

Abstract
During the past several years it has become clear that alterations in the expression of microRNA genes contribute to the pathogenesis of most, perhaps all, human malignancies. These alterations can be caused by a variety of mechanisms, including deletions, amplifications or mutations involving microRNA loci, by epigenetic silencing or by dysregulation of transcription factors targeting specific microRNAs. Since malignant cells show dependence on the dysregulated expression of microRNA genes, which in turn control or are controlled by dysregulation of multiple protein coding oncogenes or tumor suppressor genes, these small RNAs provide important opportunities for development of future microRNA based therapies.
Introduction
For the past two and a half decades it has been thought that cancer is caused by genetic and/or epigenetic alterations to protein coding genes, oncogenes and tumor suppressor genes. These alterations result mainly from somatic genetic events (1) occurring over long periods of time, particularly in solid malignancies such as lung, breast, prostate and gastrointestinal cancers. These findings have suggested the development of novel (targeted) therapies based on the specific genetic alterations involved in cancer pathogenesis. For example, in the case of chronic myelogenous leukemia (CML), the disease has been shown to be caused by a t(9;22) chromosome translocation leading to a fused BCR/ABL gene with dysregulated Abl oncogene tyrosine kinase activity (2,3). Novartis investigators have developed an inhibitor of the Abl tyrosine kinase called Imatinib, which induces complete remission in ∼95% of CML patients in chronic phase (4,5). During the past few years, other drugs affecting the activity of oncogenic proteins or the function of genes facilitating tumor growth and/or survival (for example by affecting angiogenesis), have been developed and are being used in patient treatment and clinical trials (1).
While it is very clear that tumors are initiated, for the most part, by somatic genetic alterations (1), it is also evident that many tumors display alterations in the expression of tumor suppressor genes because of epigenetic alterations, such as methylation of CpG islands in their promoters, leading to loss of function (6). Thus, therapies directed to reverse the epigenetic changes occurring in a large variety of malignancies have been developed. For example, 5-azacytidine or decitabine have been used to reactivate tumor suppressor genes silenced by methylation of promoter CpG islands (7). Histone deacetylase (HDAC) inhibitors have also been used to reactivate the expression of silenced genes (8).
Although progress has been made in identifying genetic and epigenetic causes of cancer that provide targets for therapy, several challenges remain. For example, although targeted therapies based on the identification of oncogenic mutations with causal roles in cancer have been developed, the treatment of malignancies initiated through loss of function of tumor suppressor genes is more difficult, since the lost gene function must be replaced in all the cancer cells. Identifying additional alterations that cause or contribute to malignancy is a high priority.
In 1993, Victor Ambros and colleagues discovered a gene that affected development in C. elegans, Lin 4, and found that its gene product was a small non-protein coding RNA (9).
After these seminal findings, the cloning and characterization of small, 20-22 nucleotides long, members of the non-protein coding RNA family, has led to identification of ∼1,000 microRNAs. MicroRNAs are generated in a process that typically involves the transcription of a long precursor (pri-microRNA), processing to produce a second precursor (pre microRNA) of 60-100 nucleotides, transport to the cytoplasm, and a second processing step carried out by the RNaseIII, Dicer, which results in the final miRNA products (for review see Ref. 10). In the cytoplasm, microRNA duplexes are unwound and the mature strand is incorporated into RISC (RNA-induced silencing complex). Following binding to partially complementary sites, usually in the 3′ untranslated regions (3′UTR) of mRNAs, microRNAs cause an inhibition of translation and some degree of degradation of the target mRNA (for review 11,12). Thus, microRNAs are negative regulators of gene expression, and have been found to regulate over 30% of mRNAs and to play roles in fundamental processes – such as development, differentiation, cell proliferation, apoptosis, and stress response – and range of species from C. elegans, plants and Drosophila to mammals, including humans (10,13).
In the past few years, many miRNAs have been implicated in a range of human cancers (Box 1). Both losses and gains of miRNA function have been shown to contribute to cancer development through a range of mechanisms. Here, we discuss the causes and consequences of miRNA dysregulation in cancer and the new opportunities that this knowledge provides for the development of new cancer therapeutics.
Box 1. Defining the contribution of microRNAs to cancer pathogenesis.
A straightforward approach to begin defining the contribution of microRNAs to cancer pathogenesis is by expression profiling by a variety of methods including microarrays and RT-PCR. In general it is good practice to carry out profiling by microarray and validation by RT-PCR (12). Profiling provides important information concerning the microRNAs that are dysregulated in a specific malignancy versus its normal tissue counterpart. Then it is important to discover the mechanisms of dysregulation and the potential targets of the dysregulated microRNAs. If the mechanism of dysregulation involved genetic alterations at a microRNA locus, for example a deletion or a mutation affecting processing of the microRNA or amplification, it provides strong evidence for contribution of that microRNA or microRNA cluster to cancer pathogenesis. If such genetic alterations occur quite early during the multi-step process of malignant transformation in a large fraction of tumors, it would suggest a role for that microRNA in the initiation of process, providing a valuable target for therapeutic intervention.
The discovery of microRNAs as cancer genes
During our attempts to clone a tumor suppressor gene at 13q14, a chromosomal region frequently lost in chronic lymphocytic leukemia, the most common human leukemia (14), we found that all the protein coding genes present in the region of interest were not specifically altered in CLL, suggesting that they were not involved in the disease. We then took advantage of a rare chromosome translocation involving 13q14 in CLL and mapped the breakpoint precisely to the region of loss. We also found a CLL case with a very small deletion, ∼30 kb at 13q14 (15). Interestingly, the translocation breakpoint mapped within the deleted region in the second case. These results suggested that the CLL suppressor gene was precisely within this small genomic region, where there were no protein-coding genes. We found, however, that two microRNA genes were within the deletion, miR-15a and miR-16-1, and that the translocation breakpoint cut the precursor of these two microRNA genes that reside in the same polycistronic RNA, indicating that loss of miR-15a and miR-16-1 is the long sought target of 13q14 deletion in CLL pathogenesis (15). Following this discovery, we observed loss of miR-15a and miR-16-1 in ∼70% of CLLs (15). Since most indolent CLLs exhibit loss of the genes for miR-15a and miR-16-1, we concluded that loss of miR-15a and 16-1 must be a very early event, perhaps the initiating event, in the pathogenesis of the indolent form of CLL. Indolent CLLs, however, frequently progress into an aggressive disease. A small minority of CLL cases are aggressive at presentation.
These early results suggested that miR-15a and miR-16-1 can function as tumor suppressor genes in CLL, since their loss is associated with the development of the indolent form of CLL (15). This finding prompted us to map the chromosomal location of all known microRNA genes, resulting in another surprising finding: many microRNA genes are located in chromosomal regions frequently involved in chromosomal alterations such as deletion or amplification, in many types of human cancers, for which extensive studies have failed to find activation of oncogenes or loss of tumor suppressor genes, suggesting that the role of microRNAs in cancer could extend far beyond CLL (16). Thus, the mapping of microRNA genes to specific chromosomal locations provided important clues for possible roles of those microRNA genes in cancer (16) (Figure 1).
Figure 1. MicroRnA genes map to chromosomal regions that are involved in alterations in human cancer.

The 24 human chromosomes are shown, with stars indicating the locations of microRNA-gene-containing regions that are implicated in cancer (one star = one microRNA gene). Many microRNAs map to chromosome regions that are involved in rearrangements in human cancer. For example, miR-15-a and miR-16-1 map to 13q14, a region that is frequently deleted in chronic lymphocytic leukaemia. miR-29a and miR-29b map to 7q32, a region that is deleted in myelodysplastic syndrome and acute myeloid leukaemia. The let-7g–let-7a1 cluster maps to 3p2, the let-7f-1–let-7d cluster maps to 9q22.3, let-7a-2 maps to 11q23-q24 and let-7c maps to 21q21 —all of these regions are involved in deletions in a range of solid malignancies, including lung, urothelial, breast, ovarian and cervical cancers.
These clues were confirmed by the expression profiling data of many different types of tumors (Box 1). For example, members of the let-7 family of microRNAs that map to chromosome regions deleted in multiple tumors were found to be lost in multiple different malignancies, including those of lung, breast, urothelial, ovarian and cervical cancer. Interestingly, an additional locus at 3q26 carries homologues of miR-15a and miR-16-1: miR-15b and miR-16-2. These microRNAs however, are expressed at very low levels in normal CD5 positive B cells (15). They could, however, be involved in other human malignancies. Similarly, let-7 family members map on different human chromosomes (16), are differentially expressed in different tissues and are involved in different human tumors (17,18). The miR-17-92 cluster that maps to a region amplified in lymphoma was found to be overexpressed in many different tumors (19). Consistent changes in microRNA expression were also observed in tumors without evident specific cytogenetic abnormalities, suggesting that pathways commonly dysregulated in human cancer could directly affect and dysregulate microRNA expression (19). A corollary to this interpretation is that microRNAs could be the downstream targets and effectors of pathways that regulate important processes such as development, proliferation, differentiation, apoptosis, stress response, etc. (19). If critical pathways can be dysregulated by alterations in multiple genes, many of which may not be easily targetable by small molecules and many of which may be lost, it becomes reasonable to speculate that their downstream microRNAs may become the suitable and, perhaps optimal, targets for therapeutic intervention. Thus, if the dysregulation of a critical pathway results in loss of expression of specific microRNA(s), these microRNA(s) could be used as therapeutics. If it results in overexpression of specific microRNAs, complementary (antisense) molecules could be used for treatment.
MicroRNAs as tumor suppressors
Every malignancy is heterogeneous because cancer development is the result of a multi-step process involving multiple alterations in oncogenes and tumor suppressor genes occurring during time, often years. This heterogeneity is challenge in terms of understanding the events that initiate cancer development and in identifying potential new therapeutic targets. In terms of therapies, it is very important to be able to target the earliest genetic alterations, since these alterations will be present in 100% of the cancer cells, while later alterations may be present only in a fraction of the malignant cells. Genetic analysis provides a way to define early targets. Since loss of miR-15/16 was detected in the great majority of indolent CLLs and in aggressive CLLs originated from the indolent form (15), it seems logical to infer that such genetic alterations must occur quite early during the process of leukemogenesis. This interpretation was validated by the discovery of mutations affecting the expression of these microRNAs in patients with CLL and of a mouse CLL model that also carries mutation in the same microRNA locus (20,21). Further validation is provided by the development of indolent CLL in miR-15/16 knock out mice (manuscripts in preparation).
It is becoming clear that the sequence of genetic events leading to the development of malignancy is not rigid. For example, most indolent CLLs develop a 13q14 deletion that affects the miR-15/16 locus and these leukemias over time may lose microRNA genes at 11q23 or the TP53 gene at 17p13, becoming aggressive through these changes. Aggressive CLLs, however, can start with an 11q23 deletion and then acquire a miR-15/16 deletion, suggesting that both such alterations are necessary for development of an aggressive CLL. Similarly, in the aggressive CLL mouse model, the TCL1 transgenic mouse, dysregulation of TCL1, which occurs in all human aggressive CLLs with 11q23 deletion, is followed by loss of expression of miR-15/16. The fact that these two different alterations can occur in alternative sequence, stresses even more the role of both alterations in the pathogenesis of the aggressive form of CLL. It seems likely that similar situations may occur in other malignancies.
Sequencing of microRNAs dysregulated in CLL revealed mutations in the precursor of miR-15a and miR-16-1 that affected processing of the microRNAs, in a very small fraction of CLL cases (20). Thus, not only deletions but also mutations may lead to microRNA loss of function. Interestingly, some of these mutations occurred in the germline, confirming that loss of function of miR-15a and 16-1 contributes to the development of CLL (20) and indicating that loss of miR-15a and 16-1 expression in the germline leads to a familial form of CLL. Raveche et al., have studied the genetics of an indolent form of CLL occurring in the NZB mouse strain (21). This mouse strain develops an indolent form of CLL late in life, as in humans (21). Having mapped the disease to mouse chromosome 14 in a region homologous to 13q14 in humans, the investigators sequenced miR-15/16 and found a mutation 6, not 7 as in humans (20,21), nucleotides 3′ to miR-16-1 that also seems to affect processing of the microRNA precursor (21). Thus, germline mutations affecting the processing of miR-15a and miR-16-1 can cause the indolent form of CLL in both mouse and man. The sequencing of microRNAs in human CLLs also revealed mutations, some germline, some somatic, in other microRNA loci, including miR-29 family members (20).
Targets of miRNA tumor suppressors
In 2005, Slack and his associates discovered that let-7 family members negatively regulate the expression of the RAS oncogene, indicating that loss of let-7 family members results in the constitutive overexpression of RAS, an oncogene that contributes to the pathogenesis of several types of human tumors (22). The following year, Cimmino et al. discovered that miR-15a and miR-16-1, present in the same polycistronic RNA, target BCL2 (23), an oncogene that inhibits apoptosis and is the culprit in another indolent B cell malignancy, follicular lymphoma, due to a consistent t(14;18) chromosomal translocation (24,25). Thus, it became clear that loss of microRNAs could contribute to malignant transformation through the dysfunction of cellular oncogenes such as RAS and BCL2. More recently, other targets of these microRNAs have been discovered, providing additional evidence of their role in cancer pathogenesis. For example, let-7 was also found to target HMGA2, a gene that is dysregulated in a variety of human tumors, including liposarcoma (26).
It is very clear that microRNAs have many different targets, dozens in some cases, hundreds in others, but depending on the cells involved, it seems likely that only a small number of them play a critical role in cancer pathogenesis. This has been validated by the fact that alterations in the expression of specific protein targets leads to the same phenotype. In fact, approximately 4% of CLLs carry translocations involving the BCL2 gene and an immunoglobulin locus that deregulate the oncogene (27,28). Thus, overexpression of BCL2 in CLL seems to be the critical event in the initiation of most CLLs.
Target identification can take advantage of algorithms developed for target prediction that must, however, be followed by experimental validation since in many cases the predicted targets are not real targets. Since the number of predicted targets is large, this approach is rather laborious. Alternatively, proteomics can be used to identify all the protein products that are affected by the introduction and expression of a specific microRNA in different cell types (29).
A multitude of miRNA tumour suppressors
Since these early findings, other microRNAs functioning as tumor suppressors have been discovered (see Table 1).
Table 1. MicroRNAs functioning as oncogenes or tumor suppressor genes in human cancer.
| MicroRNAs | Dysregulation in | Function | Validated Targets | Oncogene (Onc)/Tumor Suppressor (TS) | References |
|---|---|---|---|---|---|
| miR-15-a | Loss in CLL | Induces apoptosis and inhibits tumorigenesis | BCL2, Wt1 Rab9B, Mage83 | TS | 15, 20, 23, 30, 31, 32 |
| miR-16-1 | Loss in prostate cancer and loss in multiple myeloma | “Same” | “Same” | ||
| Let 7 (a,b,c,d,e,f,g,i) | Loss in lung and breast cancer and in a variety of solid and hematopoietic malignancies | Induces apoptosis and inhibits tumorigenesis | RAS, MYC, HMGA2 | TS | 22, 26, 33, 34 |
| miR-29 (a, b, c) | Loss in aggressive CLL,AML(11q23), lung and breast cancer and colangocarcinoma | Induces apoptosis, and inhibits tumorigenicity. Reactivates silenced tumor suppressor genes | TCL1. MCL1, DNMTs | TS | 30, 35, 36, 37 |
| miR-34 | Loss in pancreatic, colon, breast and liver cancer | Induces apoptosis | CDK4, CDK6 Cycline E2, EZF3, MET | TS | 33, 39, 40 |
| MiR-145 | Loss in breast cancer | Inhibits proliferation and induces apoptosis of breast cancer cells | ERG | TS | 41 |
| miR-221/222 | Loss in erythroblastic leukemia | Inhibits proliferation in erythroblasts | KIT | TS | 30 |
| miR-221/222 | Overexpression in aggressive CLL, thyroid carcinoma, hepatocellular carcinoma | Promotes cell proliferation and inhibits apoptosis in a variety of solid malignancies | P27, p57, PTEN, TIMP3 | ONC | 42, 43, 44 |
| miR-155 | Upregulated in aggressive CLL, BL, and lung, breast and colon cancers | Induces cell proliferation and leukemia/lymphomas in mice | MAF, SHIP1 | ONC | 45, 46, 47, 48, 49 |
| Cluster miR-17-92 | Upregulated in lymphomas and in breast, lung, colon, stomach and pancreas cancers | Induces proliferation | E2F1, Bim, PTEN | ONC | 19, 49, 50, 51, 52 |
| miR-21 | Upregulated in glioblastomas, and in breast, colon, pancreas, lung, prostate, liver and stomach cancer, AML(11q23); aggressive CLL | Inhibits apoptosis and increases tumorigenicity | PTEN, PDCD4, TPM1, TIMP3 | ONC | 41, 46, 53, 54, 55, 56, 57, 58, 59, 60, 61 |
| miR-372/373 | Upregulated in testicular tumors | Promotes tumorigenicity in cooperation with RAS | LATS2 | ONC | 62 |
Several of the microRNAs described as suppressors have been found to be deleted or mutated in a variety of human malignancies. For example, loss of miR-15a and miR-16-1 has also been observed in prostate cancer and in multiple myeloma (Table 1). MiR-29s have found to be deleted in a fraction of MDSs and AMLs. MiR-29 precursors have also found to be mutated in CLL (20). Let-7s map to regions of the genome involved in deletions in a number of solid and liquid malignancies (16). In addition, miR-15/16, miR-29s and Let-7s can be found to be silenced by methylation in some cases (12). In many cases, the involvement of microRNAs as suppressors has been suggested by profiling studies (Box 1). Thus alterations in microRNA genes expression have been observed in a large variety of tumor types.
Importantly, microRNAs should not be described as oncogenes or tumor suppressor genes, unless the tissue or cell type involved in their action is specified. For example, miR-221 and miR-222 target an oncogene, KIT, and inhibit growth of erythroblastic leukemia (30), thus functioning as a suppressor in erythroblastic cells, but they also target at least four important tumor suppressors, PTEN (phosphotase and tensin homolog), p27, p57 and TIMP3, and function as oncogenic microRNAs by suppressing these tumor suppressors in a variety of human solid tumors (Table 1)(41). Thus, before describing a microRNA as a tumor suppressor or an oncogene, it is necessary to specify in which cell or tissue, since cellular context is critical for the function of microRNAs (12).
MicroRNAs as oncogenes
Overexpression of microRNAs has also been observed in a large variety of human tumors, and there is evidence that a number of these function as oncogenes.
miR-155 and the miR-17-92 cluster
The first miRNAs to be found overexpressed in cancers were miR-155 and the miR-17-92 cluster of microRNAs (47-50) They are amplified at the DNA level in a variety of B cell lymphomas 47,48). The microRNA gene encoding miR-155 is embedded in a host non-protein coding genomic region named B cell integration cluster (BIC) and is located on chromosome 21q23 (47). Several groups have shown that miR-155 is overexpressed in aggressive CLL (20), pediatric Burkitt lymphoma (45), Hodgkin disease (46), primary mediastinal non-Hodgkin lymphoma (46), AML (61), lung cancer (19) and breast cancer (41). Less is known about the mechanisms of dysregulation of miR-155, with the exception of cases in which the miR-155 gene is amplified, as in lymphomas.
The miR-17-92 cluster, comprising six microRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b1 and miR-92-1), is located in a region of 800 base pairs in the non-protein coding gene C13arf25 at 13q31.3 (49). This chromosomal region was found to be amplified in follicular lymphoma and diffuse large B cell lymphoma (49). Overexpression of this cluster has been observed in a large variety of human solid tumors, including breast, colon, lung, pancreas, prostate and stomach cancers, in addition to lymphoma (19). MicroRNAs of this cluster promote proliferation, inhibit apoptosis, induce tumor angiogenesis, and cooperate with MYC to accelerate the development of lymphomas (53). Interestingly, this cluster is transactivated by MYC, an oncogene that is frequently dysregulated in malignancies of various types (50; see next section for a more detailed discussion of the cooperation of these miRNAs with MYC). Recent studies of gain and loss of function indicated that this cluster is essential for B cell proliferation and that overexpression in B cells induces a lymphoproliferative disorder in mice (52). This cluster affects cell cycle progression and proliferation through its regulation of the E2F transcription factors (50,51). Genes of the E2F family encode proteins that activate multiple genes involved in cell cycle progression from G1 to S phase (50-52). It has also been shown that E2F1 and E2F3 proteins can activate microRNA genes within the cluster, contributing to a regulatory loop (50,51); microRNAs encoded within the cluster can also downmodulate important targets such as the tumor suppressor PTEN and p21 and the apoptotic protein Bim (50).
The experiments attempting to show the oncogenic potential of the miR-17-92 cluster have not dissected, however, the role and contribution of each member of the cluster to malignant transformation. Experiments of dysregulation of each member of the cluster in transgenic mice will provide important insights into the role of each of the microRNAs contained in the cluster in tumor development. In addition, investigation of the processing of each member of the cluster in different cell types may provide interesting novel findings concerning mechanisms of their dysregulation.
The definitive proof that a microRNA can contribute directly to the pathogenesis of a malignancy has been provided by the development of transgenic mice with targeted overexpression of miR-155 in B cells (48). Mice that carried a miR-155 transgene under control of the immunoglobulin Eμ enhancer expressed high levels of miR-155 in B cells and developed, at first, a polyclonal expansion of large pre B cells, and then frank leukemia or high grade lymphoma, all transplantable into syngeneic mice. Since a few months elapse before the mice develop frank malignancy, it is likely that additional genetic alterations must occur to convert the pre-malignant into malignant cells (48). These results indicate that dysregulation of a single oncogenic microRNA can lead to the development of a malignant tumor. Overexpression of miR-155 and loss of let 7 have been found to be poor prognostic indicators for stage 1 lung adenocarcinoma (33).
A multitude of potential oncogenic microRNAs
As indicated in Table 1, several additional microRNAs have been found to be overexpressed in a variety of human tumors by profiling and may be candidate oncogenes. The formal proof of this, however, awaits results of dysregulation experiments in animal models. These overexpressed microRNAs have been found to target important tumor suppressors. For example, miR-21 has been found to target PTEN, PDCD4 (programmed cell death gene 4), TIMP1 and TIMP3 (44); the miR-17-92 cluster PTEN, E2F1 and Bim and miR-221/222 PTEN, p27, p57 and TIMP3 (Table 1).
Of particular interest is the fact that miR-21 was found to be overexpressed in most of the different solid tumors analyzed (19). In fact, overexpression of miR-21 has been observed in a large variety of human tumors, including glioblastoma, breast, lung, colon, pancreas, liver, stomach and prostate cancer and AML with 11q23 alteration (19, 41,51,54-61). In addition, overexpression of miR-21 has been observed during the development and progression of colorectal cancer (60). Inhibition of miR-21 by antisense oligonucleotides in glioblastoma resulted in cell death (56). Similarly, inhibition of miR-21 caused apoptosis in liver and breast cancer cells (57). As mentioned, miR-21 has been found to inhibit several tumor suppressor genes. MiR-221/222 have been found to be overexpressed in many hematopoietic and solid malignancies such as aggressive CLLs (20), acute leukemias (61), papillary thyroid carcinoma, hepatocellular carcinoma (43), colon, pancreas and prostate cancer (32,59,60).
MicroRNA dysregulation by transcription factors
As mentioned above, the miR-17-92 cluster, that comprises six microRNAs, is frequently amplified in lymphoma. Members of this cluster play a role in lung development and in the regulation of differentiation in hematopoietic cells. O'Donnell et al. have shown that the miR-17-92 cluster is transactivated by MYC, an oncogene that is frequently dysregulated in cancer (63). Dysregulation of MYC by chromosomal translocation and juxtaposition to immunoglobulin enhancer is the cause of Burkitt lymphoma (1). Two recent studies of gain and loss of function showed that this cluster is essential for B-cell proliferation and that a modest overexpression in B cells induces a lymphoproliferation disorder in mice (51,64). This cluster promotes cell cycle progression and proliferation, at least in part through regulation of E2F transcription factors that regulate cell cycle progression. This cluster also has anti-apoptotic effects through its inhibition of Bim, PTEN and p21 (65). Members of the miR-17-92 cluster have homologs in two other clusters, one on chromosome 7 within the MCM7 gene (miR-106b, miR-93 and miR-25) and the other on the X chromosome (miR-106a, miR18b, miR-206, miR-196-2, miR-82a-2 and miR-363). Figure 2 shows the genomic structure of the 3 microRNA clusters on chromosome 13, 7 and X. MYC induces the expression of the cluster on chromosome 7 and 13, as well as E2F1 that regulates MCM7 and C13arf25 genes that host the two clusters. At the same time, miR-25 and miR-92a-1 downregulate the pro-apoptotic Bim gene, while MYC induces it (65). Inactivation of the transforming growth factor β (TGF β) tumor suppressor pathway is an important step in the development of a variety of human tumors (66). Two of the 3 clusters (miR-106b-25 and miR-17-92) function as key modulators of TGF β signaling in many tumors, interfering with cell cycle arrest and apoptosis when overexpressed in malignant cells (65) (Figure 2).
Figure 2. Role of miR-106b-25 and miR-17-92 clusters in the control of transforming growth factor β(TGFβ) signaling.
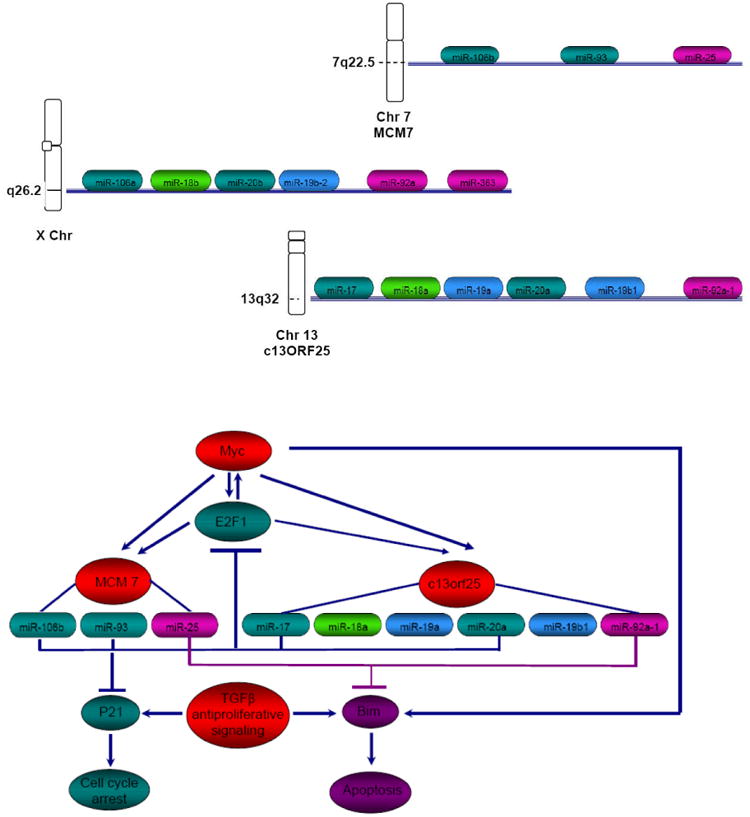
This figure shows the genomic organization of the three related microRNA clusters mapping on chromosomes (Chr) 13,7 and X. The homology between the different individual microRNAs is indicated by the same colors. MiR-106b-25/miR-17-92 regulates the Myc/E2F1/TGFβ network. Both MCM7 and c13orf25 microRNA host genes are induced by Myc and E2F1. Elevated expression of microRNAs of the miR-106 family consisting of miR-106b, miR-92, miR-17-5p and miR-20a causes reduced expression of p21, a cell cycle inhibitor, thus inhibiting TGFβ dependent cell cycle arrest. Elevated expression of miR-25/miR-92 affects TGFβ-induced apoptosis through inhibition of Bim expression. Since overexpression of Myc induces apoptosis, possibly through Bim induction, elevated expression of miR-25/92 can avoid Myc induced apoptosis in cancer cells.
It seems likely that normally these two clusters dictate the pace of TGF β response, repressing Bim and p21 expression in proliferating cells and allowing sequential expression of p21 and Bim as cells differentiate and undergo TGF β-induced cell cycle arrest and apoptosis. The work of Ventura et al. shows that the two clusters control apoptosis, since the miR-17-92/miR-106b-25 double knockout mice exhibit a much more severe phenotype compared to miR-17-92 single knockout mice (51). In cancer development, dysregulation of miR-106b-25 and/or miR-17-92 would compromise this regulatory mechanism, providing an escape from TGF β-induced cell cycle arrest and apoptosis.
Recently, a number of reports revealed that the miR-34 family (miR-34a,b,c) is induced directly by the p53 tumor suppressor (38-40) and suggested that some p53 effects could be mediated by these microRNAs. By using various cellular models, the authors compared microRNA expression in cells with high or low p53 expression and found that the expression levels of p53 correlated with the levels of expression of miR-34 family members. In addition, chromatin immunoprecipitation experiments showed that p53 binds to the miR-34 promoters (39,40). Introduction of miR-34s into cells that have lost their expression resulted in cell cycle arrest. To assess the role of each miR-34 family member, it will be necessary to knock out the expression of each member in different mouse tissues to determine whether loss of expression leads to malignant transformation or to higher susceptibility to a variety of tumors.
Breast cancer is the second leading cause of death among women in the western world and its molecular pathogenesis is still not completely understood. Several lines of evidence indicate that Estrogen Receptorα (ERα-) breast tumors, highly aggressive and non responsive to hormonal therapy, arise from ERα positive (ERα+) precursors through different molecular pathways. High levels of miR-221 and -222 were found in estrogen receptor negative (ER-) cells and breast tumor samples. MiR-221/222 overexpression in ER+ cells suppressed ERα protein and luciferase assays confirmed that estrogen receptor α (ERα) is a target of miR-221/222. ERα was also found to negatively regulate expression of miR-221 and -222 by promoter binding (67). Thus, silencing ERα for example by methylation, or by dysregulating miR-221/-222 through the activation of pathways involved in oncogenesis, such as the Met pathway, results in the constitutive activation of miR-221/222 and inhibition of the tumor suppressors p27, p57, PTEN and TIMP3, contributing to the development of the “invasive” phenotype characteristic of frankly malignant cells (44). This regulatory feedback loop seems to be involved in the development of estrogen receptor negative breast cancers (66).
Thus, microRNAs can be dysregulated by transcription factors and therefore genetic or epigenetic alterations resulting in dysregulation of transcription factors can cause microRNA dysregulation, contributing to malignant transformation (Table 2). Volinia et al. have defined microRNA expression signatures that distinguish cancerous from normal tissues (19). Interestingly, they observed that some microRNA genes were dysregulated, not just in one tumor type but in many, suggesting that these microRNAs may be downstream targets of pathways that are commonly dysregulated in cancer (19).
Table 2. Consequences of microRNA dysregulation in human cancer.
| microRNA Dysregulation | Targets | |
|---|---|---|
| ↑ microRNA overexpression | ↓ Tumor suppressors | ↓ for example, PTEN, p22, p57, TIMP3, PDCD4 |
| ↓ microRNA loss | ↑ Oncogenes | ↑ for example, BCL2, MCL1, RAS, HMGA2, MYC, MET |
| ↓ microRNA loss | ↑ DNA methyltransferases | ↓ tumor suppressors; for example, p16, FHIT, WWOX |
| ↓ microRNA loss | ↑ Chromatin silencers | ↓ tumor suppressors |
If microRNAs are downstream targets of pathways that are commonly dysregulated in human cancer (see Box 1), they become excellent targets for therapeutic intervention. It is known that proliferative signals lead to the activation of the MYC transcription factor that regulates, positively and negatively, the expression of a large number of protein coding genes and microRNA genes, including the miR-17-92 cluster (positively) and miR-15/16 (negatively). Thus, it seems quite possible that the mechanism of action of the activated oncogene is, at least in part, due to microRNA dysregulation (where overexpression of miR-17-92 causes downregulation of tumor suppressors and underexpression of miR-15/16 causes upregulation of oncogenes such as BCL2 and MCL1). Thus, genetic or epigenetic alterations in protein coding cancer genes or in microRNA genes may have similar consequences. Until now it has not been possible to target the overexpression of MYC in tumors with drugs, but it is possible to target the microRNAs that are dysregulated by MYC, for example by treating the cancer cells with anti-miR-17-92 or with miR-15/16. Recently, Bonci et al. have shown that the miR-15a/miR16-1 cluster can control prostate cancer by targeting multiple oncogenic activity (68). More generally, in tumors with alterations in protein coding cancer genes, it should be possible to cause tumor regression by using microRNAs and anti-microRNAs. In fact, recently, Kota et al. have caused regression of Myc induced hepatocellular carcinomas in mice by targeting a microRNA induced by Myc (69).
Roles of MicroRNAs in Cancer Epigenetics
Altered patterns of epigenetic modifications, in particular methylation of CpG islands, in the promoter regions of tumor suppressors is a common finding in many tumors (for review see 70). Such changes are often accompanied by aberrant patterns of histone modification. Thus silencing of tumor suppressors in cancer can be caused not only by deletion and mutations, but also by epigenetic changes.
MiRNAs as targets of epigenetic changes
The most studied epigenetic changes in cancer cells is the methylation of cytosines within the dinucleotide CpG in DNA (70). Such methylable sites, known as CpG islands, are preferentially located in the 5′ region (promoter, 5′ untranslated region and exon 1) of many genes, are non methylated in normal cells and are transcribed in the presence of the appropriate transcription factors. Methylation of the CpG island of tumor suppressors results in their silencing and contributes to malignant transformation (70). As mentioned above, the expression of microRNAs can be affected by genetic changes, such as deletion, gene amplification and mutation, and by transcription factors. In addition, the expression of microRNAs can be affected by epigenetic changes such as methylation of the CpG islands of their promoters. Saito et al. reported that miR-127 is silenced by promoter methylation in bladder tumors and that its expression could be restored by using hypomethylating agents such as 5-azacytidine (71). This microRNA targets BCL6, an oncogene involved in the development of diffuse large B cell lymphoma. Thus, silencing of miR-127 may lead to BCL6 overexpression (71). Other investigators have described additional microRNAs that are silenced by methylation in various cancers and can be reactivated by hypomethylating agents.
MiRNAs as regulators of epigenetic changes
MicroRNAs can also regulate enzymes involved in methylation of CpG islands of tumor suppressor genes; Fabbri et al showed that microRNAs of the miR-29 family target de novo DNA methyltransferases, DNMT3A and 3B, where overexpression of DNMTs is a poor prognostic indicator (36). Interestingly, introduction of miR-29 family members into lung cancer cell lines caused demethylation of the CpG islands in the promoter region of tumor suppressor genes, allowing their reactivation and loss of tumorigenicity of the lung cancer cells (36).
Similarly, a recent study by Garzon et al. has shown that miR-29s target not only DNMT3A and 3B, but also, indirectly, the DNMT1 maintenance DNA methyltransferase (72). Introduction of miR-29 into acute myelogenous leukemia cells resulted in loss of expression of the oncogene MCL1 and the three DNA methyltransferases, as well as reactivation of the p16 tumor suppressor (72); thus, treatment of the malignancies with miR-29 family members caused reversion of the epigenetic changes contributing to malignant transformation in human cancers such as lung cancer and AML.
Garzon et al. (73) have also shown that expression of the miR-29 cluster encoded at chromosome 7q32, is downregulated in acute leukemias with deletions of 7q, an alteration frequently observed in myelodysplastic syndrome (MDS) and AML. Thus, miR-29s could be considered as candidate drugs for the treatment of these malignancies. Since loss of miR-29s is observed in patients with MDS and AML, an alternative therapeutic strategy could be to stratify patients on the basis of expression of miR-29s and then treat them with 5-azacytidine or decitabine that would lead to demethylation and reactivation of the tumor suppressors. The prediction is that only those that have lost expression of miR-29s will respond. Other microRNAs are predicted to target the DNMTs responsible for methylation of CpG islands of tumor suppressors. We already have preliminary evidence that miR-148a and b, which are lost in a variety of human tumors, also target DNMTs (C.M. Croce, unpublished results). MicroRNAs are also predicted to target HDAC and other proteins involved in chromatin structure and function (74); therefore, it will not be surprising if dysregulation of these microRNAs contributes to cancer pathogenesis.
Thus, these observations indicate that alterations in the expression of microRNAs could be responsible for some of the epigenetic changes observed in cancer cells, providing novel targets for cancer therapy (Table 2).
Therapies based on MicroRNA or anti microRNA
–The studies described in the sections above have highlighted a range of examples in which microRNA genes are involved or implicated in the pathogenesis of human cancer. The fact that several microRNA genes are dysregulated in multiple types of cancer, also suggests that critical pathways involved consistently in tumorigenesis may have microRNA genes as downstream targets (19). Thus, microRNAs, in tumors in which they are lost, or antimicroRNAs (75), in tumors in which the cognate microRNAs are overexpressed, could be considered drugs capable of inducing apoptosis and/or cell cycle arrest in cancer cells that depend on microRNA dysregulation for growth and survival.
Introduction of microRNAs (if they are lost in tumors) or antisense microRNAs (if they are overexpressed in tumors) into tumors can be achieved in many different ways. They can be introduced into vectors such as adenoviruses or adeno-associated viruses (AAV), or retroviruses that can then be used to deliver their microRNA cargo to the tissue of interest (69). An alternative is the use of microRNAs or antimicroRNAs either unmodified or modified to increase their half-life in vivo. The issue of delivery is important for therapeutics. Some tissues, however, are quite good to uptake microRNAs; for example liver, bone marrow, spleen and kidney. In these cases it may not be necessary to use vectors or modified microRNAs and antimicroRNAs. In other cases where the natural uptake is modest, it may be necessary to use vectors or other delivery systems such as liposomes.
Experiments in mice clearly indicate that microRNAs or antimicroRNAs can be used for therapy. For example, miR-15/16 have been used to suppress the growth of leukemic cells lacking miR-15/16 (29), and miR-29s have been used to suppress tumorigenicity of lung cancer cells (36). MiR-29s have also been used to suppress the growth of human AMLs that show loss of these microRNAs and overexpression of MCL-1 and DNMTs in mice (73). Thus, it is quite likely that targeted therapies exploiting microRNA dysregulation in cancer can be developed for treatment of human malignancies. In addition, therapeutic experiments in mice, by using microRNAs or anti microRNAs, suggest that these treatments have quite modest side effects, if any, similarly to Imatinib treatment of CML patients. MicroRNA profiling may be used for patient stratification and for treatment selection, since microRNAs have also been found to cause drug resistance. It is interesting that some, perhaps many, of the epigenetic changes observed in cancer may be due to microRNA dysregulation. Thus, microRNAs could be used to reverse those changes. In fact, as mentioned, loss of microRNAs of the miR-29 family was discovered in lung cancer, AML and aggressive CLL. Such microRNAs have been found to target the DNA methyltransferases and to be able to reactivate silenced tumor suppressor genes (36,73). DNA methyltransferases are also targeted by other microRNAs lost in human tumors. HDAC and other proteins involved in chromatin structure and function are also predicted to be regulated by microRNAs(74). Thus, it is not farfetched to imagine that microRNAs will be used for reversing changes in chromatin structure that are associated with tumorigenesis. Furthermore, the knowledge of which microRNAs affecting epigenetic changes are lost in cancer, will allow better patient stratification and the use of already available drugs to revert those changes. For example, since miR-29s are lost in AML with 7q- and since AML cells with 7q- in vitro can be induced to re-express silenced tumor suppressor genes with miR-29, it should be possible to stratify AML patients on the basis of miR-29 expression to determine whether only those with loss of miR-29 respond to the methylation inhibitor 5-azacytidine and decitabine.
Conclusions and Future Challenges
Since the discovery of miR-15a and miR-16-1 deletions in CLL (15), the work of many laboratories around the world has shown microRNA dysregulation in all tumors studied, including the most common, such as lung, breast, prostate and gastrointestinal cancers. Such dysregulation, like dysregulation of oncogenes and tumor suppressor genes, can be caused by multiple mechanisms such as deletion, amplification, mutation, transcriptional dysregulation, epigenetic changes, etc. Since microRNAs have multiple targets, their function in tumorigenesis could be due to their regulation of very few specific targets, possibly even one, or many targets. A future challenge will be to identify all the targets of the microRNAs involved in cancer and establish their contribution to malignant transformation. An additional challenge will be the identification of all the microRNAs that are dysregulated by pathways that are consistently dysregulated in various types of human cancers. This point is of particular potential importance, since instead of focusing on the specific alterations in protein coding oncogenes or tumor suppressor genes that are components of the pathways dysregulated in cancer, that may be difficult to be addressed therapeutically, we may focus on their downstream microRNA targets. If these microRNA targets are critical for the expression of the malignant phenotype and the cancer cells depends on their dysregulation for proliferation and survival, we can expect that the use of microRNAs or antimicroRNAs will result in tumor regression. Genomic analyses for alteration in microRNA genes or for copy number alterations in various human tumors by deep sequencing is in progress but has not been completed. These studies could provide additional information concerning the involvements of microRNAs in cancer and in many other diseases. During the past few years, we have observed a shift from conventional chemotherapy to targeted therapies. MicroRNAs and antimicroRNAs will contribute extensively to the latter.
Glossary
- RNA-induced silencing complex (RISC)
(RNA-induced silencing complex). An Argonaute protein–small-RNA complex that inhibits translation of target RNAs through degradative or non-degradative mechanisms
- CpG islands
A sequence of at least 200 bp with a greater number of CpG sites than expected for its GC content. These regions are often GC rich, typically undermethylated, and are found upstream of many mammalian genes
- Indolent
In medical terms, slow to develop or heal
- Syngeneic
macrophages (immune cells that engulf foreign particles) that are transferred between genetically identical mice
- Liposomes
Artificial lipid vesicles. Liposomes fuse with the cell membrane to deliver their contents, such as DNA for gene therapy
References
- 1.Croce CM. Oncogenes and Cancer. N Engl J Med. 2008;358:502–511. doi: 10.1056/NEJMra072367. [DOI] [PubMed] [Google Scholar]
- 2.Groffen J, et al. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- 3.Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukemia. Nature. 1985;315:550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 5.Roche-Lestienne C, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to ST1S71, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–1018. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 6.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 8.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res. 2009;15:3970–3977. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
- 9.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with anti-sense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. This paper describes the discovery of the first microRNA gene. [DOI] [PubMed] [Google Scholar]
- 10.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 11.Esquela-Kerscher A, Slack FJ. Oncomirs-microRNAs with a role in cancer. Nature Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. An excellent review on the role of microRNAs in human malignancies. [DOI] [PubMed] [Google Scholar]
- 12.Calin GA, Croce CM. microRNA signatures in human cancers. Nature Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 13.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 14.Bullrich F, et al. Characterization of the 13q14 tumor suppressor locus in CLL. Identification of ALT1, an alternative splice variant of the LEV2 gene. Cancer Res. 2001;61:6640–6648. [PubMed] [Google Scholar]
- 15.Calin GA, et al. Frequent deletions and downregulation of microRNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Nat Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. The first evidence of alterations of microRNA genes and dysregulation of microRNA gene expression in human disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calin GA, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Nat Acad Sci USA. 2004;101:2944–3004. doi: 10.1073/pnas.0307323101. The first evidence that many microRNA genes are located in genomic regions involved in alterations of human cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esquela-Kerscher A, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–764. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- 18.Chin LJ. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volinia S, et al. MicroRNA expression signature of human solid tumors defines cancer gene targets. Proc Nat Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calin GA, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. The first evidence of somatic and germ line mutations in microRNA genes in disease. [DOI] [PubMed] [Google Scholar]
- 21.Raveche ES, et al. Abnormal microRNA-16 locus with symmetry to human 13q14 linked to CLL in NZB mice. Blood. 2007;109:5078–5086. doi: 10.1182/blood-2007-02-071225. The first evidence of a germ line mutation in a microRNA precursor predisposing to a malignancy in the mouse. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. The first evidence that a microRNA targets an oncogene. [DOI] [PubMed] [Google Scholar]
- 23.Cimmino A, et al. MiR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Nat Acad Sci USA. 2006;102:13944–13949. doi: 10.1073/pnas.0506654102. The first evidence that microRNAs target an oncogene involved in the inhibition of apoptosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsujimoto Y, et al. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 25.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the Bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 26.Lee YA, Dutta A. The tumor suppressor microRNA let-7 represses HMGA2 oncogene. Genes Develop. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adachi M, Cossman J, Longo D, Croce CM, Tsujimoto Y. Variant translocation of the bcl-2 gene to immunoglobulin lambda light chain gene in chronic lymphocytic leukemia. Proc Nat Acad Sci USA. 1989;86:2771–2774. doi: 10.1073/pnas.86.8.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adachi M, Tefferi A, Greipp PR, Kipps TJ, Tsujimoto Y. Preferential linkage of bcl-2 to immunoglobulin light chain gene in chronic lymphocytic leukemia. J Exp Med. 1990;171:559–564. doi: 10.1084/jem.171.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calin GA, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Nat Acad Sci USA. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Felli N, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via Kit receptor downmodulation. Proc Nat Acad Sci USA. 2005;102:18081–18086. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roccaro AM, et al. MicroRNAs 15a and 16-1 regulate tumor proliferation in multiple myeloma. Blood. 2009 doi: 10.1182/blood-2009-01-198408. Published on line April 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, Yfantis HG, Stephens RM, Croce CM. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yanaihara N, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 34.Sampson VB, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–9770. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 35.Malt JL, et al. miR-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fabbri M, et al. MicroRNA family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Nat Acad Sci USA. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. The first evidence that microRNAs can affect epigenetic changes and can cause reactivation of silenced tumor suppressor genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pekarsky Y, et al. TCL 1 expression in CLL is regulated by miR-29 and miR-181. Cancer Res. 2008;66:11590–11593. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 38.He L, et al. A microRNA component of the p53 tumor suppressor network. Nature. 2007;447:11300–11304. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raver-Shapiro W, et al. Transcriptional activation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;29:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang TC, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iorio M, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 42.le Sage C, et al. Regulation of the p27kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fornari F, et al. MiR-221 controls CDKNIC/p57 and CDRNIB/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27:5651–5660. doi: 10.1038/onc.2008.178. [DOI] [PubMed] [Google Scholar]
- 44.Garofalo M, et al. MiR-221 and 222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009 doi: 10.1016/j.ccr.2009.10.014. in press. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes, Chrom Cancer. 2004;39:167–169. doi: 10.1002/gcc.10316. [DOI] [PubMed] [Google Scholar]
- 46.Kluiver J, et al. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuses large B cell lymphomas. J Pathol. 2005;207:243–249. doi: 10.1002/path.1825. [DOI] [PubMed] [Google Scholar]
- 47.Eis PS, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costinean S, et al. Pre B cell proliferation and lymphoblastic leukemia/high grade lymphoma in Eμ -miR-155 transgenic mice. Proc Natl Acad Sci USA. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. This paper provides the first direct evidence that the dysregulation of a single microRNA can lead to cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ota A, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphomas. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 50.Mendell JT. MiRiad roles for the miR-17-82 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ventura AG, et al. Targeted deletion reveals essential and overlapping functions of the miR-17-92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao C, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He L, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cialfre SA, et al. Extensive modulation of set of microRNAs in primary gioblastoma. Biochem Biophys Res Comun. 2005;334:1351–1358. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 55.Meng F, et al. MicroRNA-21 regulates the expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan JA, Kritchevsky AM, Kasic KS. MicroRNA-21 is an anti-apoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 57.Frankel LB, et al. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283:1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 58.Zhu S, Si ML, Win H, Mo YY. MicroRNA-21 targets the tumor suppressor Tropomyosin 1 (TPM1) J Biol Chem. 2007;282:14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 59.Bloomston M, et al. MicroRNA expression pattern to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–1908. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 60.Schetter AJ, et al. MicroRNA expression profiles association with prognosis and therapeutic out come in colon adenocarcinoma. JAMA. 2008;299:425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garzon R, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008;111:3183–3189. doi: 10.1182/blood-2007-07-098749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Voorhoeve PM, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 63.O'Donnell, et al. C-Myc regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 64.Xiao C, et al. Lymphoproliferative disease and auto-immunology in mice with elevated miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:409–419. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petrocca F. E2F1–regulated microRNAs impair TGF β-dependent cell cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–286. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 66.Dergnek R, Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 67.DiLeva G, et al. A regulatory ‘miRcircuitry’ inducing miR-221 and 222 and ERα determines ERα status of breast cancer cells. J Nat Cancer Inst. 2009 in press. [Google Scholar]
- 68.Bonci D, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activity. Nature Med. 2008;14:1271–1277. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 69.Kota J, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 71.Saito Y, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 72.Garzon R, et al. MicroRNA -29b induces global DNA hypomethylation and tumor suppressor gene re-expression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garzon R, et al. MicroRNA 29 expression and function in acute myeloid leukemia. Blood. 2009 doi: 10.1182/blood-2009-03-211938. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noonan EJ, et al. MiR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene. 2009;28:1714–1724. doi: 10.1038/onc.2009.19. [DOI] [PubMed] [Google Scholar]
- 75.Krutzfeldt J, et al. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]