

Abstract
Advances in genomic research have revealed that each patient has their own unique tumor profile. While silencing RNA (siRNA) screening tests can identify which genes drive tumor cell growth, results obtained from these assays have been limited in their clinical translatability because they employ cell lines growing on flat surfaces. The Cancer BioChip System (CBCS) is a functional screening assay for identification of siRNA capable of inhibiting anchorage-independent three-dimensional (3D) cancer cell growth. Anchorage-independent growth assays are important in vitro predicators of regulators of cancer cell growth. Unique features of the CBCS include a Cancer BioChip, wherein cells incorporate different siRNAs in parallel and grow in a 3D matrix to form colonies that can be quantified using real-time imaging and an image analysis software. Thus, the CBCS can be developed as a tool for personalized identification of targeted cancer therapies.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-012-0116-8) contains supplementary material, which is available to authorized users.
Keywords: Plating Efficiency, Soft Agar Assay, Primary Breast Cancer Cell, M4A4 Cell, Transform Tumor Cell
Introduction
Functional screening of candidate drug targets requires the development of in vitro assays for the testing of gene-specific inhibitors in a clinically relevant setting. The discovery that double-stranded RNA (dsRNA), also called silencing RNA (siRNA), can cause post-transcriptional gene silencing via RNA interference (RNAi) in most eukaryotic cells has made this approach a technology of choice for inhibiting gene expression [1–4]. When introduced into the cell, siRNA interacts with PIWI family proteins to form the RNA-induced silencing complex (RISC), which recognizes and then degrades homologous mRNA, leading to post-transcriptional suppression of gene expression [5, 6]. It is now possible to design gene-specific inhibitors for any candidate gene based on its sequence and perform genome-scale gene silencing experiments for functional genomics and drug discovery [7–9].
Several studies have been reported using genome-wide loss-of-function screens for the identification and validation of cancer drug targets (see Iorns et al. [10], for review). Some used either Transfected Cell Microarrays (TCM) [11–15] or pooled shRNA libraries [16–18] for identification of shRNAs capable of altering function in cancer cell lines. Other platforms employed Lentivirus-infected cell microarrays (LICM) [19, 20] to obtain high levels of siRNA in immortalized and primary cells. While these studies supported the feasibility of high-throughput gene silencing and the influence of gene expression on various parameters of cell function, none of these studies evaluated the functional impact of siRNA on cancer cell growth in a setting that would directly translate to the in vivo milieu.
There are many limitations to TCM assays. The TCM platform was developed to treat only attached cancer cell lines, which have altered properties and biological responses that may not mimic in vivo cancer cell biology. Mammary epithelial cells behave differently when grown in a three-dimensional (3D) matrix rather than on two-dimensional flat surfaces (see Jacks and Weinberg [21], for review). Furthermore, cancer cells grown under these conditions tend to migrate, which limits the duration and throughput capability of this assay, since individual siRNAs will have to be spotted at distances that would keep neighboring cells apart. Another limitation is that it is not possible to study primary patient tumor cells with these assays, since they allow both normal and tumor cell growth. Since cellular sensitivity to treatment is influenced by in vitro growth conditions, none of these assays can be used as a stand-alone cancer drug-screening assay. Thus, it is important to perform these studies in an in vitro 3D setting that would translate to the clinic.
The most commonly used in vitro test for potential chemotherapeutic agents is the anchorage-independent growth assay, also known as soft agar, clonogenicity, human tumor colony-forming, or human tumor stem cell (HTSC) assay [22–29]. Anchorage-independent growth is usually quantified using semisolid media, such as agar. Soft agar assays are the most stringent assays for cancer drug screening, since they allow transformed cells, but not normal cells, to grow in vitro. These transformed cells exhibit stem cell-like properties, grow in suspension, and exhibit minimal contact-triggered growth inhibition. Salmon et al. [22] showed correlation between in vitro results obtained using the HTSC assay and the clinical responses of myeloma and ovarian cancer patients to a variety of chemotherapeutic agents. Larger scale testing revealed that clinically effective chemotherapeutic agents were also active in the HTSC assay with the exception of those requiring systemic activation, while clinically ineffective agents were confirmed to be true negatives with 97 % accuracy [23]. Other groups also showed the potential use of this assay in predicting patient responsiveness to chemotherapy [24–26, 28, 30].
In their current format, anchorage-independent growth assays are not amenable for large-scale screening [31, 32], since they require large numbers of cells and the plating efficiency of human tumor biopsies is very low. Subsequently, only breast, colorectal, kidney, lung, melanomas, and ovarian tumors have been tested in this manner, since these tumors produce sufficient cell yields [28]. Most importantly, current assays are capable of only testing one inhibitor at a time and are tedious, time consuming, and costly since they are not easily amenable to automation.
We have developed a high-throughput 3D assay for screening siRNA, the Cancer BioChip System (CBCS) (Falcon Genomics, Inc., Pittsburgh, PA; US Patent # 7,537,913 B2 and 8,110,375 B2). The CBCS is a kit that includes a device (“Cancer Biochip”), into which multitudes of chemical compounds to be tested (e.g., siRNA library) are embedded or overlaid on a matrix that inhibits cell attachment, along with a top matrix to be mixed with the cells (see Fig. 1). The top matrix allows 3D cell growth and limits cell migration. When cells are mixed with the matrix and cultured on the Cancer BioChip, they incorporate the underlying active agent and grow in 3D. When used, the CBCS evaluates the effect of the chemical compounds on the cells in real-time by tracking growth using commercially available imaging systems and custom-designed imaging and image analysis software. Results obtained from these studies will give information about gene function, cytotoxicity, therapeutic targets, and functional genomic profiles of cancer patients.
Fig. 1.
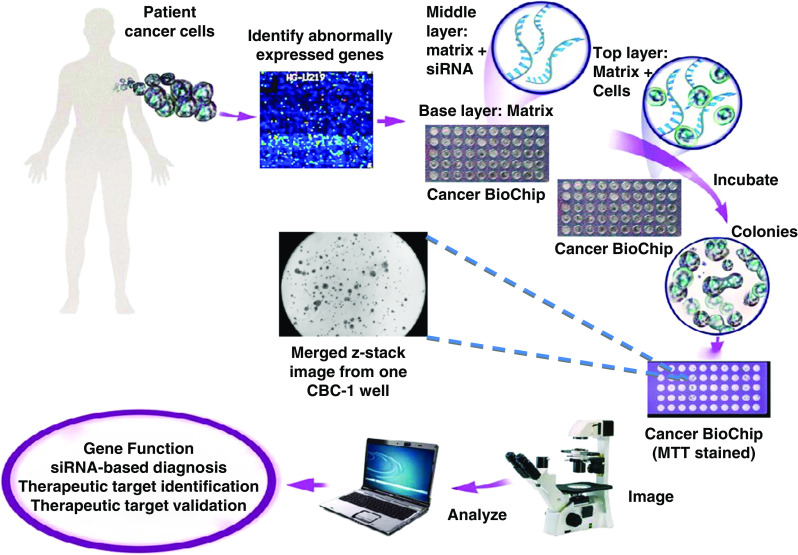
Diagram of the Cancer BioChip System (CBCS) kit and method. Abnormally expressed genes are targeted with siRNA on the Cancer BioChip to determine their effect on anchorage-independent tumor cell growth. Cells cultured on the Cancer BioChip incorporate siRNA while being suspended in a 3D matrix. Growth of siRNA-transfected colonies is monitored using customized imaging and image analysis methods. Results obtained from the CBCS lead to identification of potential therapeutic targets in a setting that has high clinical translatability. Shown is a first-generation Cancer BioChip (CBC-1) with and without staining with MTT for viability testing. The center photomicrograph depicts merged z-stack images acquired from a CBC-1 well using an inverted microscope and a 4× objective
Thus, the unique features of the CBCS are as follows: rapid, one-step, high-throughput siRNA transfection approach combined with live assessment and quantification of anchorage-independent cancer cell growth. By miniaturizing assay conditions, smaller amounts of patient tissue and reagents are needed. This allows for testing of a multitude of compounds simultaneously and in parallel without requirement of large incubator space. The ability to observe tumor cell growth in real time and over a long period of time allows for identification of early, latent, stable, and/or transient effects of each compound.
In this paper, we report on the validation of a first-generation CBCS (CBC-1) capable of simultaneous quantitative real-time assessment of the effect of 50 different siRNA mixtures on anchorage-independent tumor cell growth (Fig. 1). Matrices employed in the CBC-1 consisted of soft agar. Cells that are plated on the CBC-1 incorporate the underlying siRNA and form colonies that can be examined and quantified over a long period of time.
The CBC-1 can be used to optimize basic functions of the CBCS prior to scaling up to a high-throughput full genome screen. These include plating, transfection, silencing, and cytostatic efficiencies. In this paper, we report on each one of these parameters for the validation of the CBC-1 as a stand-alone in vitro test for identification of inhibitors of breast cancer cell growth in a clinically relevant setting.
Results
Anchorage-Independent Growth of Breast Cancer Patient Cells and Cell Lines on the CBC-1 (Plating Efficiency)
High-throughput anchorage-independent growth testing is a unique property of the CBCS. Using the CBC-1, we have determined colony growth in agar of transformed breast cancer cell lines (MDA-MB-231, M4A4, MCF7, and SKBR3) and immortalized NIH-3T3 fibroblast cells. We have observed colony formation in all cells except for the nontransformed NIH-3T3 cells (Online Resource 1). Plating efficiencies, as determined by percent of cells that formed live colonies, were between 20 and 39 % for these cell lines.
Analysis of colony formation on the CBC-1 for primary breast cancer patient cells gave a wide range of results. Since these cells consisted of a mixture of all cell types obtained from a tumor, the percent of transformed tumor cells varied among patients. In addition, the growth rate of cells obtained from different patients was different. Figure 2a shows examples of colonies that formed from five different breast cancer patients and corresponding plating efficiencies.
Fig. 2.

a Plating efficiency of primary breast cancer patient cells on the CBC-1. Shown are photomicrographs of MTT stained CBC-1 imaged at days 21–22 (patients 19, 20, and 21) and days 28–29 (patients 28 and 29) in culture. Cells were seeded on the CBC-1 at 600–700 cells per well. b Growth of primary breast cancer patient cells on the CBC-1 after 9 days in culture. Shown are bright field photomicrographs of the entire well with 100, 300, 500, 700, and 1,000 starting cell number per well. Colonies appear as black dots. c Scatter plot showing the linear relationship between starting cell numbers and number of colonies forming in each well for the patient shown in b
For each patient, a plating efficiency experiment has to be performed to identify the optimal starting cell number for each CBC-1. The number of colonies formed has to be directly related to the starting cell counts. For these experiments, we determine the plating efficiency by measuring the percentage of colonies formed, after seeding 100, 300, 500, 700, and 1,000 cells per well on the CBC-1 (n = 5). Figure 2b and c show results from one representative experiment using primary breast cancer cells. Shown are representative merged z-stack images from individual wells (Fig. 2b). Analysis of cell and colony size showed that the average cell area at day 1 was estimated to be 203 ± 12 μm2 and average colony size on day 9 was 1,504 ± 40 μm2. For this patient, we have observed a linear correlation between starting cell numbers and numbers of formed colonies at day 9 with an average plating efficiency of 51.6 ± 2.5 % (Fig. 2c). Thus, a starting cell number of 700 cells per well could be used for further testing on the CBC-1, whereby the assay would be in the linear range. These results show that we can use the CBC-1 to determine the plating efficiency of primary patient cells prior to functional genomic testing on a higher throughput CBCS.
Silencing RNA Transfection on the CBC-1 Using a Proprietary One-Step Method
The CBCS is intended to achieve efficient delivery of siRNA into primary cancer cells. Since different cells have different transfection efficiencies, it is essential to determine the method of choice for transfection for every cell type to be tested on the CBCS. This can be quickly achieved using the CBC-1. siRNA can be delivered into cells cultured on the CBC-1 using different types of transfection reagents. We tested lentiviral vector-mediated delivery using control vectors from the Mission TRC (Sigma) and Accell siRNA delivery methods (Dharmacon). The Accell siRNA method is unique, since it uses a proprietary siRNA backbone modification to drive uptake of the siRNA into the cells; as a result, there is no need for a separate delivery reagent. Another advantage of using Accell siRNA for CBCS development is that it does not require the specialized equipment needed for safe manipulation of lentiviruses (Biological Safety level 2+).
To determine transfection efficiency on the CBC-1 using either transfection method, we employed the following positive controls: the Mission TRC control vector, a lentiviral vector expressing the gene for Turbo Green Fluorescence Protein (TurboGFP) and the Accell Green siRNA control, a FAM-labeled Accell Non-Targeting siRNA. We have tested the impact of several variables on expression of the Mission TRC TurboGFP control into primary patient cells and the MDA-MB-231 breast cancer cell line. We found that the TurboGFP signal in these cells was very weak using Mission TRC TurboGFP. In both MDA-MB-231 and primary breast cancer cells, the transfection efficiency on the CBC-1 using the Mission TRC TurboGFP vector ranged between 8 and 20 % (data not shown).
On the other hand, control Accell Green Non-Targeting RNAs gave a very strong signal in most primary breast cancer cells tested on the CBC-1. Figure 3 shows representative photomicrographs of primary breast cancer cells growing on the CBC-1 in the presence of increasing doses of Accell Green (2.5, 5, and 10 μM). While we can obtain up to 65 % transfection efficiency at 2.5 μM Accell Green (65 ± 8 %), the highest transfection efficiency was observed at 5 μM (96 ± 2 %). No further increase in percent-transfected cells could be observed with the higher concentrations, and expression can be maintained at a lower level for up to 21 days (Table 1). At day 21, colonies remained viable as determined by MTT stain (data not shown). However, the signal was not transmitted to all daughter cells in the colonies. Further testing of primary patient breast cancer cells (n = 14) with Accell Green (10 μM) gave transfection efficiencies ranging between 55 and 95 % at day 2 (data not shown). In summary, we observed elevated siRNA expression in primary breast cancer cells and cell lines on the CBC-1 using the Accell delivery method, while Mission TRC TurboGFP expression was much weaker.
Fig. 3.

Representative photomicrographs showing expression of Accell Green in primary breast cancer cells. Shown are merged z-stacks for bright field (BF) and fluorescence (Accell Green) images. Treatment groups include No siRNA and Accell Green siRNA (2.5 μM, 5 μM, and 10 μM). Fluorescence Accell Green signal appears as gray areas
Table 1.
Percent transfection efficiency of Accell Green into primary breast cancer cells at 5, 12, and 21 days post-transfection
| Accell Green | Day 5 | Day 12 | Day 21 |
|---|---|---|---|
| 0.0 μM | 8 ± 2 | 13 ± 5 | 5 ± 3 |
| 2.5 μM | 65 ± 8a | 50 ± 12a | 34 ± 3a |
| 5.0 μM | 96 ± 2a,b | 90 ± 2a,b | 66 ± 7a,b |
| 10.0 μM | 82 ± 14a | 72 ± 17a | 55 ± 14a |
a p < 0.05 relative to 0.0 μM
b p < 0.05 relative to 2.5 μM using Student's t test
Silencing Efficiency of EGFP siRNA in M4A4 EGFP-Expressing Cells Growing on the CBC-1
Successful transfection does not necessarily imply successful suppression of the gene of interest. The following experiments were performed to determine whether we could achieve inhibition of gene expression on the CBC-1. We have tested whether EGFP siRNA can suppress EGFP protein expression in breast cancer cell lines that have been engineered to express EGFP (M4A4 cells, ATCC).
Although we could not see high transfection efficiencies with the Mission TRC TurboGFP particles on the CBC-1, we have observed gene silencing with both lentiviral transfection of shRNA for eGFP (Mission TRC control, Sigma; data not shown) and the Accell EGFP shRNA pool (Thermo Fisher Dharmacon). Figure 4 shows silencing of EGFP in M4A4 cells grown on the CBC-1 and transfected with increasing concentrations of Accell EGFP siRNA (2.5, 5, and 10 μM). Signal intensity for each colony was assessed by measuring average integrated density for all colonies. While maximal suppression of EGFP signal was 64 % at day 8 with the Accell EGFP siRNA (Fig. 4), it reached 93 % with the Mission TRC EGFP shRNA at day 14 (data not shown).
Fig. 4.

Suppression of EGFP signal in M4A4 breast cancer cells transfected with Accell EGFP pool siRNA on the CBC-1 after 5 days in culture. Photomicrographs show merged bright field (BF) z-stacks and merged green fluorescence z-stacks (eGFP). EGFP Signal was expressed relative to controls (n = 5). a P < 0.05 relative to control. b P < 0.05 relative to 2.5 μM. Magnification: 10×
These results prove that silencing of gene expression can be obtained even with low levels of lentiviral shRNA transfection efficiency. The most important deliverable in our assay is silencing of gene expression and this can be obtained with either lentiviral vectors or Accell siRNA.
Suppression of MCF7 Colony Formation with Estrogen Receptor α (ESR1) siRNA on CBC-1
The ultimate validation for the CBCS is the inhibition of cancer cell growth by siRNA knockdown of tumor genes. For these studies, we determined MCF7 breast cancer cell line growth response to ESR1 Accell siRNA on the CBC-1. MCF7 cells have been very well characterized in the literature and are known for their dependence on the estrogen receptor [33]. For these experiments, four different Accell siRNAs (10 μM) targeting different sequences in the ESR1 gene were tested on the CBC-1 either individually (ESR1-1, ESR1-2, ESR1-3, and ESR1-4) or together in a pool (ESR1 pool). In addition, each CBC-1 included the following controls: no cells, no siRNA, and Accell nontargeting siRNA (10 μM). Figure 5a shows representative merged z-stack images from MCF7 cells after 7 days of culture on the CBC-1 in the presence of each of the abovementioned conditions. The number of colonies (size greater than 1,500 μm2) as well as the average size of all cells was measured at different time points in culture. Measurement of relative change in colony numbers and change in average cell size from days 2 to 7 showed that ESR1-2, ESR1-3, ESR1-4, and ESR1 pools suppressed colony formation on the CBC-1 (Fig. 5b, c), while ESR1-1 was ineffective. While untransfected cells (No siRNA) and cells transfected with nontargeting siRNA showed increase in number of colonies and average cell size over time, those transfected with ESR1 Accell siRNAs were suppressed except for ESR1-1. The highest level of colony suppression was obtained with either ESR1-4 or ESR1 pools. These results show that we can demonstrate suppression of breast cancer colony growth on the CBC-1 using siRNAs for genes known to be involved in breast cancer.
Fig. 5.
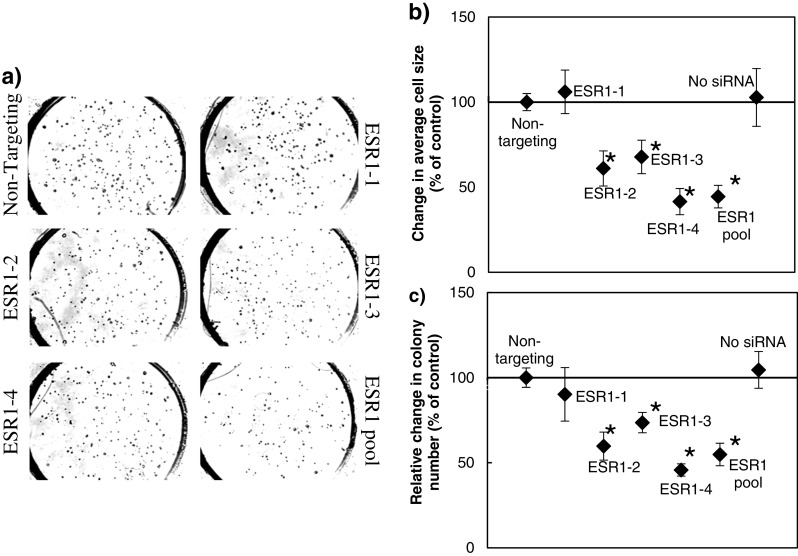
a Suppression of MCF7 cell growth on the CBC-1 with Accell ESR1 siRNA. Four different Accell siRNA (ESR1-1, ESR1-2, ESR1-3, and ESR1-4) sequences and a pool of all four were tested at 10 μM. Accell nontargeting was used as a negative control. Shown are photomicrographs of merged z-stack images from different wells on the CBC-1 at day 7. Cells per colonies appear as dark dots. b Shown are changes in average MCF7 cell size, and c relative change in colony number between days 2 and 7 normalized to control (n = 5). Asterisk indicates conditions that are significantly different from control (p < 0.05)
Discussion
The CBCS is a functional genomic assay that identifies inhibitors of anchorage-independent 3D cell growth. In this paper, we developed a first-generation Cancer BioChip, CBC-1, which can be employed for initial determination of plating, transfection, silencing, and cytotoxic efficiencies for every cell type prior to high-throughput screening. We show that we can grow primary patient cells on the CBC-1 with varying plating efficiencies. We show a linear correlation between the number of cells seeded on the CBC-1 and the number of colonies formed. We also compared two siRNA delivery methods on the CBC-1 and found higher transfection efficiency into primary patient cells using Accell siRNA as compared to lentiviral vector-mediated delivery. However, both siRNA delivery methods led to suppression of target gene expression. We also demonstrate the effectiveness of different ESR1 siRNAs, individually and as a pool, at inhibiting MCF7 colony growth at different time points and in real time. These studies validate the utility of the CBC-1 at identifying inhibitors of primary patient cells that can grow in an anchorage-independent fashion. These cells are thought to be the tumor-initiating cells and should be targeted for therapy. Such data could translate into clinically relevant results for cancer target identification and personalized formulation of therapy.
There are many advantages for using soft agar as opposed to other matrices on the CBCS. Soft agar assays are considered to be optimal for testing primary tumor cells for the following reasons:
Unlike collagen and other mammalian cell-derived matrices which can alter cellular properties, agar is a polysaccharide derived from marine algae and does not influence cellular behavior. Matrices consisting of Extracellular Matrix (ECM) components, such as collagen and Matrigel, allow all types of cells to grow and influence their in vitro behavior. In fact, nonmalignant Human Mammary Epithelial Cells (HMEC), differentiate into mammary acini when cultured in 3D collagen or laminin gels [34, 35].
Soft agar assays selectively allow transformed tumor cells to grow. When cells become transformed, they acquire the innate capability of dividing in suspension, do not need cell–cell contact, do not require attachment to a surface, and can recapitulate the tumor phenotype when transplanted in SCID nude mice [36]. The most widely used model for testing carcinogenic compounds in vitro is the NIH-3T3 soft agar assay. NIH-3T3 fibroblast cells cannot form colonies in soft agar unless they become transformed. Transformation could be obtained by treatment with carcinogens, infection with tumor viruses, or genetic manipulation [37–39].
Soft agar assays also provide an environment for low cellular attachment, which is optimal for growth of “mammospheres” in the presence of selective media. These mammospheres are considered to be “tumor-initiating cells” with CD44+CD24−/low phenotype [35, 40]. These cells are thought to be responsible for tumor recurrence and metastasis.
Another important feature of using agar on the CBCS is that it limits migration of the cells and keeps them immobilized on top of the tested siRNA, which allows for higher throughput testing of compounds embedded at higher density within the Cancer BioChip. Thus, the soft agar matrix used on the CBC-1 is a stringent environment for high-throughput chemosensitivity testing of transformed tumor cells obtained from primary biopsies.
While TCM siRNA screening methods have been used for the identification of genes essential for viability and proliferation of breast cancer cells, these assays utilize cell lines growing on flat surfaces. For these assays, attachment of the cells is essential for the incorporation of underlying siRNA. Thus, targets obtained from these studies were most often individually validated using anchorage-independent growth assays. In some instances, cells responded differently upon validation, which further emphasizes the need for siRNA screening of cells growing in an anchorage-independent 3D fashion [41].
There have been several previous attempts at developing high-throughput anchorage-independent growth assays. Some involved using 96-well plates with automated colony counting at 1 week using the CytoFluor® Series 4000 Multi-Well Plate Reader [42]. The use of Alamar blue for colony readout also allowed for automated colony counting using the CytoFluor® Series 4000 Multi-Well Plate Reader. However, the need for a separate cell transfection step prior to plating cells in agar increases assay time and results in further cell loss. In addition, the use of Alamar blue for cell staining limits the ability to obtain a real-time chemosensitivity response, and it would not allow for the identification of drugs that might lead to resistance. The 96-well format supports the testing of a larger number of inhibitors than previously possible but is still unable to provide efficient real-time siRNA screening in 3D.
Other large-scale siRNA screens in 3D employed the pooled shRNA library dropout approach [43, 44]. For these assays, primary cells are virally transduced with shRNA libraries and then cultured in 3D to identify inhibitors of cellular transformation. While these assays identified potential tumor suppressor genes, they could not be employed as an in vitro diagnostic test for identification of patient-specific therapies. For these assays, every cell has to be transduced with at least one viral replicon, and cells that do not grow in these assays are correlated with shRNA lethality. Since the cells are transduced with shRNA prior to plating, this approach will not work for screening of inhibitors of primary patient cells, which do not grow at 100 % plating efficiency. Cell death under these conditions could be attributed to either low plating efficiency or shRNA lethality. Since our approach employs a transfection method that occurs in cells growing in an anchorage-independent environment, lack of growth under these conditions is solely attributed to siRNA transfection. Therefore, it is essential to have on-agar transfection/transduction of siRNA/shRNA in order to perform functional genomic screens for inhibitors of anchorage-independent cell growth.
In summary, the CBCS is a technology for screening siRNA capable of inhibiting anchorage-independent and 3D primary patient tumor cell growth. We validate a first-generation CBCS that can be employed for determination of plating efficiency of patient cells, siRNA transfection method for high siRNA incorporation, silencing efficiency of target genes, and cytostatic efficiency. Our results show that we can grow patient cells that incorporate underlying siRNA at high efficiency. We also show high specificity and efficiency of siRNA suppression of target genes and inhibition of tumor cell growth. Thus, the CBC-1 can be used for functional genomic profiling of primary patient cells and identification of more efficient and targeted patient-specific treatment modalities.
Methods
Silencing RNA
Accell siRNA (Thermo Fisher Dharmacon, Lafayette, CO) or Mission TRC shRNA-expressing lentiviral vectors (Sigma, Saint Louis, MO) were employed in this study. Accell siRNA controls included: Accell Non-Targeting and Accell Green Non-Targeting siRNA. For Accell ESR1 siRNAs, the sequences were: ESR1-1: CCGUAAUGAUUCUAUAAUG, ESR1-2: GCCUGGUGAUUAUUCAUUU, ESR1-3: GGAAGGUUUUACAUUAUUC, and ESR1-4: UAUUCAUGUUAAGAUACUA. A pool of all four sequences was also tested. The Mission TRC shRNA-expressing lentiviral vectors were: Mission pLKO.1-puro Control Transduction Particles, Mission TurboGFP Control Transduction Particles, and Mission eGFP shRNA Control Transduction Particles. Lentiviral vectors at a titer of 106 particles/ml, which gives 5,000 particles per well, were mixed with Protamine Sulfate (6 μg/ml) and agar prior to application on the CBC-1.
Preparation of the CBC-1
A CBC-1 (Falcon Genomics, Inc., Pittsburgh, PA) that consisted of CultureWell chambered coverslips (Grace Bio-labs, Inc., Bend, OR), with each spot being 3 mm in diameter and 1 mm deep was used. The matrix consisted of soft agar (Noble Agar, Difco, BD & Co, Sparks, MD). After application of a base agar (0.8 % in delivery media) layer in each well, siRNA mixed with agar (0.2 % in delivery media) was applied, followed by cancer cells mixed with top agar (0.4 % in delivery media). The CBC-1 was incubated in a CO2 incubator at 37 °C overnight, covered with complete growth medium on the following day, and fed twice a week thereafter. Colony formation was monitored using an inverted microscope at different time intervals up to 20–29 days. At the end of this incubation period, cell viability was determined using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) stain.
Patient Cell Culture
Primary breast cancer cells were procured from a commercial supplier (Celprogen, San Pedro, CA) or prepared in-house from fresh patient tumor tissue collected by the Cooperative Tissue Network (CHTN), which is funded by the National Cancer Institute, NIH. All patient tissues were procured by CHTN according to current regulations and guidance for repositories from the Office of Human Research Protections (OHRP, DHHS). The Institutional Review Board (IRB) protocol for this study was authorized and approved by the Allegheny Singer Research Institute-WPAHS IRB. Patient tumor samples were shipped from CHTN affiliated institutions in RPMI-1640 containing antibiotics, 10 % fetal calf serum, and 2.5 μg/ml fungizone and maintained cold during shipment. Upon arrival to our facility, tissue was immediately processed for preparation of tumor cells. We used collagenase types 3 and 1 (Worthington, Lakewood, NJ) sequentially to digest the tissue and isolate cells. We then cultured the cells in flasks coated with Geltrex (Invitrogen) in the presence of HuMEC medium (Invitrogen).
Cell Lines
Breast cancer cell lines, including MDA-MB-231, M4A4, MCF7, SKBR3, and the immortalized fibroblast cells NIH-3T3, were obtained from the American Type Culture Collection (ATCC, Manassas, VA). All cells were cultured according to ATCC guidelines in medium (DMEM (Hyclone Laboratories, South Logan, UT) for MCF7, NIH-3T3, and M4A4 cells; RPMI (Sigma) for SKBR3 cells; Leibovitz's L-15 Medium (Sigma) for MDA-MB-231 cells) containing 10 % fetal bovine serum, 1 % antibiotics-antimycotics (Invitrogen, Carlsbad, CA; Catalogue # 15240-062) at 37 °C in the presence of 5 % CO2. Cells were passaged using Trypsin–EDTA (0.05 %; Hyclone) when they reached 70–80 % confluency in culture.
Imaging and Image Analysis
Individual wells were imaged on an inverted Motic AE31 microscope using high-resolution cooled CCD cameras, CoolSnap K4 (Photometrics, Inc.), or QiCam (Qimaging®). Several images were acquired along the z-axis and were processed to create z-stacks, which were subsequently merged using ImageJ (National Institute of Health).
Image analysis was performed using custom-designed image analysis macros in ImageJ. These macros measure number of cells/colonies, their average size, and the total area occupied by cells for each condition in an automated fashion. Change in average size from day 2 to later days is calculated and expressed as percent of controls. Relative change in colony number was also calculated by subtracting the number of colonies between day 2 and later time points, normalized to total cell counts at day 2, and expressed as a percent of controls. Growth curves were obtained by plotting the total area at different time points. Plating efficiency was determined by calculating the number of live colonies as a percent of total measured cells at day 2.
Determination of both transfection and silencing efficiencies requires measurement of the presence or absence of a fluorescence signal in each cell or colony. Since cells grow in a 3D matrix on the CBC-1, imaging of each cell and identification of its level of fluorescence signal requires acquisition of z-stacks in both bright field and fluorescence, respectively. For fluorescent signal analysis, z-stacks were first merged using maximum intensity for the fluorescence images and minimum intensity for the bright field images. Background signal was subtracted from the merged fluorescence images. Particles ranging between 300 and 10,000 μm2 were identified as regions of interest (ROI) in the merged bright field images. ROIs were then overlaid on the merged fluorescence image followed by measurement of the fluorescence intensity.
To determine transfection efficiency, background intensity of the fluorescence signal was calculated by averaging the maximum signal per colony from all colonies observed in the control wells (n = 5 wells per experiment). Cells that expressed a signal intensity that is equivalent to the average background signal plus two times its standard deviation (average background signal +2* SD) were considered to be transfected by either Accell Green or Mission TurboGFP. Percent transfection efficiency was determined by dividing the number of transfected colonies by total number of cells observed in each well and multiplying by 100.
For silencing efficiency, the average integrated fluorescent signal density for all M4A4 colonies was measured in the presence or absence of Accell EGFP siRNA or Mission TRC EGFP shRNA. Data was expressed as percent EGFP signal relative to control.
Statistical Analysis
For plating efficiency experiments, a linear regression analysis was performed to determine the percent colonies formed at different starting concentrations of cells on the CBC-1. The slope of the linear regression determined plating efficiency. ANOVA followed by Student's t test was used to compare between different experimental conditions.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Growth of breast cancer cell lines on the CBC-1. Shown are representative merged z-stack images from CBC-1 wells where M4A4, MCF-7, MDA-MB-231, and SKBR3 have been growing for 18, 14, 21, and 15 days, respectively. Colonies appear as dark blobs (PDF 310 kb)
Acknowledgments
This work was supported through a Phase 1 SBIR grant from the National Cancer institute: 1 R43 CA141962-01A2, (PI: R.A.A.) and the Technology Development Fund from the Pittsburgh Life Sciences Greenhouse.
Conflict of interest
R.A.A, J.N.M, and A.L.D are employed by Falcon Genomics, Inc. M.P.Z was a previous intern of Falcon Genomics, Inc.
References
- 1.Jorgensen RA, Cluster PD, English J, Que Q, Napoli CA. Chalcone synthase cosuppression phenotypes in petunia flowers: comparison of sense vs. antisense constructs and single-copy vs. complex T-DNA sequences. Plant Mol Biol. 1996;31:957–973. doi: 10.1007/BF00040715. [DOI] [PubMed] [Google Scholar]
- 2.Matzke AJ, Neuhuber F, Park YD, Ambros PF, Matzke MA. Homology-dependent gene silencing in transgenic plants: epistatic silencing loci contain multiple copies of methylated transgenes. Mol Gen Genet. 1994;244:219–229. doi: 10.1007/BF00285449. [DOI] [PubMed] [Google Scholar]
- 3.Caplen NJ, Fleenor J, Fire A, Morgan RA. dsRNA-mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene. 2000;252:95–105. doi: 10.1016/S0378-1119(00)00224-9. [DOI] [PubMed] [Google Scholar]
- 4.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans . Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 5.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 6.Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci U S A. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng L, Liu J, Batalov S, Zhou D, Orth A, Ding S, Schultz PG. An approach to genomewide screens of expressed small interfering RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:135–140. doi: 10.1073/pnas.2136685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, McCombie WR, Elledge SJ, Hannon GJ. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 9.Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, Agami R, Ge W, Cavet G, Linsley PS, Beijersbergen RL, Bernards R. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 10.Iorns E, Lord CJ, Turner N, Ashworth A. Utilizing RNA interference to enhance cancer drug discovery. Nat Rev Drug Discov. 2007;6:556–568. doi: 10.1038/nrd2355. [DOI] [PubMed] [Google Scholar]
- 11.Kumar R, Conklin DS, Mittal V. High-throughput selection of effective RNAi probes for gene silencing. Genome Res. 2003;13:2333–2340. doi: 10.1101/gr.1575003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziauddin J, Sabatini DM. Microarrays of cells expressing defined cDNAs. Nature. 2001;411:107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]
- 13.Hannon GJ, Conklin DS. RNA interference by short hairpin RNAs expressed in vertebrate cells. Methods Mol Biol. 2004;257:255–266. doi: 10.1385/1-59259-750-5:255. [DOI] [PubMed] [Google Scholar]
- 14.Mousses S, Caplen NJ, Cornelison R, Weaver D, Basik M, Hautaniemi S, Elkahloun AG, Lotufo RA, Choudary A, Dougherty ER, Suh E, Kallioniemi O. RNAi microarray analysis in cultured mammalian cells. Genome Res. 2003;13:2341–2347. doi: 10.1101/gr.1478703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baghdoyan S, Roupioz Y, Pitaval A, Castel D, Khomyakova E, Papine A, Soussaline F, Gidrol X. Quantitative analysis of highly parallel transfection in cell microarrays. Nucleic Acids Res. 2004;32:e77. doi: 10.1093/nar/gnh074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 17.Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, Westbrook TF, Liang AC, Chang K, Hackett JA, Harper JW, Hannon GJ, Elledge SJ. Cancer proliferation gene discovery through functional genomics. Science. 2008;319:620–624. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silva JM, Marran K, Parker JS, Silva J, Golding M, Schlabach MR, Elledge SJ, Hannon GJ, Chang K. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319:617–620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 20.Bailey SN, Ali SM, Carpenter AE, Higgins CO, Sabatini DM. Microarrays of lentiviruses for gene function screens in immortalized and primary cells. Nat Methods. 2006;3:117–122. doi: 10.1038/nmeth848. [DOI] [PubMed] [Google Scholar]
- 21.Jacks T, Weinberg RA. Taking the study of cancer cell survival to a new dimension. Cell. 2002;111:923–925. doi: 10.1016/S0092-8674(02)01229-1. [DOI] [PubMed] [Google Scholar]
- 22.Salmon SE, Hamburger AW, Soehnlen B, Durie BG, Alberts DS, Moon TE. Quantitation of differential sensitivity of human-tumor stem cells to anticancer drugs. N Engl J Med. 1978;298:1321–1327. doi: 10.1056/NEJM197806152982401. [DOI] [PubMed] [Google Scholar]
- 23.Shoemaker RH, Wolpert-DeFilippes MK, Kern DH, Lieber MM, Makuch RW, Melnick NR, Miller WT, Salmon SE, Simon RM, Venditti JM, et al. Application of a human tumor colony-forming assay to new drug screening. Cancer Res. 1985;45:2145–2153. [PubMed] [Google Scholar]
- 24.Bertelsen CA, Sondak VK, Mann BD, Korn EL, Kern DH. Chemosensitivity testing of human solid tumors. A review of 1582 assays with 258 clinical correlations. Cancer. 1984;53:1240–1245. doi: 10.1002/1097-0142(19840315)53:6<1240::AID-CNCR2820530604>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 25.Moon TE, Salmon SE, White CS, Chen HS, Meyskens FL, Durie BG, Alberts DS. Quantitative association between the in vitro human tumor stem cell assay and clinical response to cancer chemotherapy. Cancer Chemother Pharmacol. 1981;6:211–218. doi: 10.1007/BF00256973. [DOI] [PubMed] [Google Scholar]
- 26.Von Hoff DD, Casper J, Bradley E, Sandbach J, Jones D, Makuch R. Association between human tumor colony-forming assay results and response of an individual patient's tumor to chemotherapy. Am J Med. 1981;70:1027–1041. doi: 10.1016/0002-9343(81)90859-7. [DOI] [PubMed] [Google Scholar]
- 27.Fiebig HH, Schmid JR, Bieser W, Henss H, Lohr GW. Colony assay with human tumor xenografts, murine tumors and human bone marrow. Potential for anticancer drug development. Eur J Cancer Clin Oncol. 1987;23:937–948. doi: 10.1016/0277-5379(87)90339-7. [DOI] [PubMed] [Google Scholar]
- 28.Suggitt M, Bibby MC. 50 years of preclinical anticancer drug screening: empirical to target-driven approaches. Clin Cancer Res. 2005;11:971–981. [PubMed] [Google Scholar]
- 29.Blumenthal RD. An overview of chemosensitivity testing. Methods Mol Med. 2005;110:3–18. doi: 10.1385/1-59259-869-2:003. [DOI] [PubMed] [Google Scholar]
- 30.Salmon SE, Alberts DS, Durie BG, Meyskens FL, Jones SE, Soehnlen B, Chen HS, Moon T. Clinical correlations of drug sensitivity in the human tumor stem cell assay. Recent Results Cancer Res. 1980;74:300–305. doi: 10.1007/978-3-642-81488-4_36. [DOI] [PubMed] [Google Scholar]
- 31.Fiebig HH, Maier A, Burger AM. Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur J Cancer. 2004;40:802–820. doi: 10.1016/j.ejca.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Selby P, Buick RN, Tannock I. A critical appraisal of the “human tumor stem-cell assay”. N Engl J Med. 1983;308:129–134. doi: 10.1056/NEJM198301203080304. [DOI] [PubMed] [Google Scholar]
- 33.Pratt SE, Pollak MN. Estrogen and antiestrogen modulation of MCF7 human breast cancer cell proliferation is associated with specific alterations in accumulation of insulin-like growth factor-binding proteins in conditioned media. Cancer Res. 1993;53:5193–5198. [PubMed] [Google Scholar]
- 34.Bissell MJ, Weaver VM, Lelievre SA, Wang F, Petersen OW, Schmeichel KL. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res. 1999;59:1757–1763s. [PubMed] [Google Scholar]
- 35.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blair DG, Cooper CS, Oskarsson MK, Eader LA, Vande Woude GF. New method for detecting cellular transforming genes. Science. 1982;218:1122–1125. doi: 10.1126/science.6293052. [DOI] [PubMed] [Google Scholar]
- 37.Shalloway D, Coussens PM, Yaciuk P. Overexpression of the c-src protein does not induce transformation of NIH 3T3 cells. Proc Natl Acad Sci U S A. 1984;81:7071–7075. doi: 10.1073/pnas.81.22.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schafer R, Griegel S, Dubbert MA, Willecke K. Unstable transformation of mouse 3T3 cells by transfection with DNA from normal human lymphocytes. EMBO J. 1984;3:659–663. doi: 10.1002/j.1460-2075.1984.tb01863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keath EJ, Caimi PG, Cole MD. Fibroblast lines expressing activated c-myc oncogenes are tumorigenic in nude mice and syngeneic animals. Cell. 1984;39:339–348. doi: 10.1016/0092-8674(84)90012-6. [DOI] [PubMed] [Google Scholar]
- 40.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 41.Bauer JA, Ye F, Marshall CB, Lehmann BD, Pendleton CS, Shyr Y, Arteaga CL, Pietenpol JA. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast Cancer Res. 2010;12:R41. doi: 10.1186/bcr2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ke N, Albers A, Claassen G, Yu DH, Chatterton JE, Hu X, Meyhack B, Wong-Staal F, Li QX. One-week 96-well soft agar growth assay for cancer target validation. Biotechniques. 2004;36:826–828. doi: 10.2144/04365ST07. [DOI] [PubMed] [Google Scholar]
- 43.Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, Zhao JJ, Roberts TM, Mandel G, Hannon GJ, Depinho RA, Chin L, Elledge SJ. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–848. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 44.Kolfschoten IG, van Leeuwen B, Berns K, Mullenders J, Beijersbergen RL, Bernards R, Voorhoeve PM, Agami R. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell. 2005;121:849–858. doi: 10.1016/j.cell.2005.04.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth of breast cancer cell lines on the CBC-1. Shown are representative merged z-stack images from CBC-1 wells where M4A4, MCF-7, MDA-MB-231, and SKBR3 have been growing for 18, 14, 21, and 15 days, respectively. Colonies appear as dark blobs (PDF 310 kb)