.1.71]. It contains physical and genetic interactions for many yeast proteins that were derived from high-and low-throughput experiments in the literature. The subgraph of BioGRID corresponding to interactions between TFs, as well as their degree distribution, is shown in Supplementary Figure S12. From the 59 predicted TFIs, we validate 13 of them as listed in BioGRID (a positive predictive value of 22%). A complete list of predicted and validated pairs can be obtained with the Supplementary Code. Validated TFI predictions had a significantly lower correlation distance than unvalidated TFIs (Wilcoxon's test, P-value 0.004). This shows that interacting TF pairs are more closely related (considering our interaction measure and distance function) than unvalidated predictions. This is further investigated through an ROC plot (Figure 2A). The AUC (76%) shows a strong deviation from random predictions (diagonal) and shows that the profile correlation measure can serve as a proxy for predicting interactions. To better assess the performance of the clustering and prediction algorithm, we verified the overrepresentation of validated prediction using Fisher's test (P-value <10−5, odds ratio 5.291, with a 95% confidence interval [2.67;10]). When testing only genetic or physical interactions from BioGRID (P-values <10−5 and 0.003, respectively), we find a bias toward prediction of genetic interactions, as defined by BioGRID.
Figure 2.

Validation of OHC predictions (A) ROC curve using TFIs in BioGRID as benchmark, colored area and horizontal lines are confidence intervals of sensitivity and specificity, respectively. (B) Overlap between predicted and validated pairs between all datasets. D1: total mRNA fraction D2: labeled mRNA fraction, D3: mRNA data from Mitchell et al. and D4: Pol II ChIP-chip occupancy measurements; predictions across datasets agree well, dataset D4 having the most distinct predictions. Each intersect is shown as a circle, with radius proportional to the intersect size. The black box shows the intersect used as novel predictions. The numbers in parentheses indicate the subset of interactions that are validated by BioGRID. (C) Pairwise comparison of expression or occupancy values for all genes. Numbers in lower part indicate Spearman's correlation between datasets. D1 and D3 have the highest correlation as they are both total mRNA expression measurements. D2 has a very good but lower correlation with D1 and D3. This is due to subtle differences when measuring newly synthesized mRNA. D4 has a very weak positive correlation as Pol2 occupancy as an indicator of gene activity is very different from mRNA expression.
We tested the consistency of predictions on incomplete TF-target annotations by removing an increasing precentage of TFs from the annotation. We measured the agreement of predictions on the smaller TF annotation with predictions made on the original annotation Supplementary Figure S16). In addition, we measured the performance as the number of validated pairs according to BioGRID. Expectedly the drop in agreement is stronger than the drop in performance, because removing one TF from a pair will regroup the remaining TF with another with high probability, thus changing the predictions. Simultaneously performance decreases more slowly, showing that the regrouping of TFs leads to new validated pairs. After removal of 20% of TFs, performance merely drops from 22% to 18%.
OHC is stable on a wide range of gene activity data
To test the stability of our method we applied it to the mRNA expression data of the labeled fraction from the same osmotic stress experiment used previously (termed dataset D2, see ‘Materials and Methods’ section). Both datasets are similar (Spearman's ρ = 0.85, Figure 2C) and we expect similar results. On this dataset, we predict 60 pairwise interactions, 11 validated by the BioGRID database (18% prediction accuracy; predicted pairs: Nrg1p-Nrg2p, Fhl1p-Ifh1p, Stp1p-Stp2p, Msn2p-Msn4p, Mbp1p-Swi4p, Ecm22p-Upc2p, Cbf1p-Met28p Ndt80p-Sum1p, Arg80p-Arg81p, Hap3p-Hap5p and Mga2p-Spt23p). The validated interactions highly agree between both datasets, eight pairs being validated by both runs (Figure 2B). The interactions Ace2p-Swi5p, Ecm22p-Mot3p, Pdr1p-Pdr3p, Mbp1p-Skn7p and Flo8p-Phd1p found in the first dataset are lost in the second, the interactions Mbp1p-Swi4p, Ecm22p-Upc2p and Cbf1p-Met28p in the second are not present in the first dataset. Comparison of all predicted interactions (Figure 2B) features an overlap of 23 pairwise interactions (38%).
Reproducibility was tested by running the method on another osmotic stress dataset from (31) (mRNA expression measurement 30 min after addition of NaCl) termed D3 (Spearman's ρ = 0.88; Figure 2C). The method predicts 60 pairwise interactions and 14 validated interactions (23%). The overlap with the previous two datasets is 26 and 23 pairs for datasets D1 and D2, respectively. Validated interactions agree strongly; they overlap at 12 and 8 validated interactions for D1 and D2, respectively (Figure 2B). It is interesting to notice that the datasets D3/D1 agree more closely than D3/D2 and D1/D2. This might be due to the fact that D1 and D3 measure the total mRNA at the extraction timepoint and thus include mRNAs transcribed before the onset of stress and not yet degraded, contrary to D2 which corresponds to the labeled mRNA fraction and thus contains only mRNAs transcribed after the onset of stress. Indeed, D1/D3 have a higher correlation than D1/D2 and D3/D2 (Figure 2C).
To show that the method also works on proxies of gene activity other than mRNA expression measurements, we used the Pol II ChIP-chip data from (24) (termed D4). On this dataset, the method predicts 57 interactions, 12 of which can be validated (21% accuracy). Its performance is thus comparable to the performance on mRNA expression data. The predictions vary strongly as there are only 12, 10 and 12 predicted interactions shared with the datasets D1, D2 and D3, respectively (Figure 2B). This is due to a low correlation between the datasets D1 and D3 varying between 0.16 and 0.3 (Spearman's rank correlation, see Figure 2C). Despite the low correlation, a core of eight interactions is shared between all datasets (including four novel predictions) and shows that the method is robust enough to adapt to various measures of gene activity.
The method can also readily be applied to data from other organisms. Supplementary Figure S15 and accompanying text describe the application to human pancreatic cancer data. Using a TF annotation containing 153 TFs, OHC predicts 57 putative interactions of which 7 can be validated (12% positive predictive value). The difference in performance compared with yeast data might be attributed to an incomplete annotation of human TFIs in BioGRID.
OHC finds cis and trans TFIs
We distinguish between two main types of combinatorial TFIs: cis-regulatory interactions and trans-regulatory interactions (36). Cis interactions are mediated by a specific TF binding site configuration at the cis-regulatory region of a gene, possibly resulting in cooperative or competitive binding of TFs. Competitive binding occurs when two TFs share a common or overlapping binding motif. Cooperative binding of TFs occurs if two TFs are required to bind simultaneously to be functional, or if the binding of the second TF is enhanced by the binding of the first TF, which is the case, e.g. for nucleosome-mediated cooperativity (37).
Trans interactions are defined as direct protein–protein interactions of both TFs prior to DNA binding, either by forming a protein complex or by complex formation with other co-factors involved in Pol II recruitment and transcription initiation.
TF pairs predicted by our method on dataset D1 and validated by BioGRID include the following types of interaction: Ace2p-Swi5p (38) and Sum1p-Ndt80p (39) undergo competitive cis-regulatory interactions, the former having identical binding sites, the latter having overlapping binding sites. Mot3p-Ecm22p (40), Mbp1p-Skn7p (41), Arg80p-Arg81p (42), Hap3p-Hap5p (43) and Pdr1p-Pdr3p (44) are all examples of trans-regulatory protein interactions forming prior to DNA binding. The pair Ifh1p-Fhl1p represents a special type of trans interaction. Fhl1p is by default bound to the promoter of ribosomal protein genes without influencing transcription. The phosphorylation of Ifh1p enables the binding and activation of Fhl1p (45).
Three interactions [Msn2p-Msn4p (46), Mga2p-Spt23p (47) and Stp1-Stp2p (48)] could not be categorized unambiguously. They consist of homologous or functionally redundant proteins, implying that both cis and trans interactions could serve as regulatory mechanism. We call these interactions ‘ambiguous’.
OHC provides a compendium of condition-specific TFIs
Absolutely no changes to the model are required when applying the method to large datasets containing gene activity measurements under diverse conditions. Consequently, we ran the method on mRNA expression data from 173 experiments [data compiled by Gash et al. (25)] which is grouped into 16 conditions with at least five experiments. Clustering the experiments according to the correlation of the expression profiles across conditions recovers the grouping into 16 conditions defined above. Similarly, clustering the predictions made by OHC on each experiment according to the number of common TFIs between experiments recovers the condition classes as well (Supplementary Figure S13). This demonstrates that predictions by OHC are truly condition specific and reproducible.
We compiled a compendium of confident condition-specific TFIs. For each condition, we selected the OHC interactions that are found in more than half of the experiments for that condition. This compendium is provided as Supplementary Data. The graph representation of this compendium (Figure 3) is sparsely connected with many isolated pairs. The number of conditions for a pair of TF is encoded by edge width, indicating the specificity of the interaction. Due to false negatives and the limited variety of environmental conditions in (25), our network is far from being complete, and too sparse to be conclusive about its topological properties, such as edge degree distribution and connectivity. Yet, it highlights an important organizational feature of signaling pathways, namely a functional hierarchy, where information is flowing from general to specific regulators: some TF pairs interact in more than one condition. Most of them are either protein complexes (e.g. Hap2p-Hap3p-Hap5p), form heterodimer (e.g. Arg80-Arg81p and Ino2p-Ino4p) or are highly similar or homolog TF (e.g. Nrg1p-Nrg2p, Msn2p-Msn4p, Mga2-Spt23p and Upc2p-Ecm22p). This is the reason why the aforementioned interactions are detected in multiple conditions; simply because the activation of one interaction partner leads to complex formation. The other TFs that interact with different partners, need both to be active under the same condition for an interaction to be predicted. This is the case for the interaction between Skn7p and Stb5p which is exclusively predicted by OHC under diamide treatment, which seems plausible as both have a role in the oxidative stress response (49,50). Skn7p is the more general TF while Stb5 is diamide specific. Indeed, STB5 null mutants have a decreased resistance to diamide (51). Another interesting finding is the TF Tye1p which, as our model postulates, regulates glycolysis together with Gcr1p under H2O2 exposure and together with Rgt1p when cells are exposed to dithiothreitol. Condition-specific transcriptional control is achieved by activating Tye1p under several oxidative stress inducing agents and specifically pairing it with TFs only active under one such agent. OHC helps in discovering this type of combinatorial gene regulation.
Figure 3.

Components of the network of confident TFIs predicted in more than half of the experiments from each condition. Edge width is proportional to the number of conditions in which the edge was found and the components are sorted in increasing edge width from left to right. TF pairs on the right, that are found in many conditions are either protein complexes or homologous or highly similar proteins. TFIs on the left are highly condition specific and only occur in a single condition.
Novel predictions of TFIs can be validated experimentally
Novel predictions are defined as consensus predictions between datasets D1, D2 and D3 (indicated by a black box in Figure 2B). We left dataset D4 out because of the low correlation with the rest of the data. This gives us eight novel predictions namely the pairs: Cin5p-Yap6p, Gcn4p-Arr1p, Zap1p-Spt2p, Sko1p-Sok2p, Hsf1p-Aft1p, Sip4p-Cdc14p, Cup2p-Yrr1p and Rim101p-Otu1p.
Cin5p and Yap6p bind competitively
We realized both Cin5p and Yap6p have very similar binding motifs (Figure 4A) according to the YeTFaSCo database (52), choosing the motifs with high expert confidence. They are derived from ChIP-chip data by Harbison et al. (6) and MacIsaac et al. (7) for CIN5 and YAP6, respectively.
Figure 4.

(A) Motifs of CIN5 and YAP6 from YeTFaSCo database. Both motifs are very similar, which is confirmed by motif search, as a test for co-occurrence of both motifs is significant (see text). (B) Growth assay on YPD plates and YPD plates containing 1.2 M NaCl after incubating for 4 days at 30°C. Only cell growth at a dilution of 1:100 is shown and the relevant parts have been extracted from the image. The full image of the experiment including replicates and negative controls can be found as Supplementary Figure S11. Growth phenotype is not affected in YPD medium. Wild-type cells have decreased phenotype under osmotic stress and arr1Δ mutants show strong decrease in growth phenotype, while gcn4Δ mutants and double mutants do not. This shows the synthetic rescue of the effect of the knockout of ARR1 in the double mutant. (C) Hypothetical model explaining the observations from the growth assay experiments (B). This model is focused on the genes that respond positively to osmotic stress (symbolized by the line with arrowhead). A double inhibition chain of Arr1p ⊣ Gcn4p leads to the observed phenotypes under osmotic stress: in wild-type cells, the inhibitory effect of Gcn4p is prevented by Arr1p and the cells grow normally. The same observation is made when knocking out GCN4 as the logic of regulation does not change. The knockout of ARR1 relieves the inhibition on Gcn4p, which in turn downregulates the target genes. We speculated that this is causing problems with osmo-adaptation, leading to a reduced cell growth. The double mutant rescues that phenotype as the genes are only driven by the osmotic stress (as in wild type). (D) Log-expression values of candidate genes responsible for synthetic rescue across all arrays. All candidates are affected by the knockout of ARR1. Four candidate genes are uncharacterized ORFs, two are proteins of unknown function and the rest has a variety of different roles in different pathways. The genes have not yet been linked to osmotic stress.
We searched for both motifs using these position-specific weight matrices (PWMs) and the MEME suite (53) (FIMO version 4.7.0 using default parameters for P-value and q-value thresholds) on intergenic regions defined by (6). Testing for co-occurence of both motifs on all intergenic regions is highly significant (P-value <10−5). We found 135 intergenic regions where both motifs have one or several matches. In this set, we find 149 competitive matches, where the distance between both motif occurrences is 0 and 36 cases having five or more nucleotides between motif occurrences. Motif search also shows that the TFs can bind alone as for some intergenic regions only a match for a single TF falls below the P-value threshold. As there are protein-binding microarray [PBM (9)] derived motifs for each TF, we deduce that both proteins can bind DNA on their own. The motif similarity from ChIP-chip data is thus not due to a protein complex between Cin5p and Yap6p and we conclude that both TFs bind competitively to the promoter of their common target genes.
The other novel predictions do not show such a clear evidence for an interaction based on their motifs, so we decided to perform experimental validation for one additional pair. We chose the pair Gcn4p-Arr1p as both interaction partners have the largest target sets among all predicted pairs (as defined by YEASTRACT, 1260 and 743 target genes for Gcn4p and Arr1p, respectively).
GCN4/ARR1 show a synthetic rescue phenotype
To validate the interaction between GCN4 and ARR1, we performed a classical genetic interaction screen (Figure 4B and Supplementary Figure S11). We assayed the growth of a wild-type strain as well as single and double deletion strains in rich medium (YPD) and under osmotic stress (YPD + 1.2 M NaCl). The single deletions had no effect in rich media due to the condition specificity of the prediction. While wild type and gcn4Δ grew normal under osmotic stress, arr1Δ showed a strong decrease in cell growth. The growth defect is rescued in the double deletion strain gcn4Δ/arr1Δ. This indicates an interaction between both proteins, though the experimental design cannot distinguish between a cis or trans interaction.
Our current working hypothesis on the mechanism of the interaction is shown in Figure 4C. We expect most genes commonly regulated by both TFs to be salt stress responders (because of the condition specificity of OHC). We know from previous experiments (24) that Gcn4p acts as a repressor under osmotic stress. By positioning Arr1p upstream of and inhibiting Gcn4p, this model explains our observations from the growth assay experiments. The removal of Arr1p from the system probably leads to genes important for osmo-adaptation to be repressed by Gcn4p, reducing cell growth rate. The removal of Gcn4p has no noticable effect in this model. The double knockout reestablishes conditions close to wild type, where genes are only regulated by the osmotic stress response.
We performed mutant cycle analysis [see (54)] to elucidate the mechanism of this interaction. Briefly, transcriptional profiling was done for single and double deletion strains, before and after exposure to osmotic stress conditions (0.8 M NaCl; see Supplementary Figure S14 for a comparison of all profiles). For each gene, its expression under osmotic stress was explained by a linear model accounting for an effect of the GCN4 deletion, an effect of the ARR1 deletion and their interaction effect. We selected the genes whose interaction effect was positive and larger than log21.5 (45 genes). Then, we filtered this group for genes showing a decrease in expression in the arr1Δ arrays and an expression similar to wild type in the double mutant (leaving 37 genes). The genes should be salt stress responders and thus should show a 2-fold increase of their wild-type expression under osmotic stress relative to wild-type expression in synthetic complete medium. This criterion reduced the candidate set to nine genes (Figure 4D). The filtering criteria were chosen in accordance to the expected model (Figure 4C). When shuffling the arrays and applying the same criteria we find at most two genes, showing that the result is not random. Four of the nine candidate genes are uncharacterized ORFs (YDR366C, YJL107C, YMR034C and YGR066C), Bop2p and Spg4p are proteins of unknown function. The other candidates are involved in a variety of biological processes such as heme degradation, pheromone-induced signaling, survival at high temperature or as a membrane protein [Saccharomyces Genome Database (SGD) (55)]. This suggests a novel function of these genes as a part of the osmotic stress response pathway, albeit their roles are unclear and a Blastn/Blastp homology search did not help reveal their function.
DISCUSSION
OHC has been established as a method to predict condition-specific TFIs; its implementation, which does not require any parameter adjustments by the user, is provided as a software package for R. It takes advantage of the increasingly reliable and comprehensive resources on gene-specific transcriptional regulators. OHC is data-inexpensive; ‘two’ genome-wide gene activity measurements (under normal and stress conditions) are already sufficient. With this sparse input, we derive a robust interaction measure that is stable on many different types of gene activity data. Despite its modest sensitivity, its predictions are relevant due to their high specificity.
Applied on osmotic stress data and TF-target relations from YEASTRACT, OHC predicts 59 interactions. 23 of the interactions can be validated by BioGRID (22%). Although gene activity data are available for many different conditions in all organisms, it may be difficult to find a mapping of TFs to a set of target genes suitable for OHC in other organisms. For the yeast S. cerevisiae, there are fortunately several options available, the most important being the YEASTRACT database (29) and the dataset provided by MacIsaac et al. (7). When we run the method using the latter, we predict 38 interactions, only 6 of which can be validated by BioGRID (16% prediction accuracy). While the annotation from MacIsaac et al., based on ChIP-chip data, is of high quality, it does not suit our purpose, as it contains assignments made under standard experimental conditions. YEASTRACT contains many TF-target gene assignments under different stress conditions and knockout strains.
It is important to note that the predictions made by OHC are entirely different from predictions based on target genes set alone. Indeed, a straightforward Fisher test for target gene overlap does not find the same TFIs as OHC (data not shown). In particular, the method can and does predict interactions between TFs that have no overlap in target genes and thus no interaction score. This is possible because we predict interactions based on profile similarity which takes into account the interaction scores with all other TFs. We found three TFIs without target gene overlap: Kar4p-Stb1p, Rds1p-YJL206C and Cbf1p-Mig2p.
In silico validation of the method is based on all interactions between TFs submitted to BioGRID. As this repository is not exhaustive, the performance measurements in this article represent conservative estimates. Moreover, entries in BioGRID are biased toward interactions present under normal growth conditions and frequently studied stress conditions, as these account for the large part of the studies that contributed to BioGRID. We selected one novel candidate pair (Gcn4p-Arr1p) for in vivo validation by growth assays under osmotic stress. The growth defect in the arr1Δ strain showed a synthetic rescue phenotype in the arr1Δ/gcn4Δ double deletion strain. Subsequent gene expression analysis revealed nine candidate genes potentially involved in the synthetic rescue, not previously connected to osmotic stress.
Application to a large dataset comprising 16 conditions showed that different pairs are detected in each condition. We compiled a compendium of confident condition-specific interactions, where each pair has to be predicted in at least half of the experiments for each condition (stability). This provides a resource for studying functionally relevant condition-specific TFIs. Since different interactions are predicted in different conditions, we confirm that TF combinatorics drive adaptation to environmental challenges.
This method can be extended in several ways: first, the linear model from which the interaction score is derived can be replaced by a more elaborate physical model of TF activation, as has been attempted by (56,57). Currently these models fall short of describing TF competition adequately (58,59). Nonetheless, we speculate that inclusion of chromatin structure, in particular nucleosome positioning, in the interaction score will improve the method. Second, OHC can be generalized to other organisms, as reliable TF-target annotations will become available. Finally, the screening principle introduced here lends itself to generalization: the only property of TFs that enters the model is that each TF splits the genes into two disjoint sets (targets versus non-targets), i.e. each TF defines a binary property on the set of genes. It is therefore straightforward to perform a condition-specific interaction screen on any collection of binary properties, such as pathway membership [e.g. KEGG (60)] or functional annotation [e.g. GO (61)].
ACCESSION NUMBERS
E-MEXP-3566.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–16 and Supplementary Algorithm.
AVAILABILITY
The method is implemented in the R package OneHandClapping available from CRAN (http://cran.r-project.org/web/packages/OneHandClapping/index.html). A short introduction into using the package and generating some results from this publication are found as Supplementary Code (code.R).
FUNDING
LMUexcellent guest professorship ‘Computational Biochemistry’ (to A.T.); Boehringer Ingelheim Fonds and the Elite Network of Bavaria (to M.S.); the Deutsche Forschungsgemeinschaft [SFB646, TR5, FOR1068]; NRM; European Molecular Biology Organization (EMBO); an Advanced Investigator Grant of the European Research Concil, a LMUexcellent research professorship ‘Molecular Systems Biology of gene regulation’; LMUinnovativ project Bioimaging Network (BIN) and the Jung-Stiftung (to P.C.). Funding for open access charge: the Deutsche Forschungsgemeinschaft [SFB646].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- 2.Das D, Banerjee N, Zhang MQ. Interacting models of cooperative gene regulation. Proc. Natl Acad. Sci. USA. 2004;101:16234–16239. doi: 10.1073/pnas.0407365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 4.Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EEM. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 5.Herrgård MJ, Covert MW, Bernhard Reconciling gene expression data with known genome-scale regulatory network structures. Genome Res. 2003;13:2423–2434. doi: 10.1101/gr.1330003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacIsaac K, Wang T, Gordon DB, Gifford D, Stormo G, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113+. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee E, Bussemaker HJ. Identifying the genetic determinants of transcription factor activity. Mol. Syst. Biol. 2010;6:412–412. doi: 10.1038/msb.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukherjee S, Berger MF, Jona G, Wang XS, Muzzey D, Snyder M, Young RA, Bulyk ML. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nature Genet. 2004;36:1331–1339. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beyer A, Workman C, Hollunder J, Radke D, Möller U, Wilhelm T, Ideker T. Integrated assessment and prediction of transcription factor binding. PLoS Comput. Biol. 2006;2:e70+. doi: 10.1371/journal.pcbi.0020070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollunder J, Friedel M, Beyer A, Workman CT, Wilhelm T. DASS: efficient discovery and P-value calculation of substructures in unordered data. Bioinformatics. 2007;23:77–83. doi: 10.1093/bioinformatics/btl511. [DOI] [PubMed] [Google Scholar]
- 12.Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat. Genet. 2001;29:153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee N, Zhang MQ. Identifying cooperativity among transcription factors controlling the cell cycle in yeast. Nucleic Acids Res. 2003;31:7024–7031. doi: 10.1093/nar/gkg894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bar-Joseph Z, Gerber GK, Lee TI, Rinaldi NJ, Yoo JY, Robert F, Gordon DB, Fraenkel E, Jaakkola TS, Young RA, et al. Computational discovery of gene modules and regulatory networks. Nat. Biotechnol. 2003;21:1337–1342. doi: 10.1038/nbt890. [DOI] [PubMed] [Google Scholar]
- 15.Beer MA, Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- 16.Sabatti C, James GM. Bayesian sparse hidden components analysis for transcription regulation networks. Bioinformatics. 2006;22:739–746. doi: 10.1093/bioinformatics/btk017. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Zhang X-S, Xia Y. Predicting eukaryotic transcriptional cooperativity by Bayesian network integration of genomewide data. Nucleic Acids Res. 2009;37:5943–5958. doi: 10.1093/nar/gkp625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 20.Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagê N, Robinson M, Raghibizadeh S, Hogue CWV, Bussey H, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 21.Mani R, St, Hartman JL, Giaever G, Roth FP. Defining genetic interaction. Proc. Natl Acad. Sci. USA. 2008;105:3461–3466. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–1081. doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 23.Bandyopadhyay S, Mehta M, Kuo D, Sung M-K, Chuang R, Jaehnig EJ, Bodenmiller B, Licon K, Copeland W, Shales M, et al. Rewiring of genetic networks in response to DNA damage. Science. 2010;330:1385–1389. doi: 10.1126/science.1195618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller C, Schwalb B, Maier K, Schulz D, Dümcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dolken L, et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 2011;7:458–458. doi: 10.1038/msb.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium TEP. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 27.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE Project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negre N, Brown CD, Ma L, Bristow CA, Miller SW, Wagner U, Kheradpour P, Eaton ML, Loriaux P, Sealfon R, et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teixeira MC, Monteiro P, Jain P, Tenreiro S, Fernandes AR, Mira NP, Alenquer M, Freitas AT, Oliveira AL, Sa-Correia I. The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 2006;34(Suppl. 1):D446–451. doi: 10.1093/nar/gkj013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilcox RR. Introduction to Robust Estimation and Hypothesis Testing. 2nd edn. San Diego, CA: Academic Press; 2005. [Google Scholar]
- 31.Mitchell A, Romano GH, Groisman B, Yona A, Dekel E, Kupiec M, Dahan O, Pilpel Y. Adaptive prediction of environmental changes by microorganisms. Nature. 2009;460:220–224. doi: 10.1038/nature08112. [DOI] [PubMed] [Google Scholar]
- 32.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004;99:909+. [Google Scholar]
- 34.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoif S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tirosh I, Reikhav S, Levy AA, Barkai N. A yeast hybrid provides insight into the evolution of gene expression regulation. Science. 2009;324:659–662. doi: 10.1126/science.1169766. [DOI] [PubMed] [Google Scholar]
- 37.Mirny LA. Nucleosome-mediated cooperativity between transcription factors. Proc. Natl Acad. Sci. USA. 2010;107:22534–22539. doi: 10.1073/pnas.0913805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voth WP, Yu Y, Takahata S, Kretschmann KL, Lieb JD, Parker RL, Milash B, Stillman DJ. Forkhead proteins control the outcome of transcription factor binding by antiactivation. EMBO J. 2007;26:4324–4334. doi: 10.1038/sj.emboj.7601859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierce M, Benjamin KR, Montano SP, Georgiadis MM, Winter E, Vershon AK. Sum1 and Ndt80 proteins compete for binding to middle sporulation element sequences that control meiotic gene expression. Mol. Cell. Biol. 2003;23:4814–4825. doi: 10.1128/MCB.23.14.4814-4825.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies BSJ, Rine J. A role for sterol levels in oxygen sensing in Saccharomyces cerevisiae. Genetics. 2006;174:191–201. doi: 10.1534/genetics.106.059964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouquin N, Johnson AL, Morgan BA, Johnston LH. Association of the cell cycle transcription factor Mbp1 with the Skn7 response regulator in budding yeast. Mol. Biol. Cell. 1999;10:3389–3400. doi: 10.1091/mbc.10.10.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amar N, Messenguy F, El Bakkoury M, Dubois E. ArgRII, a component of the ArgR-Mcm1 complex involved in the control of arginine metabolism in Saccharomyces cerevisiae Is the sensor of Arginine. Mol. Cell. Biol. 2000;20:2087–2097. doi: 10.1128/mcb.20.6.2087-2097.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McNabb DS, Xing Y, Guarente L. Cloning of yeast HAP5: a novel subunit of a heterotrimeric complex required for CCAAT binding. Genes Dev. 1995;9:47–58. doi: 10.1101/gad.9.1.47. [DOI] [PubMed] [Google Scholar]
- 44.Mamnun YM, Pandjaitan R, Mahé Y, Delahodde A, Kuchler K. The yeast zinc finger regulators Pdr1p and Pdr3p control pleiotropic drug resistance (PDR) as homo- and heterodimers in vivo. Mol. Microbiol. 2002;46:1429–1440. doi: 10.1046/j.1365-2958.2002.03262.x. [DOI] [PubMed] [Google Scholar]
- 45.Rudra D, Zhao Y, Warner JR. Central role of Ifh1p-Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J. 2005;24:533–542. doi: 10.1038/sj.emboj.7600553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martínez-Pastor MT, Marchler G, Schüller C, Marchler-Bauer A, Ruis H, Estruch F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE) EMBO J. 1996;15:2227–2235. [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang S, Skalsky Y, Garfinkel DJ. MGA2 or SPT23 is required for transcription of the Δ9 fatty acid desaturase gene, OLE1, and nuclear membrane integrity in Saccharomyces cerevisiae. Genetics. 1999;151:473–483. doi: 10.1093/genetics/151.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wielemans K, Jean C, Vissers S, André B. Amino acid signaling in yeast: post-genome duplication divergence of the Stp1 and Stp2 transcription factors. J. Biol. Chem. 2010;285:855–865. doi: 10.1074/jbc.M109.015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krems B, Charizanis C, Entian KD. The response regulator-like protein Pos9/Skn7 of Saccharomyces cerevisiaeis involved in oxidative stress resistance. Curr. genet. 1996;29:327–334. doi: 10.1007/BF02208613. [DOI] [PubMed] [Google Scholar]
- 50.Larochelle M, Drouin S, Robert F, Turcotte B. Oxidative stress-activated zinc cluster protein Stb5 has dual activator/repressor functions required for pentose phosphate pathway regulation and NADPH production. Mol. Cell. Biol. 2006;26:6690–6701. doi: 10.1128/MCB.02450-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hector RE, Bowman MJ, Skory CD, Cotta MA. The Saccharomyces cerevisiae YMR315W gene encodes an NADP(H)-specific oxidoreductase regulated by the transcription factor Stb5p in response to NADPH limitation. New Biotechnol. 2009;26:171–180. doi: 10.1016/j.nbt.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 52.de Boer CG, Hughes TR. YeTFaSCo: a database of evaluated yeast transcription factor sequence specificities. Nucleic Acids Res. 2012;40(D1):D169–D179. doi: 10.1093/nar/gkr993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Suppl. 2):W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Capaldi AP, Kaplan T, Liu Y, Habib N, Regev A, Friedman N, O'Shea EK. Structure and function of a transcriptional network activated by the MAPK Hog1. Nature Genet. 2008;40:1300–1306. doi: 10.1038/ng.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buchler NE, Gerland U, Hwa T. On schemes of combinatorial transcription logic. Pro. Natl Acad. Sci. USA. 2003;100:5136–5141. doi: 10.1073/pnas.0930314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raveh-Sadka T, Levo M, Segal E. Incorporating nucleosomes into thermodynamic models of transcription regulation. Genome Res. 2009;19:1480–1496. doi: 10.1101/gr.088260.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilman A, Arkin AP. GENETIC ‘CODE’: representations and dynamical models of genetic components and networks. Ann. Rev. Genomics Hum. Genet. 2002;3:341–369. doi: 10.1146/annurev.genom.3.030502.111004. [DOI] [PubMed] [Google Scholar]
- 59.Wasson T, Hartemink AJ. An ensemble model of competitive multi-factor binding of the genome. Genome Res. 2009;19:2101–2112. doi: 10.1101/gr.093450.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.