

Abstract
For over thirty years it has been known that enteroendocrine cells derive from common precursor cells in the intestinal crypts. Until recently relatively little was understood about the events that result in commitment to endocrine differentiation or the eventual segregation of over 10 different hormone expressing cell types in the gastrointestinal tract. Enteroendocrine cells arise from pluripotent intestinal stem cells. Differentiation of enteroendocrine cells is controlled by the sequential expression of three basic helix loop helix transcription factors, Math1, Neurogenin 3, and NeuroD. Math1 expression is required for specification and segregation of the intestinal secretory lineage (Paneth, goblet, and enteroendocrine cells) from the absorptive enterocyte lineage. Neurogenin 3 represents the earliest stage of enteroendocrine differentiation and in its absence enteroendocrine cells fail to develop. Subsequent expression of NeuroD appears to represent a later stage of differentiation for maturing enteroendocrine cells. Enteroendocrine cell fate is inhibited by the Notch signaling pathway, which appears to inhibit both Math1 and Neurogenin 3. Understanding enteroendocrine cell differentiation will become increasingly important for identifying potential future targets for common diseases like diabetes and obesity.
Keywords: enteroendocrine cells, basic helix loop helix proteins, MATH1, neurogenin3, neuroD1, intestinal stem cells
Introduction
The mammalian gastrointestinal tract contains the largest population of hormone producing cells in the body [1]. Unlike most endocrine organs where endocrine cells are the predominant cell type, enteroendocrine cells account for less than 1% of the epithelial cells lining the stomach, small intestine, and colon and are scattered diffusely as individual cells throughout the mucosa [2]. The first gut hormones were discovered at the beginning of the 20th century as biological activities in extracts of intestinal tissue with physiological effects on digestive organ function. Bayliss and Starling in 1903 showed that canine intestinal extracts injected into another animal stimulated secretion of pancreatic juice. They proposed that that the gut contained a substance, secretin that was secreted into the blood to act on a distant target organ. Their discovery represented the birth of modern endocrinology.
Over the next 60 years, many additional biological activities in intestinal extracts were discovered. However, the next major advance in the field that established the peptide nature of gut hormones culminated in the purification of secretin in 1963 by Mutt and Jorpes. Further advances in peptide chemistry as well as recombinant DNA technology led to the discovery of more peptide hormones in the gut.
The availability of new powerful genetic approaches for analyzing cell lineages in mice, as well as major discoveries in the identification of intestinal stem cells, have resulted in major advances in understanding differentiation of enteroendocrine cells. This paper will review our current understanding of the development and differentiation of gut endocrine cells. Topics considered will include the relationship between enteroendocrine cells and intestinal stem cells and transcriptional programs that drive specification and differentiation of enteroendocrine cells, and mechanisms that control gut hormone gene expression.
Origin of enteroendocrine cells: Neural crest- or endoderm?
With the emergence of immunohistochemical techniques in the 1960’s, gastrointestinal endocrine cells were found to express markers for neuronal differentiation including those involved in the biosynthesis of neurotransmitters as well as showing ultrastructural properties common to neurons. These observations led to the “APUD” hypothesis, suggesting that enteroendocrine cells arose from cells that migrated from the neural crest to the gut [3]. Subsequent embryonic cell tracing techniques have clearly established that gastrointestinal endocrine cells are derived from the endoderm and not neuroectoderm [4–8]. However, the original observations that enteroendocrine cells share features with neurons has assumed new significance with recent discoveries showing that gut endocrine differentiation [9–12]is regulated by basic helix loop helix transcription factors similarly to differentiation in the nervous system [13–15]. As will be discussed later, gut endocrine differentiation is also under control of the Notch signaling pathway.
Common origin for enteroendocrine cells
The discovery of over 15 gut hormones produced by enteroendocrine cells, along with characterization each cell type and its origins became a topic of considerable interest. It was unclear if each endocrine cell type expressed one or multiple hormones. In addition, little was known about the precursor cells that gave rise to enteroendocrine cells. Classic studies of Cheng and Leblond from 1974 identified self renewing properties of the four epithelial cell types of the intestine, including enteroendocrine cells and proposed that they arise from common multipotential cells in the intestinal crypts [2]. Continuous infusions of tritiated thymidine first labeled cells deep in the crypt followed by more differentiated cells up the crypt-villus axis including enterocytes, goblet cells, enteroendocrine cells, and Paneth cells (Fig. 2). Cell labeling kinetics indicated that enterocytes, goblet cells, and enteroendocrine cells differentiated as cells migrate up the crypt-villus axis turning over every 3–4 days whereas Paneth cells migrated downwards to the crypt base with a much slower turnover of approximately 21 days [2]. Unlike many endocrine cells in different glands that differentiate early in life and turnover slowly, enteroendocrine cells actively self-renew and differentiate throughout the life of an animal from a large reservoir of stem cells. As mature enteroendocrine cells migrate to the tips of the villi, they presumably undergo apoptosis and are extruded into the lumen [16].
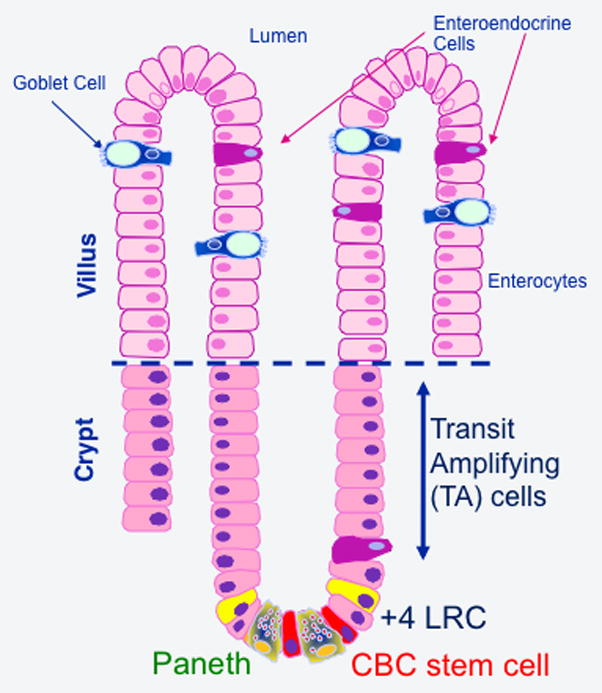
Fig. 2. Differentiation and functional anatomy within the crypt-villus unit.
Rapidly cycling crypt base columnar (CBC) and slowing cycling LRC+4 stem cells give rise to all four epithelial lineages. Enterocytes, goblet cells, and enteroendocrine cells differentiate as cells exit the lower crypt and a proliferative transit amplifying (TA) zone as they migrate up the crypt-villus axis. Some enteroendocrine cells differentiate just above the crypt base without without going through a TA population. In contrast, Paneth cells migrate to the crypt base and become intercalated between CBC stem cells.
Additional evidence for a common multipotential enteroendocrine precursor cell included the identification of individual cells coexpressing more than one hormone. In the small intestine, a subpopulation of substance P expressing cells in villi coexpressed serotonin and secretin whereas proliferating substance P cells in the crypts did not, leading to speculation that serotonin and secretin cells arise from substance P cells [17, 18]. In the colon, peptide YY, GLP-1, cholecystokinin, and neurotensin are coexpressed in L-type enteroendocrine cells. Colonic serotonin expressing cells coexpress substance P but never the preceding four hormones, leading to the hypothesis that there are two major branches for enteroendocrine differentiation in the colon [19].
Expression of stable reporter genes in transgenic mice provided further indirect support for the existence of multipotential enteroendocrine progenitor cells. Expression of a human growth hormone reporter (hGH) under control of the L-FABP gene showed labeling of multiple lineages of enteroendocrine cells [17]. Cell lineage ablation studies involving expression of a toxic gene in transgenic mice further established relationships between different enteroendocrine cell types. Expression of herpes simplex virus 1 thymidine kinase (HSVTK) in secretin-expressing cells of transgenic mice rendered cells susceptible to the antiviral drug ganciclovir [20]. Treatment of transgenic mice with the drug resulted in depletion of secretin cells as well as most cells expressing CCK and L cells expressing peptide YY and GLP-1. In addition, the numbers of cells expressing serotonin, GIP, substance P, and somatostatin were significantly reduced as well, revealing previously unappreciated relationships between these different cell types and secretin cells. The results further suggested that many cell lineages may express secretin at levels below the detectable limit for immunostaining [20].
Current model of enteroendocrine cell differentiation
Earlier studies on enteroendocrine cell differentiation were hampered by significant limitations. Most histochemical studies and labeling kinetics were largely descriptive and did not identify genes required for enteroendocrine cells to differentiate. The paucity of markers for the precursor cells was a second major limitation for progress. Since then, several factors have contributed to major advances in understanding enteroendocrine differentiation. New lines of knockout transgenic mice have identified genes that are required for enteroendocrine cells to differentiate. Increased use of recombination-based cell lineage analysis along with the discovery of better markers for intestinal stem cells has further enhanced identification of cells from which the enteroendocrine lineage arises.
In many developmental systems, expression of one or more specific transcription factors sequentially activates lineage specific gene expression programs. Differentiation may be highly dependent on the timing and context of transcription factor expression. A number of signaling pathways, most notably Wnt, Notch, BMP, and hedgehog often regulate transcriptional activation of specific lineage differentiation programs.
Enteroendocrine cells arise from pluripotent intestinal stem cells, which give rise to the four epithelial cell types of the small intestine (Fig. 1). As cells exit the stem cell compartment at the crypt base, they are fated either to become absorptive enterocytes or to cells of the secretory lineages (goblet cells, Paneth cells, and enteroendocrine cells). The progenitor cells rapidly proliferate, undergoing a number of cell divisions in the transit-amplifying zone of the upper crypt before terminally differentiating in the villus compartment. Three basic helix loop helix transcription factors related to the Drosophila protein, atonal, are involved in different stages differentiation of enteroendocrine cells and will be considered in more detail later. MATH1 appears to specify the secretory lineage whereas Neurog3 is obligatory for endocrine lineage specification, with NeuroD playing a role in later events in differentiation (Table 1).
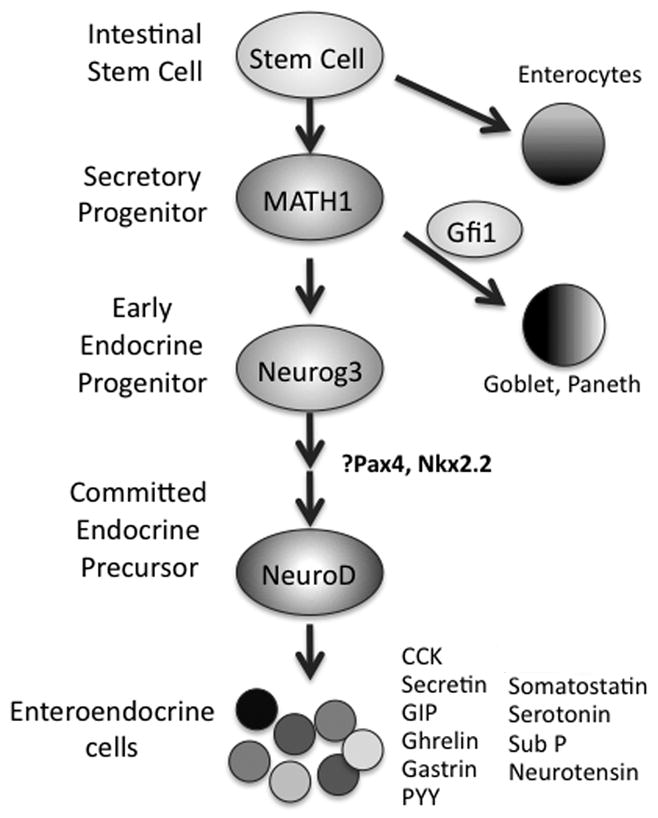
Fig. 1. Enteroendocrine cell differentiation from progenitors.
Enteroendocrine cells arise from pluripotent stem cells that give rise to secretory progenitor cells that express the transcription factor, Math1. GFI1 expression drives differentiation to the goblet and Paneth cell lineages. Neurog3 represents the first specific step to initiate endocrine cell fate. Additional transcription factors, including Pax4 and Nkx2.2 are switched on as enteroendocrine cells mature. Once cells express NeuroD, they become restricted to an enteroendocrine cell fate before giving rise to various mature enteroendocrine cells.
Table 1.
Intestinal phenotype of MATH1, Neurog3, and NeuroD1 knockout mice
| Survival | Gut Endocrine Phenotype | Alternate names | |
|---|---|---|---|
| MATH1 | Perinatal | Absent goblet, paneth, and enteroendocrine cells | Atoh1, mammalian atonal homologue 1 |
| Neurogenin3 | Perinatal | No enteroendocrine cells | Ngn3, Neurog3 |
| NeuroD1 | perinatal/longer depending on background | Secretin and chole-cystokinin cells absent. | BETA2 |
Intestinal Stem Cells
Studies of Cheng and Leblond suggested the existence of multipotential cells that give rise to each intestinal lineage, based on cell labeling kinetics of cells mutagenized to alter cell surface lectin expression. True pluripotency could not be established in this system due to the absence markers for cell lineage analysis. Studies of stem cells in other organs, like bone marrow and skin, indicated that adult stem cells were relatively quiescent and long-lived. The technique of long-term label retention, which identified cells that incorporated and retained BrDU, was used to localize putative hematopoietic and skin cells. Using this technique, label-retaining cells in the small intestine were localized, on average, to a position 4 cells from the Paneth cells at the crypt base and are referred to +4 LRCs. Although +4 LRCs exhibited many properties expected of stem cells functional stem cell activity such as pluripotency has only recently been demonstrated based on their telomerase activity.
Recently, an orphan G protein couple receptor, LGR5, a Wnt target gene upregulated in colorectal cancer, was identified as a marker for cells that fulfill the properties predicted for intestinal stem cells. LGR5 expression was mainly confined to crypt base columnar cells flanked by Paneth cells. Lineage tracing the descendants of LGR5+ cells using mice expressing Cre recombinase under control of the LGR5 gene and Cre indicator mice showed LGR5+ cells give rise to all four epithelial lineages, including enteroendocrine cells. LGR5+ cells self renew, are long-lived (60 days), and are relatively resistant to radiation. Surprisingly, LGR5+ cells are rapidly cycling, dividing every 24 hours, in contrast to quiescent stem cell populations in other organs as well as the +4 LRCs in the intestine. Isolated LGR5+ cells can be cultured to form organoid bodies containing all intestinal lineages in structures bearing a resemblance to intestinal crypts in the absence of mesenchymal cells [21].
Segregation of the enteroendocrine lineage from intestinal secretory progenitor cells
The Math1 gene encodes a basic helix loop helix protein that is the mouse homologue of the Drosophila atonal. Math1 is expressed in the gastrointestinal tract during development and has been identified in both the immature crypts and villi of the intestinal epithelium but not in the stomach [12]. Math1 appears to be essential for specification of the intestinal secretory progenitor as Math1−/− mice fail to develop all three secretory cell types (goblet, Paneth, and enteroendocrine cells) [12]. Expression of a β-galactosidase gene knocked into one Math1 allele, suggested that Math1 expressing cells become goblet, enteroendocrine, or Paneth cells and suggests that each of these three lineages arises from a Math1-expressing precursor cell [12]. Further evidence that Math 1 directs intestinal progenitors to the secretory lineage was shown in mice expressing Math1 in the intestine under the control of the villin promoter. Widespread expression of Math1 resulted in a striking expansion of secretory lineage cell number with a dramatic loss in absorptive enterocytes [22]. The corepressor protein, Gfi1, a downstream target of Math1 appears to direct secretory lineage cells to the goblet and Paneth cell lineages whereas in its absence these two lineages are reduced with an increase in enteroendocrine cells [23].
Neurogenin 3 is required for differentiation of enteroendocrine cells
Expression of a second atonal-related bHLH protein, Neurogenin 3 (Neurog3), appears to be essential for initiating the endocrine differentiation program in the gastrointestinal tract [9, 10, 24]. Neurog3 −/− mice express Math1, suggesting that Neurog3 is a downstream target of Math1 in the transcription factor cascade controlling endocrine differentiation [9]. Neurog3 −/− mice fail to develop any endocrine cells in the small intestine. However, in the glandular stomach, small numbers of serotonin cells, enterochromaffin like cells, and ghrelin-expressing cells differentiate in the absence of Neurog3, indicating that these lineages are not dependent upon Neurog3 expression [10]. Interpretation of these results has been somewhat difficult because Neurog3 null mice die within 3 days of birth prior to the differentiation of most gastric endocrine cells.
Neurog3 expression is detected as early as e12.5 in the developing murine intestine and is restricted to proliferating, immature cells in the crypts of the adult intestine as well as the glandular stomach [9, 10]. For many years, Neurog3 was not identified in cells expressing endocrine differentiation markers in either the pancreas or the intestine, leading to the suggestion that it was transiently expressed in endocrine progenitor cells, switching off prior to terminal differentiation [25]. Although loss of function studies in transgenic mice suggested that Neurog3 expression was essential for enteroendocrine cell specification, the absence of Neurog3 expression in hormone producing cells made it uncertain whether the effects of Neurog3 were cell autonomous. Expression of β-galactosidase in transgenic mice under control of 6.9 kb Neurog3 gene flanking sequence showed transgene expression in some but not all endocrine cells in the intestine and the stomach [9]. The discrepancy between transgene reporters and the endogenous gene expression may have resulted from greater sensitivity for detecting for β-galactosidase. It is also possible that the transgene did not completely recapitulate the expression of Neurog3. While these studies suggested that some Neurog3+ cells become endocrine cells, it was unclear whether all enteroendocrine cells arise from Neurog3+ cells.
Cell fate of Neurog3-expressing cells
We established the cell-fate of Neurog3 expressing cells in the GI using a Neurog3-Cre mouse where Cre recombinase was inserted into a BAC containing the Neurog3 gene to ensure that sufficient sequences were included to recapitulate the expression of the endogenous gene [26]. Crossing the Neurog3-Cre mouse with the ROSA26 Cre indicator strain enabled all cells that expressed Cre to undergo recombination at the ROSA26 reporter, with excision of a LoxP-Stop-LoxP (LSL) cassette. The resultant transcriptional read through with expression of β-galactosidase allowed us to identify all cells that arose from Neurog3+ as well as their descendants irrespective of their cell fate. One powerful advantage of this strategy is the ability to identify cells that transiently expressed Neurog3 at an earlier stage of differentiation.
As anticipated, almost all cells expressing the endocrine marker, chromogranin A (ChgA) in Neurog3-Cre; ROSA26 mice also stained for β-galactosidase activity, indicating that they arose from Neurog3-expressing precursor cells [26]. This observation also established that the effects of Neurog3 on endocrine progenitor cells were cell autonomous. Surprisingly, we found that up to 40% of duodenal Paneth cells expressed the β-galactosidase indicator. We also observed that up to 15% of goblet cells in the duodenum expressed β-galactosidase, although percentage of β-gal+ goblet cells gradually decreased in the distal small intestine to zero in the colon. In rare instances (1:250) we identified entire crypt-villus units stained for β-galactosidase activity.
The labeling of Paneth and some goblet cells by lineage tracing indicated that the Neurog3-Cre transgene was expressed in a subpopulation of intestinal secretory progenitor cells. The occasional labeling of entire crypt-villus units suggested that the reporter gene in these cases was expressed in pluripotent (stem) cells. These latter observations are consistent with low-level expression of Neurog3 with other nonendocrine pancreas lineage markers in single cells isolated from prepancreatic endoderm [27]. The labeling of nonendocrine cells in the intestine probably occurs from very low levels of Cre expression that efficiently catalyze a recombination at the ROSA26 locus. It is likely that Neurog3 protein is expressed at very low levels, below the threshold to drive endocrine differentiation in cells destined to nonendocrine cell fates. Others have subsequently shown that cells that express Neurog3 protein do not contribute to nonendocrine cell types [28].
The ability of Neurog3 to direct an endocrine differentiation in the intestine was confirmed by driving expression throughout the intestinal epithelium under control of the villin gene, resulting in increased number of enteroendocrine cells and increased expression of several hormones and proendocrine transcription factors [29]. The increase in enteroendocrine cell number was associated with reduced numbers of goblet cells with little change in the total number of secretory cells, suggesting that Neurog3 can redirect differentiation of a bipotential secretory progenitor cell to an enteroendocrine cell fate.
Recent development in understanding Neurog3 function in the intestine
Earlier studies on the role of Neurog3 in intestinal function and homeostasis were limited by difficulties in direct detection of Neurog3 expression by immunohistochemistry and by death of Neurog3 null mice in the perinatal period. The ability to detect this protein by immunostaining has improved considerably. Clonal labeling studies identified Neurog3+ cells most commonly at positions 5–8 in the crypt [28]. Surprisingly, only 1/3 of the Neurog3+ cells were slowly cycling with the remainder being post mitotic. About 1/3 of Neurog3+ cells expressed of endocrine differentiation markers like chromogranin A in contrast to earlier studies, which suggested that Neurog3 expression was extinguished with the onset of hormone expression in enteroendocrine cells. Thus it appears that the majority of Neurog3+ cells may be precursors that do not express detectable levels of mature endocrine cell markers and none contribute directly to nonendocrine lineages [28]. A very recent study identified Neurog3+ cells near the crypt base at positions +1 and +2 differentiation markers as well as staining for the putative stem cell markers Lgr5, DCAMKL1, and CD133. Cells expressing markers of endocrine differentiation appeared to be postmitotic throughout the crypt [30]. These observations may indicate that the enteroendocrine lineage differentiates directly from the stem cell pool without going through a transit amplifying population, potentially explaining their low frequency in the intestinal mucosa.
Neurog3−/− mice die shortly after birth, presumably from severe diabetes and/or neurological deficits. Unfortunately, the stomach and intestine and their respective enteroendocrine cells are not fully differentiated at that stage. Neurog3 was selectively deleted from all cells in the intestine by crossing mice with a conditional Neurog3 null allele with a villin-Cre line [31]. This approach made it possible to examine the functional importance of enteroendocrine cells since single and double hormone knockouts often show minimal phenotypes due to functional redundancy and overlapping functions with nontargeted hormones. Mice with Neurog3 deleted from the intestine showed profound growth retardation, fat malabsorption, with increased crypt proliferation and accelerated cell turnover, establishing the importance of intestinal Neurog3 expression and enteroendocrine cells for intestinal homeostasis and normal growth and development. Of note, these findings are similar to congenital malabsorptive diarrhea reported in infants with mutations in the Neurogenin 3 gene associated with greatly reduced numbers of enteroendocrine cells [32].
The bHLH protein NeuroD and later stages of enteroendocrine differentiation
The cell type restricted bHLH transcription factor NeuroD1, also known as BETA2, was originally discovered as a factor important for activating insulin gene transcription and neuronal differentiation [33, 34]. NeuroD binds to E box sequences (CANNTG) as part of a heterodimeric complex with ubiquitously expressed bHLH proteins like E12/E47. Several observations suggest that NeuroD is a downstream target of Neurog3. NeuroD expression is absent in Neurog3 −/− mice. In addition, Neurog3 binds to the NeuroD gene promoter and increases NeuroD transcription [9, 35]. Subsequent work showed that NeuroD is also an important regulator of secretin gene transcription [36]. NeuroD−/− mice fail to develop secretin and cholecystokinin cells in the intestinal tract [37], suggesting a potential role for this protein for the expression of both secretin and cholecystokinin.
Although most enteroendocrine cells express NeuroD [38], the presence of most enteroendocrine cell types in NeuroD−/− mice indicates that they do not require NeuroD for hormone expression. These findings were confirmed by crossing NeuroD-Cre mice [39] to ROSA26 indicator mice, showing expression of β-galactosidase in most enteroendocrine cells with no expression in nonendocrine lineages of the intestine (Leiter, A., unpublished observations). Thus cells that express NeuroD appear to be truly endocrine restricted in contrast to Neurog3+ cells. In addition to its effects on secretin gene transcription, NeuroD induces cell cycle arrest, possibly by increasing expression of p21, an inhibitor of cyclin dependent kinases [40]. In NeuroD null mice, cells expressing the null allele show reduced p21 expression with increased expression of cell proliferation markers, suggesting that NeuroD may function to coordinate expression of secretin with cell cycle arrest and terminal differentiation.
Since the secretin gene is the only confirmed direct target of NeuroD in enteroendocrine cells identified thus far, our laboratory has focused most of our efforts to characterizing its transcriptional regulation. Transcription of the secretin gene is restricted to S type enteroendocrine cells and is under control of a single a proximal enhancer between −30 and −200 (Fig. 3). The enhancer contains two Sp1 binding sites and another upstream site important for transcriptional activity, in addition to a NeuroD binding E box. The absence of additional enhancer elements for this gene made it difficult to explain its restricted expression when only a single transcription factor that bound to this enhancer, NeuroD, was relatively restricted in its expression.
Fig. 3. Functional cooperation between NeuroD and other proteins occupying the secretin gene enhancer.
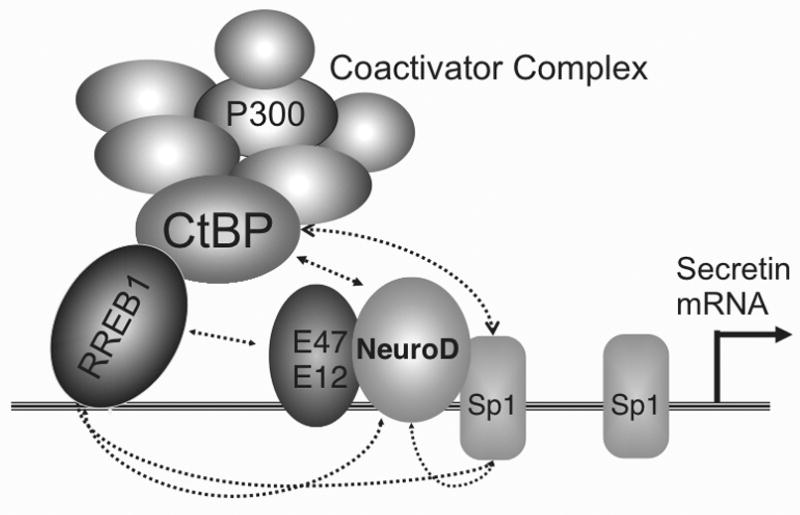
Four DNA binding proteins bind to a proximal enhancer in the secretin gene within 200 bp of the transcription start site. RREB1 binds directly to the enhancer and recruits CtBP and other proteins in multiprotein coactivator complex. Dotted lines indicate direct protein-protein interactions.
The functional importance of Sp1 in tissue specific expression of the secretin gene was examined in further detail [41]. NeuroD and Sp1 appeared to have a much greater than additive effect on NeuroD-dependent transcription. Examination of DNA-Protein complexes that formed between the enhancer and NeuroD, E12, and Sp1 by EMSA revealed the presence of a slow migrating higher order complex in addition to the NeuroD/E12 heterodimer and Sp1 containing complex. The complex was comprised of all three proteins and was present in far greater excess than predicted, suggesting cooperative binding activity. NeuroD appeared to stabilize the binding of Sp1 to its binding site immediately adjacent to the E box and thereby further increase transcription. This mechanism was confirmed with chromatinized DNA using ChIP assays to show loss of Sp1 occupancy in the absence of NeuroD.
We identified a previously described large zinc finger protein, RREB1 as another protein that bound to the enhancer by expression cloning. RREB1 was originally discovered as a DNA binding protein involved in RAS signaling but was subsequently found to be devoid of transactivating function [42, 43]. Subsequent studies showed that RREB1 did not directly transactivate the secretin gene. However, it potentiated NeuroD dependent transcription, an activity that depended on its ability to both bind to DNA as well as to physically associate with NeuroD [44]. These studies suggested that RREB1 might represent a novel class of transcription modifier protein as a coactivator that must bind to DNA for its effects. RREB1 contains two consensus motifs, PXDLS, for binding to the corepressor protein CtBP. RREB1 has been subsequently identified as a component of a large multiprotein corepressor complex containing several chromatin and histone modifying activities in addition to CtBP [45, 46]. The recruitment of proteins like CtBP that are normally associated with large corepressor complexes to the actively transcribed secretin gene is not well understood and is currently under investigation (Fig. 3).
Role of Notch signaling in enteroendocrine differentiation
The Notch signaling pathway is used throughout metazoan evolution to enable adjacent cells to adopt alternate cell fates. When Notch ligands from one cell bind to the Notch receptors on an adjacent cell, the intracellular domain of the receptor (NICD) is cleaved, allowing it to migrate to the nucleus where it interacts with the DNA binding protein RBP-Jκ (also known as CSL) [47] to form a complex that binds to and activates the promoters of the hairy/enhancer of split (HES) bHLH transcriptional repressors that serve as effectors of the Notch pathway [48]. HES proteins bind to GC rich sequences known as N boxes in promoters to repress the expression of a number of bHLH transcription factors important for cell fate decisions [49]. Since intestinal secretory lineage cell fate, including enteroendocrine cells, depends on three bHLH protein, Math1, Neurog3, and NeuroD, it is not surprising that these lineages are regulated by Notch.
Both gain and loss of function studies suggest that Notch signaling inhibits enteroendocrine differentiation. Disruption of the Notch pathway in Hes1 knockout mice was the first study to implicate Notch in enteroendocrine differentiation. Hes1−/− mice show increased numbers of enteroendocrine and goblet cells, suggesting that precocious secretory lineage differentiation occurred in the absence of Notch signaling with increased expression of proendocrine bHLH transcription factors [50]. Somewhat different results were seen in mice where RBP-jκwas conditionally deleted from the intestine or when animals were treated with γ secretase inhibitors [51]. Disruption of Notch in this context resulted in massive expansion of the goblet cell lineage at the expense of absorptive enterocytes with little effect on the number of enteroendocrine cells.
Conditional activation of Notch throughout the intestinal epithelium resulted in marked reduction in the number of goblet and enteroendocrine cells [52, 53], confirming the inhibitor effects of Notch on enteroendocrine cells. The studies done thus far did not directly address which of the three bHLH proteins involved in enteroendocrine cell fate are targeted by Notch. However, the effects on two secretory lineages suggest that Math11 is a likely target. Conditional activation of Notch in Neurog3+ cells phenocopied the perinatal mortality and absence of enteroendocrine cells seen in Neurog3−/− mice whereas conditional Notch activation in NeuroD+ cells had no effect on enteroendocrine cell differentiation, suggesting that Neurog3 but not NeuroD is a direct target of Notch (Leiter, A., unpublished observations). Consistent with our findings, the Neurog3 promoter has several Hes1 binding sites and transfection studies indicate that Hes1 directly inhibits Neurog3 expression [54].
Role of Wnt signaling in enteroendocrine differentiation
Enteroendocrine cells failed to develop with generalized disruption of Wnt signaling in TCF4 null mice or in Villin-DKK1 mice, initially raising the question whether Wnt activity is required for their specification [55, 56]. The absence of enteroendocrine cells following disruption of canonical Wnt signaling in the intestine may be the result of stem cell depletion rather than a lineage specific requirement. To determine if Wnt signaling was required for differentiation of enteroendocrine cells, we disrupted Wnt signaling in enteroendocrine precursors by conditionally deleting β-catenin in Neurog3+ cells. The enteroendocrine cells of these mice appeared normal with no effect on the number and subtypes [39], suggesting that Wnt-β catenin signaling was not required for either specification or terminal differentiation of the enteroendocrine lineage.
Abnormal Wnt signaling may be associated with gut neuroendocrine tumors [57]. Since Wnt signaling is the hallmark of self-renewing tissues and most enteroendocrine cells are relatively quiescent, it is not certain whether they respond to this pathway. Conditional activation of Wnt signaling by deleting exon 3 of β-catenin in Neurog3-expressing cells resulted in widespread adenomatous polyposis in the small intestine with many polyps expressing serotonin, suggesting that early enteroendocrine precursors retain the capacity to respond to Wnt. Surprisingly, conditional activation of β-catenin in NeuroD+ cells had no effect on the intestine, suggesting that at this stage of differentiation, maturing enteroendocrine cells do not respond to Wnt effectors [39]. These observations are consistent with other findings described earlier that suggest that expression of Neurog3 and NeuroD represent distinct, early and late stages in the differentiation of enteroendocrine cells.
Future questions
Many questions remain regarding how different endocrine hormone producing cells differentiate in the intestine. While Math1 and neurogenin3 are important for the global initiation of endocrine differentiation in the intestine, the signals and pathways involved in defining specific endocrine cell types remain largely unknown. Answers to these questions will depend in part on the analysis of how gut hormone gene expression is regulated and finding suitable models for further investigation. Furthermore, the hormones of the gastrointestinal tract have been increasingly implicated in the regulation of energy homeostasis, food intake, and insulin secretion. Targeting enteroendocrine cells may become an important therapeutic strategy in the future for treating and/or preventing common human diseases, including diabetes and obesity.
Acknowledgments
This work was supported in part by NIH grants DK43673, DK52870, and DK90000 to A.B.L., and the University of Massachusetts Diabetes and Endocrinology Research Center. Support was also provided by grants from the Caring for Carcinoid Foundation and the Worcester Foundation to ABL.
References
- 1.Rehfeld JF. The new biology of gastrointestinal hormones. Physiol Rev. 1998;78:1087–1108. doi: 10.1152/physrev.1998.78.4.1087. [DOI] [PubMed] [Google Scholar]
- 2.Cheng H, Leblond CP. Origin, differentiation, and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian theory of the origin of of the origin of the four epithelial cell types. American Journal of Anatomy. 1974;141:537–562. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 3.Pearse AG. The calcitonin secreting C cells and their relationship to the APUD cell series. J Endocrinol. 1969;45(Suppl):13–14. [PubMed] [Google Scholar]
- 4.Andrew A, Kramer B, Rawdon BB. The origin of gut and pancreatic neuroendocrine (APUD) cells--the last word? J Pathol. 1998;186:117–118. doi: 10.1002/(SICI)1096-9896(1998100)186:2<117::AID-PATH152>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 5.Andrew A. An experimental investigation into the possible neural crest origin of pancreatic APUD (islet) cells. J Embryol Exp Morphol. 1976;35:577–593. [PubMed] [Google Scholar]
- 6.Fontaine J, Le Lievre C, Le Douarin NM. What is the developmental fate of the neural crest cells which migrate into the pancreas in the avian embryo? Gen Comp Endocrinol. 1977;33:394–404. doi: 10.1016/0016-6480(77)90055-7. [DOI] [PubMed] [Google Scholar]
- 7.Le Douarin NM, Teillet MA. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J Embryol Exp Morphol. 1973;30:31–48. [PubMed] [Google Scholar]
- 8.Le Douarin NM. On the origin of pancreatic endocrine cells. Cell. 1988;53:169–171. doi: 10.1016/0092-8674(88)90375-3. [DOI] [PubMed] [Google Scholar]
- 9.Jenny M, Uhl C, Roche C, et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. Embo J. 2002;21:6338–6347. doi: 10.1093/emboj/cdf649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. 2002;16:1488–1497. doi: 10.1101/gad.985002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naya FJ, Huang HP, Qiu Y, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science. 2001;294:2155–2158. doi: 10.1126/science.1065718. [DOI] [PubMed] [Google Scholar]
- 13.Anderson DJ. Lineages and transcription factors in the specification of vertebrate primary sensory neurons. Curr Opin Neurobiol. 1999;9:517–524. doi: 10.1016/S0959-4388(99)00015-X. [DOI] [PubMed] [Google Scholar]
- 14.Anderson DJ, Groves A, Lo L, et al. Cell lineage determination and the control of neuronal identity in the neural crest. Cold Spring Harb Symp Quant Biol. 1997;62:493–504. [PubMed] [Google Scholar]
- 15.Sommer L, Ma Q, Anderson DJ. neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurosci. 1996;8:221–241. doi: 10.1006/mcne.1996.0060. [DOI] [PubMed] [Google Scholar]
- 16.Simon TC, Gordon JI. Intestinal epithelial cell differentiation: new insights from mice, flies and nematodes. Curr Opin Genet Dev. 1995;5:577–586. doi: 10.1016/0959-437x(95)80026-3. [DOI] [PubMed] [Google Scholar]
- 17.Roth KA, Hertz JM, Gordon JI. Mapping enteroendocrine cell populations in transgenic mice reveals an unexpected degree of complexity in cellular differentiation within the gastrointestinal tract. Journal of Cell Biology. 1990;110:1791–1801. doi: 10.1083/jcb.110.5.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aiken KD, Roth KA. Temporal differentiation and migration of substance P, serotonin, and secretin immunoreactive enteroendocrine cells in the mouse proximal small intestine. Dev Dyn. 1992;194:303–310. doi: 10.1002/aja.1001940406. [DOI] [PubMed] [Google Scholar]
- 19.Roth KA, Kim S, Gordon JI. Immunocytochemical studies suggest two pathways for enteroendocrine cell differentiation in the colon. Am J Physiol. 1992;263:G174–180. doi: 10.1152/ajpgi.1992.263.2.G174. [DOI] [PubMed] [Google Scholar]
- 20.Rindi G, Ratineau C, Ronco A, Candusso ME, Tsai M, Leiter AB. Targeted ablation of secretin-producing cells in transgenic mice reveals a common differentiation pathway with multiple enteroendocrine cell lineages in the small intestine. Development. 1999;126:4149–4156. doi: 10.1242/dev.126.18.4149. [DOI] [PubMed] [Google Scholar]
- 21.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 22.VanDussen KL, Samuelson LC. Mouse atonal homolog 1 directs intestinal progenitors to secretory cell rather than absorptive cell fate. Dev Biol. 2010;346:215–223. doi: 10.1016/j.ydbio.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shroyer NF, Wallis D, Venken KJ, Bellen HJ, Zoghbi HY. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 2005;19:2412–2417. doi: 10.1101/gad.1353905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwitzgebel VM, Scheel DW, Conners JR, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127:3533–3542. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 26.Schonhoff SE, Giel-Moloney M, Leiter AB. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol. 2004;270:443–454. doi: 10.1016/j.ydbio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Chiang MK, Melton DA. Single-cell transcript analysis of pancreas development. Dev Cell. 2003;4:383–393. doi: 10.1016/s1534-5807(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 28.Bjerknes M, Cheng H. Neurogenin 3 and the enteroendocrine cell lineage in the adult mouse small intestinal epithelium. Dev Biol. 2006;300:722–735. doi: 10.1016/j.ydbio.2006.07.040. [DOI] [PubMed] [Google Scholar]
- 29.Lopez-Diaz L, Jain RN, Keeley TM, et al. Intestinal Neurogenin 3 directs differentiation of a bipotential secretory progenitor to endocrine cell rather than goblet cell fate. Dev Biol. 2007;309:298–305. doi: 10.1016/j.ydbio.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sei Y, Lu X, Liou A, Zhao X, Wank SA. A stem cell marker-expressing subset of enteroendocrine cells resides at the crypt base in the small intestine. Am J Physiol Gastrointest Liver Physiol. 2011;300:G345–356. doi: 10.1152/ajpgi.00278.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellitzer G, Beucher A, Lobstein V, et al. Loss of enteroendocrine cells in mice alters lipid absorption and glucose homeostasis and impairs postnatal survival. J Clin Invest. 2010;120:1708–1721. doi: 10.1172/JCI40794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Cortina G, Wu SV, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med. 2006;355:270–280. doi: 10.1056/NEJMoa054288. [DOI] [PubMed] [Google Scholar]
- 33.Naya FJ, Stellrecht CMM, Tsai M-J. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 34.Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 35.Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol. 2000;20:3292–3307. doi: 10.1128/mcb.20.9.3292-3307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mutoh H, Fung B, Naya F, Tsai M-J, Nishitani J, Leiter AB. The basic helix-loop-helix transcription factor BETA2/NeuroD is expressed in mammalian enteroendocrine cells and activates secretin gene expression. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3560–3564. doi: 10.1073/pnas.94.8.3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naya FJ, Huang H, Qiu Y, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/NeuroD-deficient mice. Genes and Development. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ratineau C, Petry MW, Mutoh H, Leiter AB. Cyclin D1 represses the basic helix-loop-helix transcription factor, BETA2/NeuroD. J Biol Chem. 2002;277:8847–8853. doi: 10.1074/jbc.M110747200. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Giel-Moloney M, Rindi G, Leiter AB. Enteroendocrine precursors differentiate independently of Wnt and form serotonin expressing adenomas in response to active beta-catenin. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11328–11333. doi: 10.1073/pnas.0702665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mutoh H, Naya FJ, Tsai M-J, Leiter AB. The basic helix loop helix protein BETA2 interacts with p300 to coordinate differentiation of secretin-expressing enteroendocrine cells. Genes and Development. 1998;12:820–830. doi: 10.1101/gad.12.6.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ray SK, Leiter AB. The basic helix-loop-helix transcription factor NeuroD1 facilitates interaction of Sp1 with the secretin gene enhancer. Molecular & Cellular Biology. 2007;27:7839–7847. doi: 10.1128/MCB.00438-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujimoto-Nishiyama A, Ishii S, Matsuda S, Inoue J-I, Yamamoto T. A novel zinc finger protein, Finb, is a transcriptional activator and localized in nuclear bodies. Gene. 1997;195:267–275. doi: 10.1016/s0378-1119(97)00172-8. [DOI] [PubMed] [Google Scholar]
- 43.Thiagalingam A, Bustros AD, Borges M, et al. RREB-1, a novel zinc finger protein, is involved in the differentiation response to ras in human medullary thyroid carcinomas. Molecular and Cellular Biology. 1996;16:5335–5345. doi: 10.1128/mcb.16.10.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ray SK, Nishitani J, Petry MW, Fessing MY, Leiter AB. Novel transcriptional potentiation of BETA2/NeuroD on the secretin gene promoter by the DNA-binding protein Finb/RREB-1. Mol Cell Biol. 2003;23:259–271. doi: 10.1128/MCB.23.1.259-271.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 46.Shi Y, Sawada J, Sui G, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 47.Tamura K, Taniguchi Y, Minoguchi S, et al. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 48.Jarriault S, Le Bail O, Hirsinger E, et al. Delta-1 activation of notch-1 signaling results in HES-1 transactivation. Mol Cell Biol. 1998;18:7423–7431. doi: 10.1128/mcb.18.12.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sasai Y, Kageyama R, Tagawa Y, Shigemoto R, Nakanishi S. Two mammalian helix-loop-helix factors structurally related to Drosophila hairy and Enhancer of split. Genes Dev. 1992;6:2620–2634. doi: 10.1101/gad.6.12b.2620. [DOI] [PubMed] [Google Scholar]
- 50.Jensen J, Pedersen EE, Galante P, et al. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 51.van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 52.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 53.Stanger BZ, Datar R, Murtaugh LC, Melton DA. Direct regulation of intestinal fate by Notch. Proc Natl Acad Sci U S A. 2005;102:12443–12448. doi: 10.1073/pnas.0505690102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee JC, Smith SB, Watada H, et al. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes. 2001;50:928–936. doi: 10.2337/diabetes.50.5.928. [DOI] [PubMed] [Google Scholar]
- 55.Korinek V, Barker N, Moerer P, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 56.Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17:1709–1713. doi: 10.1101/gad.267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujimori M, Ikeda S, Shimizu Y, Okajima M, Asahara T. Accumulation of beta-catenin protein and mutations in exon 3 of beta-catenin gene in gastrointestinal carcinoid tumor. Cancer Res. 2001;61:6656–6659. [PubMed] [Google Scholar]