

Abstract
The aim of this study was to investigate the dynamics of four of the most validated biomarkers for Alzheimer's disease (AD), cerebro-spinal fluid (CSF) Aβ 1–42, tau, hippocampal volume, and FDG-PET, in patients at different stage of AD. Two hundred twenty-nine cognitively healthy subjects, 154 mild cognitive impairment (MCI) patients converted to AD, and 193 (95 early and 98 late) AD patients were selected from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database. For each biomarker, individual values were Z-transformed and plotted against ADAS-cog scores, and sigmoid and linear fits were compared. For most biomarkers the sigmoid model fitted data significantly better than the linear model. Aβ 1–42 time course followed a steep curve, stabilizing early in the disease course. CSF tau and hippocampal volume changed later showing similar monotonous trends, reflecting disease progression. Hippocampal loss trend was steeper and occurred earlier in time in APOE ε4 carriers than in non-carriers. FDG-PET started changing early in time and likely followed a linear decline. In conclusion, this study provides the first evidence in favor of the dynamic biomarker model which has recently been proposed.
Keywords: Alzheimer's disease, Alzheimer's Disease Neuroimaging Initiative dataset, Biomarker dynamics, Cerebro-spinal fluid Aβ 42, Tau, Hippocampal volume, FDG-PET
1. Introduction
Most of the Alzheimer disease (AD) research between the years 2000 and 2010 has been focused on finding biomarkers which could be reliably used to diagnose AD, monitor its progression, and predict its onset. A number of fluid and imaging biomarkers have been identified and validated (Hampel et al., 2008; Shaw et al., 2007), the most studied to date being Aβ plaque deposition (assessed either in terms of reductions in cerebro-spinal fluid (CSF) Aβ 1–42 or increased amyloid positron emission tomography (PET) tracer retention), CSF tau, fluoro-deoxy-glucose (FDG) uptake on PET, and structural magnetic resonance imaging (MRI).
Evidence from several past studies strongly supports the notion that amyloid Pittsburg Compound B (PIB)-PET (Klunk et al., 2004; Rowe et al., 2007; Edison et al., 2007, Ikonomovic et al, 2008) and low CSF Aβ 1–42 (Clark et al., 2003; Fagan et al., 2006; Schoonenboom et al., 2008; Strozyk et al., 2003; Tapiola et al., 2009) are valid biomarkers for brain Aβ plaque load. Increased CSF tau, despite being not specific to AD, is an indicator of tau pathological changes and neuronal injury, and correlates with clinical disease severity (Arai et al., 1995; Blennow et al., 1995; Buerger et al., 2006; Hansson et al., 2006; Shaw et al., 2009; Tapiola et al., 2009). FDG-PET measures brain metabolism and is a valid indicator of the synaptic dysfunction that accompanies neurodegeneration in AD (Hoffman et al., 2000; Jagust et al., 2007; Minoshima et al., 1997). Structural MRI measures cerebral atrophy, which is not specific to AD but strictly correlates with the disease severity even at latest stages, and can be considered a valid biomarker of neurodegeneration (Bobinski et al., 2000; Frisoni et al., 2010; Gosche et al., 2002; Silbert et al., 2003; Zarow et al., 2005); among all MRI-based markers, hippocampal volume has been widely shown to be one of the most reliable (Jack et al., 2000; Schuff et al., 2009; Van de Pol et al., 2006).
Biomarkers have allowed to further understand the pathology underlying AD, pointing out that a dichotomous view (people with AD pathology have dementia, people without AD pathology have not), common in the past, cannot hold any more, and should be replaced by a more dynamic picture, in which pathologic and clinical changes occur gradually over time. Several studies have shown that biomarker abnormalities precede clinical symptoms. Autopsy brain studies found no strict relationship between quantitative measures of cortical amyloid deposition and the duration and severity of Alzheimer disease (Ingelsson et al., 2004). Jack and colleagues showed that many of normal controls are PIB positive, suggesting that plaque deposition occurs before neurodegeneration (Jack et al., 2008a), and showed that PIB retention (i.e. amyloid load) increase occurs in prodromal AD, being almost stable in time in the clinical phases of the disease (Jack et al., 2009).
The availability of several validated biomarkers opens the discussion about how to choose among them: which marker is better to use to diagnose AD? Which better predicts AD? All these markers are validated enough to be used in active therapeutic trials or large longitudinal observational studies, but which is better to use to track cognitive decline or monitor new drugs therapeutical efficacy?
Jack and colleagues (Jack et al., 2010) pointed out that individual biomarkers, reflecting individual aspects of the Alzheimer pathology, develop on their own time course, and do not become abnormal or steady simultaneously.
The open challenge, now, is to try to order biomarker changes in time. This would enable us to express the disease process in terms of a series of testable biological indicators, and thus to identify biomarkers which could be best used in clinical trials to select patients and measure disease-modifying drug effects, or even be used in future prevention trials. Furthermore, understanding the temporal order of each biomarker would make it possible to use a given marker for staging AD in vivo.
Among the most validated biomarkers described above, Aβ 1–42 deposition was reported to change first, as early as 20 years before symptoms appear, but quickly reach a plateau by the time a person has dementia (Jack et al., 2008a; Jack et al., 2009). Structural changes become appreciable later in the disease process, but correlate with cognitive progression as dementia worsens (Jack et al., 2008a; Jack et al., 2009; Vemuri et al., 2009). The synaptic dysfunction marker FDG-PET and the neurodegeneration marker CSF tau are supposed to lie between Aβ 1–42 and MRI (Jack et al., 2010; Reiman et al., 1998), but there is lack of evidence about it. Furthermore, long-term biomarker dynamics has been hypothesized to be nonlinear, likely sigmoid shape (Jack et al., 2010).
The aim of this study is to use the Alzheimer's Disease Neuroimaging Initiative (ADNI) dataset to investigate the dynamics of four of the most validated AD biomarkers (CSF Aβ 1–42, CSF tau, hippocampal volume, and FDG uptake on PET) in a cohort of cognitively healthy subjects and AD patients at different stage of disease.
2. Methods
2.1. Subjects
Data used in the preparation of the current paper were obtained from the ADNI database (www.loni.ucla.edu/ADNI). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies, and nonprofit organizations, as a US $60 million, 5 year public-private partnership. The ADNI primary goal has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials. The Principle Investigator of this initiative is Michael W. Weiner MD, VA Medical Center and University of California, San Francisco. ADNI is the result of efforts of many coinvestigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the USA and Canada. The initial goal of ADNI was to recruit 800 adults, ages 55 to 90, to participate in the research – approximately 200 cognitively normal older individuals to be followed for 3 years, 400 people with MCI to be followed for 3 years, and 200 people with early AD to be followed for 2 years. For up-to-date information see www.adni-info.org.
At baseline, all subjects were given the American National Adult Reading Test and the following cognitive measures were examined: digit span, category fluency, Trail Making A and B, Digit Symbol Substitution Test of the Wechsler Adult Intelligence Scale–Revised, Boston Naming Test, Auditory Verbal Learning Test, clock drawing, Neuropsychiatric Inventory Q, AD Assessment Scale–Cognitive Subscale, and Functional Assessment Questionnaire (Cummings et al., 2005; Kaplan et al., 1982; Reitan, 1958; Rey, 1964; Rosen et al., 1984; Wechsler, 1987); they underwent blood drawing (for APOE genotyping) and structural MR. Subsets of subjects underwent lumbar puncture (for CSF sampling), FDG-PET or PIB-PET.
Healthy controls (HC) were all those in the ADNI database with available ADAS-Cog score (n = 229). We then considered ADNI patients with baseline diagnosis of MCI who had progressed to AD during the ADNI project observation time (n = 154, conversion time 6 to 36 months). We finally considered all ADNI AD patients, and we divided them in two groups of similar size according to the Mini Mental State Examination (MMSE) score: early AD, with MMSE score above the 50th percentile (MMSE > 23, n = 95), and late AD, with MMSE score below the 50th percentile (MMSE ≤ 23, n = 98).
2.2. Cerebrospinal fluid measurements
Methods for CSF acquisition and biomarker measurement used in the ADNI study have been reported previously (Shaw et al., 2009). In brief, CSF was collected, transferred to polypropylene tubes, and frozen on dry ice within an hour after collection. Samples were divided into aliquots at the University of Pennsylvania ADNI Biomarker Core Laboratory, stored at −80 °C, and measured using the multiplex xMAP Luminex platform (Luminex, Corp, Austin, TX) with Innogenetics (INNOBIA AlzBio3, Ghent, Belgium) immunoassay kit-based reagents as previously described (Olsson et al., 2005). The reagents included monoclonal antibodies specific for Aβ 1–42 (4D7A3), t-tau (AT120) and p-tau phosphorylated at threonine 181 (AT270), and analyte-specific detector antibodies (HT7, 3D6). In the current study we considered only Aβ 1–42 and t-tau measurements.
2.3. FDG-PET
FDG-PET scanning was performed on multiple PET instruments of differing resolutions. FDG-PET scans were collected as 6 5-minute frames from 30 to 60 minutes after injection of approximately 5 mCi of tracer. Scans were corrected with either segmented transmission data or CT scans, depending on instrumentation.
All scans underwent quality control at University of Michigan and were preprocessed to make them more uniform and make PET images from different systems more similar according to the following procedure: raw PET images from all sites were converted to the standard DICOM format; separate frames were coregistered lessening the effects of patient motion, and recombined into a coregistered dynamic image set; coregistered frames were averaged to create a single 30 minute PET image; each resulting image was reoriented into a standard 160 × 160 × 96 voxel image grid having 1.5 mm cubic voxels and oriented such that the anterior-posterior axis of the subject is parallel to the AC-PC line, and intensity normalized using a subject-specific mask with an average voxel intensity of one; each image was finally filtered with a scanner-specific filter function to a common uniform isotropic resolution of 8 mm FWHM. More detailed information can be found at www.loni.ucla.edu/ADNI/Data/ADNI_Data.shtml.
All pre-processed PET data were analyzed at the University of Utah (Norman Foster laboratory). Pet images were resampled into a Talairach atlas registration using Neurostat stereo v 8.0, and metabolic glucose activity pixel values were extracted and projected onto surface maps using 3D–SSP. Detailed information are available at https://www.loni.ucla.edu/twiki/pub/ADNI/ADNIPostProc/UUtah_Analysis.pdf.
The average cerebral metabolic rate of glucose consumption (CMRglc) in frontal, parietal and temporal cortices normalized to pons was computed and used in the current study as measure of cerebral metabolism.
2.4. Magnetic resonance imaging
ADNI MRI scans were collected at multiple sites using either a GE, Siemens, or Philips 1.5-T system. Two high-resolution T1-weighted volumetric MP-RAGE scans were collected for each subject. Parameter values varied depending on scanning site and can be found at www.loni.ucla.edu/ADNI/Research/Cores/. Each MRI underwent a quality control evaluation at Mayo Clinic. Examinations were evaluated for the presence of structural abnormalities; presence and severity of common artifacts (e.g. blurring due to head motion) were indicated, and one of the two MPRAGE scans was recommended for use.
MPRAGE images underwent specific preprocessing correction steps: a system specific correction of image geometry distortion due to gradient nonlinearity, an image intensity nonuniformity correction using the B1 calibration scans, and a further nonuniformity correction using N3 histogram peak sharpening algorithm; the need to perform such preprocessing steps varied with manufacturer and system RF coil configuration. More detailed information can be found at www.loni.ucla.edu/ADNI/Data/ADNI_Data.shtml.
Left and right hippocampal volumes were semiautomatically computed at University of California, San Francisco, using a commercially available high dimensional brain mapping tool (Medtronic Surgical Navigation Technologies (SNT), Louisville, CO) based on fluid image transformation (Christensen et al., 1997) and previously validated (intra-class coefficient > 0.94) (Hsu et al., 2002). The software requires to manually place 2 global landmarks on AC and PC location for data reslice along AC-PC plane, and 44 local landmarks surrounding the left and right hippocampus; once scans are fully landmarked, they are processed by Medtronics algorithms, which produce hippocampal boundaries and volumes; boundaries are checked by qualified reviewers and in case of failure can be manually edited.
In the current study, individual left and right hippocampal volumes were averaged to have a single hippocampal volume measure.
2.5. Statistical analysis
All statistical analyses were carried out using the R statistical software (www.r-project.org/).
Significance of difference among the four groups was assessed by one-way ANOVA for all continue variables, and by the nonparametric χ2 test for categorical variables, i.e. gender and APOE. Post-hoc Tukey HSD test was used to estimate the between-group differences. For all comparisons, the significance threshold was set at 0.05.
Mean and standard deviation from the group of healthy controls were used to Z-transform all subjects and patients individual biomarker values at baseline (Aβ 1–42, t-tau, FDG metabolism and hippocampal volumes) according to the following formula: Z-biomarkeri(subjectj) = (biomarkeri (subjectj) – mean-biomarkeri(CN))/SD-biomarkeri(CN), to have standardized measures.
2.6. Biomarker dynamics
To investigate biomarker dynamics, all subjects and patients were ordered based on their Alzheimer's disease assessment scale-cognitive (ADAS-Cog) score (classic 70 point total), which was considered as a surrogate marker of time since AD developed (AD stage). As each biomarker has been shown to change over time, with rates of change following a nonlinear time course, likely sigmoid shaped (Carlson et al., 2008; Chan et al., 2003; Jack et al., 2008b; Jack et al., 2010; Ridha et al., 2006), for each biomarker individual Z scores, after being polarized to have increasing Z scores for increasing disease stage, were plotted against ADAS-Cog scores and the three parameters (asym, xmid and scal) of the generic sigmoid curve (y = asym/(1+ exp ((xmid-x)/scal)), Figure 1) were fitted using a nonlinear least square algorithm (nls function of the R software); for each of the fitted parameters, 95% confidence intervals were computed, and R2 was used as a measure of goodness of fit. The statistical difference between parameters estimated for different biomarkers was assessed looking at the overlap of the 84% confidence intervals, which were shown to give an approximate α = 0.05 test (95% intervals giving very conservative results) under the assumption of approximately equal standard errors (Payton et al., 2003).
Fig. 1.

Schematic representation of the generic sigmoid curve (y = asym/ (1+exp (xmid-x)/scal)). Asym is the asymptote, xmid the inflection point x-value (distance from origin), and one/scal is the angular coefficient of the tangent (i.e. the slope) at point of inflection.
For each biomarker, the sigmoid fit was compared with the linear fit. Goodness of fits was first assessed comparing sum of squares. In case linear sum of squares was higher than sigmoid one (suggesting sigmoid fit could be better than linear one), an F test was run to compare the relative increase in sum of squares with the relative increase in degree of freedom (linear model having 1 degree of freedom more than sigmoid model): F ratio was computed using the following formula:
with SS = sum of square, and d.f. = degrees of freedom, and, in case F ratio was higher than 1 (further suggesting that sigmoid fit could be better), the pertinent p value was computed (F test, numerator d.f. = linear d.f. – sigmoid d.f., denominator d.f. = sigmoid d.f.) to find out whether the sigmoid fit was significantly better than the linear one.
Subjects and patients included in the study were then divided into two groups according to APOE genotype (ε4 carriers and ε4 non carriers), biomarker dynamics was further investigated, and fitted sigmoid curves were computed for each of the two groups.
3. Results
Based on the criteria described in the previous section, 229 healthy controls (age = 76 ± 5 years, 48% females), 154 MCI patients converted to AD (age = 74 ± 7 years, 39% females, conversion time = 17 ± 8 [6–36] months), and 193 AD patients (95 early AD, aged 75 ± 7 years, 45% females, and 98 late AD, aged 75 ± 8 years, 49% females) from ADNI dataset were included in this study.
Table 1 shows main sociodemographic, clinical and neuropsychological features of the four groups of subjects enrolled in the study: the groups did not significantly differ in age and gender but differed in education, healthy subjects and MCI patients having higher education than AD patients; as expected, significant differences in MMSE and ADAS-Cog scores were found, reflecting different stage of cognitive impairment among the groups; APOE ε4 prevalence was found to be significantly different among the groups due to the large difference between cognitively healthy group (27% carriers) and the other three groups (MCI converted to AD: 69%, early AD: 68%, and late AD: 66% carriers), confirming that all patients included in the study, despite being at different stage of the disease, are affected by AD. In all tests of the neuropsychological battery, a significant difference (p < 0.0001) among the groups was observed.
Table 1. Sociodemographic, clinical and neuropsychological features of ADNI healthy controls, MCI converted to AD, early and late AD patients.
| Healthy controls (n = 229) | MCI converted AD (n = 154) | Early AD (n = 95) | Late AD (n = 98) | p | |
|---|---|---|---|---|---|
| Age, years | 76 ± 5 | 74 ±7 | 75 ±7 | 75 ± 8 | 0.52 |
| Gender, females | 110 (48%) | 60 (39%) | 43 (45%) | 48 (49%) | 0.29 |
| Education, years | 16 ± 3 | 16 ±3 | 15 ±3 | 14 ± 3 | < 0.0001 |
| MMSE | 29 ± 1 | 27 ±2 | 25 ± 1 | 22 ± 2 | < 0.0001 |
| ADAS-Cog, classic 70 | 6±3 | 13 ±4 | 16 ±5 | 20 ± 7 | < 0.0001 |
| Clock drawing test | 4.6 ± 0.7 | 3.9 ± 1.1 | 3.7 ± 1.2 | 3.1 ± 1.3 | < 0.0001 |
| AVLT (immediate recall) | 43.0 ± 9.8 | 27.2 ± 6.4 | 24.1 ± 7.3 | 21.9 ± 8.2 | < 0.0001 |
| Digit span − forward | 8.8 ± 2.0 | 8.3 ± 2.0 | 7.8 ± 1.7 | 7.3 ±2.1 | < 0.0001 |
| Digit span − backward | 7.2 ± 2.2 | 6.0 ± 1.8 | 5.2 ± 1.9 | 4.6 ± 1.9 | < 0.0001 |
| Category fluency (anim.) | 19.9 ± 5.6 | 15.4 ± 4.9 | 13.3 ± 4.6 | 11.4 ± 5.0 | < 0.0001 |
| Category fluency (veg.) | 14.7 ± 3.9 | 10.0 ± 3.2 | 8.4 ± 3.3 | 7.2 ± 3.3 | < 0.0001 |
| Trail making test A | 36 ± 13 | 50 ± 26 | 64 ± 35 | 70 ± 39 | < 0.0001 |
| Trail making test B | 89 ± 44 | 153 ± 82 | 177 ± 92 | 198 ±98 | < 0.0001 |
| Digit symbol | 45.7 ± 10.2 | 34.0 ±11.2 | 29.0 ± 12.6 | 23.8 ± 13.5 | < 0.0001 |
| Boston naming | 27.8 ± 3.0 | 25.0 ± 4.3 | 23.6 ± 5.9 | 20.3 ± 7.6 | < 0.0001 |
| AVLT (30 min delayed) | 7.4 ± 3.7 | 1.6 ± 2.4 | 0.8 ± 1.5 | 0.7 ± 1.7 | < 0.0001 |
| ANART | 9.7 ± 9.1 | 13.6 ± 9.2 | 14.2 ± 10.0 | 16.9 ± 10.3 | < 0.0001 |
| APOE, carriers | 61 (27%) | 107 (69%) | 65 (68%) | 62 (63%) | < 0.0001 |
p denotes difference significance among all groups on one-way ANOVA (continuous variables) or χ2 test (categorical variables).
Values are mean ± standard deviations (continuous variables) or frequencies (categorical values, i.e. gender and APOE).
MMSE, Mini Mental State Examination; ADAS-Cog, Alzheimer's disease assessment scale-cognitive; ANART, American national adult reading test; AVLT, auditory verbal learning test–version A; Digit symbol, WAIS-R digit symbol substitution test.
About half of the healthy subjects (114/229) and patients (181/347) considered in the current study underwent lumbar puncture thus having CSF Aβ 1–42 and tau data available; half of subjects and patients underwent FDG-PET imaging, and slightly less had data available due to technical failures; all of them underwent MR imaging and more than a half had hippocampal volume available (Table 2).
Table 2. AD biomarkers in ADNI healthy controls, MCI converted to AD, early and late AD patients.
| Healthy controls | MCI converted AD | Early AD | Late AD | p | |
|---|---|---|---|---|---|
| Tau | |||||
| n | 114 | 79 | 57 | 45 | |
| pg/mL | 69.7 ± 30.4 | 110.3 ± 51.1 | 113.9 ± 61.2 | 125.8 ± 57.6 | < 0.0001 |
| Z-score | 0.00 ± 1.00 | 1.41 ± 1.75 | 1.54 ± 2.09 | 1.94 ± 1.97 | |
| Aβ 1–42 | |||||
| n | 114 | 79 | 57 | 45 | |
| pg/mL | 205.6 ± 55.1 | 141.9 ± 43.9 | 144.3 ± 45.8 | 141.3 ± 33.9 | < 0.0001 |
| Z-score | 0.00 ± 1.00 | 1.25 ± 0.82 | 1.21 ± 0.86 | 1.27 ± 0.63 | |
| FDG-PET | |||||
| n | 102 | 67 | 53 | 44 | |
| CRMglc | 1.39 ± 0.12 | 1.29 ±0.11 | 1.29 ± 0.10 | 1.23 ± 0.12 | < 0.0001 |
| Z-score | 0.00 ± 1.00 | 0.84 ± 0.94 | 0.87 ± 0.88 | 1.35 ± 1.02 | |
| Hippocampal volume | |||||
| n | 159 | 119 | 63 | 66 | |
| mL | 2155 ± 297 | 1739 ± 363 | 1654 ± 356 | 1599 ± 323 | < 0.0001 |
| Z-score | 0.00 ± 1.00 | 1.43 ± 1.25 | 1.73 ± 1.23 | 1.92 ± 1.11 |
p denotes difference significance among all groups on one-way ANOVA.
Values are mean ± standard deviations.
Mean and standard deviation from the subgroup of cognitive normal subjects were used to compute Z scores.
Hippocampal volumes were averaged over left and right.
FDG-PET is the average cerebral metabolic rate of glucose consumption in frontal, parietal and temporal cortices normalized to pons.
Tau was significantly different among groups (p < 0.0001), healthy controls having the lowest mean value and patients at increasing stage of AD having increasing mean values; Z-scores reflected the monotonous increase. All AD groups were found to be significantly different in CSF tau concentration from healthy controls on post-hoc analysis, while no significant difference was found between AD groups. Aβ 1–42 was significantly different among groups (p < 0.0001), and healthy controls Aβ 1–42 mean value was much higher than patient ones; patients at different disease stage had similar Aβ 1–42 mean values, both in terms of absolute values and Z-score, suggesting that Aβ 1–42 load could be almost disconnected from the disease stage. On post-hoc analysis, all AD groups were found to be significantly different in Aβ 1–42 concentration from healthy controls, while no significant difference was found between AD groups. Mean CRMglc in frontal, parietal and temporal cortices was significantly different among groups (p < 0.0001); healthy controls had the highest CRMglc mean values, MCI converted to AD and early AD had comparable values while late AD had the lowest ones (both in terms of absolute values and Z scores), as mean CRMglc mostly decreased in the presymptomatic stage and continued to slightly decrease even at latest stages. On post-hoc analysis, all AD groups were significantly different in CRMglc mean values from healthy controls, MCI converted to AD were significantly different from late AD (p = 0.04), and early AD were almost significantly different from late AD (p = 0.08), while no difference was found between MCI converted to AD and early AD. Hippocampal volume was significantly different among groups (p < 0.0001), healthy controls having the highest mean value and patients at increasing stage of AD having decreasing mean values; Z-scores reflected the monotonous increase (table 2). All AD groups were significantly different in hippocampal volume from healthy controls on post-hoc analysis, MCI converted to AD were significantly different from late AD (p = 0.03), while no difference was found between early AD and either MCI converted to AD or late AD.
Figure 2 shows, for each biomarker, the individual Z scores plotted against ADAS-Cog scores; fitted sigmoid curves are overlapped. Table 3 shows the fitted sigmoid parameters, representing the asymptote (asym), the inflection point x-value (xmid), and the steepness (one/scal) of the sigmoid curve, with 95% confidence intervals. As Z scores are quite dispersed, R2 coefficients, measuring the goodness of fit, are quite low; the sigmoid curve seems to fit Aβ 1–42 and hippocampal volume (R2 = 0.256 and 0.279, respectively) better than tau and FDG-PET (R2 = 0.137 and 0.201, respectively). Comparing the fitted sigmoid curves, the FDG-PET inflection point x-value was found to be significantly different from all the other ones.
Fig. 2.
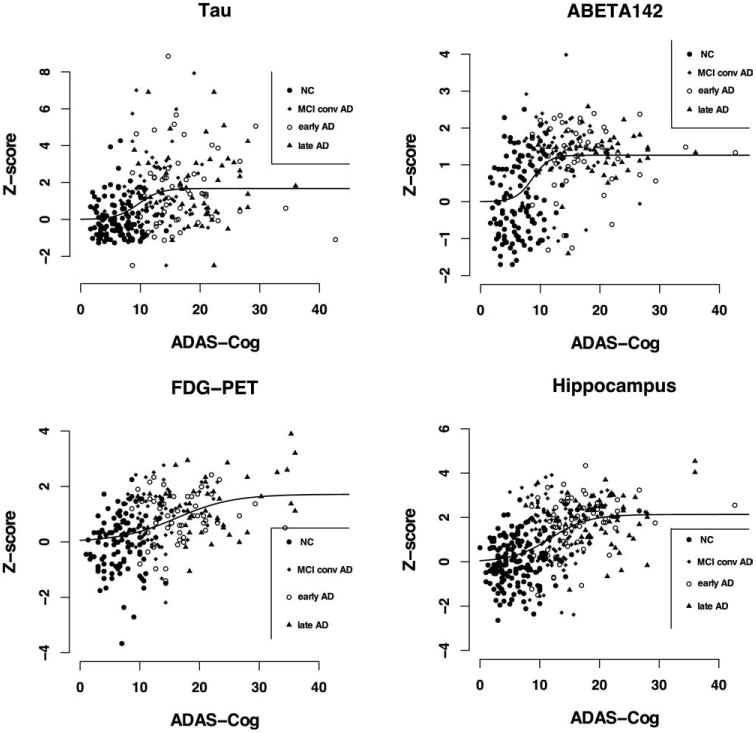
For each biomarker, individual Z scores are plotted against ADAS-Cog scores, and the fitted sigmoid curve is displayed. Full circles denote healthy controls, full squares MCI patients converted to AD, empty circles early AD, and full triangles late AD patients. Sigmoid fitting was better than linear fitting for Tau, Aβ 1–42 and hippocampus (for the latter: sigmoid nonsignificantly better than linear); linear fitting was better for FDG-PET.
Table 3.
Fitted sigmoid parameters. Individual Z scores were plotted against ADAS-Cog scores and the three parameters (asym, xmid and scal) of the generic sigmoid curve (y = asym/(1 + exp((xmid-x)/scal))) were fitted using a nonlinear least square algorithm. Sigmoid was better than linear fit for CSF Aβ 1–42, tau and hippocampal volume (for the latter: sigmoid nonsignificantly better than linear); linear fitting was better for FDG-PET.
| asym | xmid | scal | R2 | |
|---|---|---|---|---|
| Tau | 1.68 [1.31–2.05] | 9.67 [7.72–11.62] | 1.85 [0.14–3.56] | 0.137 |
| Aβ 1–42 | 1.26 [1.09–1.43] | 8.71 [7.58–9.83] | 1.40 [0.41–2.40] | 0.256 |
| FDG-PET | 1.72 [1.07–2.37] | 16.13 [11.16–21.09]* | 4.77 [1.88–7.65] | 0.201 |
| Hippocampal volume | 2.13 [1.76–2.51] | 11.82 [9.98–13.66] | 3.18 [1.78–4.57] | 0.279 |
Values are fitted parameters [95% confidence interval].
Denotes significant difference versus all other estimated parameters.
For all biomarkers except FDG-PET, the linear was higher than the sigmoid sum of squares (Aβ 1–42: 268.93 v. 246.21; tau: 860.33 v. 828.09; hippocampal volume: 576.71 v. 573.23) and the sigmoid fit was significantly better than the linear for Aβ 1–42 and tau (Aβ 1–42: F = 26.94, p < 0.0001; tau: F = 11.37, p < 0.001, hippocampal volume: F = 2.45, n.s.). For FDG-PET, the linear sum of squares was slightly lower than the sigmoid one (255.11 and 260.05, n.s.), suggesting that the linear fit could be better than that of the sigmoid model.
Among the 576 subjects and patients included in the study, 295 were APOE ε4 carriers and 281 non carriers. Of these, 150 ε4 carriers and 145 non carriers had valid CSF Aβ 1–42 and tau values, 218 ε4 carriers and 189 non carriers had valid hippocampal volume values, and 135 ε4 carriers and 131 non carriers had valid FDG-PET data. Fitted sigmoid curves could not be computed for CSF tau and FDG-PET in APOE ε4 carriers, and for Aβ 1–42 in non carriers due to failure of the fit to converge.
The sigmoid fit of hippocampal volume in APOE ε4 carriers was found to be significantly steeper than the one related to non carriers, and the points of inflection of the two curves on the x-axis were significantly different, indicating that carriers developed hippocampal atrophy earlier in the disease course (Figure 3).
Fig. 3.

Individual hippocampal volume Z scores are plotted against ADAS-Cog for both APOE ε4 carriers (full circles), and non-carriers (empty circles). The fitted sigmoid curves for the whole population (thin dotted), APOE ε4 carriers (thick solid), and non-carriers (thick dashed) are displayed. The parameters of each of the three fitted sigmoids are reported in the bottom table. Values are fitted parameters [95% confidence interval], and * denotes significant difference in APOE ε4 carriers vs non-carriers.
4. Discussion
In the current study we considered a group of healthy controls and three groups of Alzheimer's patients with increasing cognitive impairment, which could be considered as representative of the neurobiological continuum of the disease, and we investigated the dynamics of four of the most validated AD biomarkers: CSF Aβ 1–42, CSF tau, hippocampal volume and FDG uptake on PET.
Both CSF tau and hippocampal volume showed a monotonous trend, patients with increasingly severe AD showing increasing values of tau and decreasing hippocampal volumes. This is in line with previous evidence that both CSF tau (Arai et al., 1995; Blennow et al., 1995; Buerger et al., 2006; Tapiola et al., 2009) and hippocampal volume (Jack et al., 2000; Schuff et al., 2009) are valid biomarkers of neurodegeneration, and they both correlate with disease severity during the whole time course of the disease (Arai et al., 1995; Hansson et al., 2006; Jack et al., 2000; Schuff et al., 2009; Shaw et al., 2009; Tapiola et al., 2009; Vemuri et al., 2009).
As expected, AD patients at different disease stage showed similar Aβ 1–42 load (much lower than healthy controls), in agreement with previous evidence that plaque deposition occurs before neurodegeneration and by the time a person has dementia it becomes almost disconnected from the disease duration and severity (Chételat et al., 2010; Ingelsson et al., 2004; Jack et al., 2008a; Jack et al., 2009). As for FDG-PET, preclinical and early AD showed comparable CRMglc mean values, while late AD showed further reduced metabolism. This is in line with previous evidence that alterations in glucose metabolism, reflecting synaptic function and density, already occurs at a preclinical stage (de Leon et al., 2001; Jagust et al., 2006; Mosconi et al., 2009; Reiman et al., 1996; Small et al., 1995), and accompany neurodegeneration, progressive reductions correlating with disease severity (Hoffman et al., 2000; Minoshima et al., 1997; Mosconi et al., 2009).
To our knowledge, this is the first study testing the model of AD biomarker dynamics recently proposed by Jack Jr and colleagues (Jack et al., 2010) on real data. The ongoing ADNI, which has been recently shown to have successfully recruited a large cohort of healthy controls, MCI and AD patients very similar to those seen in MCI and mild AD clinical trials (Petersen et al., 2010), could be considered at present as the gold standard dataset for the study of Alzheimer's disease, and thus the best data choice. In the model, each biomarker was hypothesized to follow a nonlinear and sigmoid-shaped time course. The generic sigmoid function (y = asym/(1 + exp((xmid-x)/scal)), Figure 1) is defined by three parameters: the horizontal asymptote (asym), which gives an indication of the time (disease stage) when the biomarker stabilizes, the x-value at inflection point (xmid), representing the time when maximum variation occurs (most relevant time when to monitor the biomarker to have indications of disease progression), and the steepness of the curve (1/scal), which is a measure of the rate of change.
In this study we showed that different sigmoid curves could actually be used to describe each biomarker time course. Despite individual Z scores being quite dispersed, and thus fitted curves having low R2 coefficients, fitted sigmoid curves are still meaningful: Aβ 1–42 fitted sigmoid is steep, has its maximum variation and stabilizes early in time (i.e. in nonpathological ADAS-Cog range), in line with previous evidence of plaque deposition mainly occurring in the preclinical phase (Ingelsson et al., 2004; Jack et al., 2008a; Jack et al., 2009) and with Jack's model (Jack et al., 2010); CSF tau and hippocampal volume fitted sigmoids have similar shapes (in terms of asymptote, rate of change and steepness), in agreement with the notion that they are both markers of neurodegeneration and their change reflects disease progression during the whole time course, and they could be considered late biomarkers (Jack et al., 2010); FDG-PET fitted sigmoid starts to increase early in time, in line with previous evidence of preclinical alterations in glucose metabolism (Jagust et al., 2006; Mosconi et al., 2009), and seems to reach a steady state only at latest stages of the disease, reflecting metabolism reduction occurring during the whole time course of the disease (Jack et al., 2010).
Furthermore, for three of the four biomarkers under study (CSF Aβ 1–42, tau and hippocampal volume) the sigmoid model was found to fit biomarker dynamics better than the linear model (significantly better for both Aβ 1–42 and tau). This is an interesting finding, which provides the first evidence to the model proposed by Jack and colleagues (Jack et al., 2010).
It would be now extremely important to compare biomarker dynamics, and order biomarker changes in time to express the disease process in terms of a series of testable biological indicators. However, before fitted curves could be directly compared, some considerations need to be introduced, showing that significant differences reported in Table 3 should be taken with caution. Although each biomarker sigmoid curve was fitted on individual Z-transformed scores to standardize different biomarker values making them comparable, the use of such Z scores has an intrinsic problem. For each biomarker, Z scores were computed based on mean and standard deviations of the healthy controls group, and thus strongly depend on the variability of the biomarker within healthy controls. As variability is due both to measurement error and biological variability, and different biomarkers may have different measurement errors, the variability due to measurement error should ideally be eliminated before computing z-scores. Studies assessing and comparing the reliability of each biomarker (i.e. reproducibility studies with repeated measures) are needed to remove the effect of measurement error and compute corrected Z-scores that reflect biological variability only. This is likely the reason why, directly comparing FDG-PET versus hippocampal volume fitted curves, FDG-PET seems to have a later rise, in contrast to previous findings (Reiman et al., 1998).
The sigmoid curves describing hippocampal volume change fitted for APOE ε4 carriers and non-carriers were found to be significantly different: the one related to APOE ε4 carriers was significantly steeper than the one related to non carriers, and its point of inflection x-values was significantly shifted to the left, suggesting than in APOE ε4 carriers hippocampal atrophy occurs earlier in time.
This finding is in line with a recent study suggesting that APOE ε4 may alter the relationship between biomarkers and cognitive state (Vemuri et al., 2010), and with previous evidence that APOE ε4 carriers have smaller hippocampal volume (Reiman et al., 1998) and faster hippocampal loss (Schuff et al., 2009); this finding further supports the validity of the model proposed by Jack and colleagues (Jack et al., 2010).
To set patients along the disease stage continuum, either a biological or a clinical strategy could be adopted. For the purpose of studying biomarker dynamics no biological variable could be chosen without falling into logical recursivity, and a clinical variable (e.g. ADAS-Cog score) thus needed to be used.
Biomarker differences among groups of AD patients at different disease stages have been interpreted as biomarker changes occurring during the disease course. Current findings, achieved by cross-sectional analysis, should be considered as preliminary, and need to be verified through a truly longitudinal analysis.
It should be noted that biomarker dynamics has not been assessed on the same subject cohort as, for each biomarker, different subgroups of subjects had data available. It was not feasible to include in the study only subjects with all four biomarkers available as the sample size would have notably decreased (just 105 out of 576 subjects included in the current study had all biomarkers available) and the power of the analysis would have thus been severely reduced. As differences in subgroups used to assess biomarker dynamics could have potentially affected the study, current findings need to be verified on a larger sample with all biomarker data available.
In the current study we did not include stable MCI patients: to assess the biomarker dynamics, as homogeneous as possible groups at different time of the Alzheimer's disease course are needed, and the highly heterogeneous stable MCI group, including patients with incipient AD and with different underlying pathologies, patients who will indeed remain stable and who will revert to cognitively normal status, would have biased the study. Additional analyses on biomarker dynamics assessed including stable MCI (available online at www.centroalzheimer.it/public/Supplemental_analyses_stableMCI.doc) confirmed the main findings of the current study, despite the shape of the biomarker dynamics, likely due just to the biased composition of the additional group, was found to be closer to linear.
In the current study we did not consider PIB-PET, despite being one of the most validated AD biomarkers, for two main reasons: ADNI PIB processed data available at present are fewer than the other biomarkers; furthermore, PIB-PET has been shown to be substantially related to CSF Aβ 1–42, the two measures of brain Aβ deposition producing similar results (Jagust et al., 2009).
All patients included in the study had incipient or mild AD, and biomarker dynamics was thus investigated on a relatively narrow disease continuum. It will be interesting to investigate in the future the dynamic changes both rightward (i.e. later in the disease time course), and leftward (i.e. in the presymptomatic phase), ideally following healthy people in time throughout the whole course of the disease and modeling the thresholds where clinical symptoms occur. It will also be interesting to investigate the effect of co-occurring diseases and conditions on biomarker dynamics. Furthermore, as structural loss and synaptic dysfunction do not occur at the same time throughout the brain (Buckner et al., 2005; Frisoni et al., 2009), it will be interesting to investigate the structural and functional variations in disease-specific cerebral regions (e.g. posterior cingulate, medial temporal, lateral temporal and frontal) during the whole disease time course.
In conclusion, in this study we used the ADNI dataset to provide the first evidence in favor of the dynamic biomarker model proposed by Jack and colleagues (Jack et al., 2010), showing that most of the biomarker' dynamics follow a sigmoid trend. Aβ 1–42 time course was found to follow a steep curve, stabilizing early in the disease course; CSF tau and hippocampal volume changed later in time and showed similar monotonous trends, reflecting disease progression during the whole disease time course. Hippocampal volume loss was found to be steeper and to occur earlier in time in APOE ε4 carriers than in non-carriers, proving additional evidence of validity of the model. Despite providing only partial support for a temporal shift between different types of pathological brain changes in AD, these findings suggest that, as an early marker, Aβ 1–42 could be used for clinical trials as inclusion criteria, to select patients with preclinical AD, while markers of neural degeneration and dysfunction (e.g. CSF tau, hippocampal volume and FDG-PET) could be used as outcome measures to investigate the drug effect on neurodegeneration.
Acknowledgments
Data collection and sharing for this project was funded by the ADNI (National Institutes of Health, Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly, and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer, Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., and Wyeth, as well as nonprofit partners the Alzheimer's Association and Alzheimer's Drug Discovery Foundation, with participation from the US Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fni-h.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH Grants P30 AG010129, K01 AG030514, and the Dana Foundation.
Footnotes
Data used in the preparation of this article were obtained from the ADNI database (www.loni.ucla.edu/ADNI). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators is available at: www.loni.ucla.edu/ADNI/Collaboration/ADNI_Authorship_list.pdf).
Disclosure statement: The authors report no actual or potential conflicts of interest.
References
- Arai H, Terajima M, Miura M, Higuchi S, Muramatsu T, Machida N, Seiki H, Takase S, Clark CM, Lee VM, Trojanowski JQ, Sasaki H. Tau in cerebrospinal fluid: a potential diagnostic marker in Alzheimer's disease. Ann Neurol. 1995;38:649–652. doi: 10.1002/ana.410380414. [DOI] [PubMed] [Google Scholar]
- Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol. 1995;26:231–245. doi: 10.1007/BF02815140. [DOI] [PubMed] [Google Scholar]
- Bobinski M, de Leon MJ, Wegiel J, Desanti S, Convit A, Saint Louis LA, Rusinek H, Wisniewski HM. The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer's disease. Neuroscience. 2000;95:721–725. doi: 10.1016/s0306-4522(99)00476-5. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, laRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerger K, Ewers M, Pirttilä T, Zinkowski R, Alafuzoff I, Teipel SJ, deBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain. 2006;129:3035–3041. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- Carlson NE, Moore MM, Dame A, Howieson D, Silbert LC, Quinn JF, Kaye JA. Trajectories of brain loss in aging and the development of cognitive impairment. Neurology. 2008;70:828–833. doi: 10.1212/01.wnl.0000280577.43413.d9. [DOI] [PubMed] [Google Scholar]
- Chan D, Janssen JC, Whitwell JL, Watt HC, Jenkins R, Frost C, Rossor MN, Fox NC. Change in rates of cerebral atrophy over time in early-onset Alzheimer's disease: longitudinal MRI study. Lancet. 2003;362:1121–1122. doi: 10.1016/S0140-6736(03)14469-8. [DOI] [PubMed] [Google Scholar]
- Chételat G, Villemagne VL, Bourgeat P, Pike KE, Jones G, Ames D, Ellis KA, Szoeke C, Martins RN, O'Keefe GJ, Salvado O, Masters CL, Rowe CC. Australian Imaging Biomarkers and Lifestyle Research Group. Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol. 2010;67:317–324. doi: 10.1002/ana.21955. [DOI] [PubMed] [Google Scholar]
- Christensen GE, Joshi SC, Miller MI. Volumetric transformation of brain anatomy. IEEE Trans Med Imaging. 1997;16:864–877. doi: 10.1109/42.650882. [DOI] [PubMed] [Google Scholar]
- Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, Morris JC, McKeel DW, Jr, Farlow M, Weitlauf SL, Quinn J, Kaye J, Knopman D, Arai H, Doody RS, deCarli C, Leight S, Lee VM, Trojanowski JQ. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol. 2003;60:1696–1702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory. Elsevier Saunders; Philadelphia: 2005. [DOI] [PubMed] [Google Scholar]
- de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, Tsui W, Kandil E, Scherer AJ, Roche A, Imossi A, Thorn E, Bobinski M, Caraos C, Lesbre P, Schlyer D, Poirier J, Reisberg B, Fowler J. Prediction of cognitive decline in normal elderly subjects with two- [(18)F] fluoro-2-deoxy-d-glucose/ poitronemission tomography (FDG/PET) Proc Natl Acad Sci USA. 2001;98:10966–10971. doi: 10.1073/pnas.191044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F] FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, laRossa GN, Spinner ML, Klunk WE, Mathis CA, deKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Lorenzi M, Caroli A, Kemppainen N, Någren K, Rinne JO. In vivo mapping of amyloid toxicity in Alzheimer disease. Neurology. 2009;72:1504–1511. doi: 10.1212/WNL.0b013e3181a2e896. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Fox NC, Jack CR, Jr, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6:67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosche KM, Mortimer JA, Smith CD, Markesbery WR, Snowdon DA. Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology. 2002;58:1476–1782. doi: 10.1212/wnl.58.10.1476. [DOI] [PubMed] [Google Scholar]
- Hampel H, Burger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer's disease. Alzheimers Dement. 2008;4:38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- Hoffman JM, Welsh-Bohmer KA, Hanson M, Crain B, Hulette C, Earl N, Coleman RE. FDG PET imaging in patients with pathologically verified dementia. J Nucl Med. 2000;41:1920–1928. [PubMed] [Google Scholar]
- Hsu YY, Schuff N, Du AT, Mark K, Zhu X, Hardin D, Weiner MW. Comparison of automated and manual MRI volumetry of hippocampus in normal aging and dementia. J Magn Reson Imaging. 2002;16:305–310. doi: 10.1002/jmri.10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, deKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Petersen RC, Xu Y, O'Brien PC, Smith GE, Ivnik RJ, Boeve BF, Tangalos EG, Kokmen E. Rates of hippocampal atrophy correlate with change in clinical status in aging and Alzheimer's disease. Neurology. 2000;55:484–489. doi: 10.1212/wnl.55.4.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008a;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Weigand SD, Shiung MM, Przybelski SA, O'Brien PC, Gunter JL, Knopman DS, Boeve BF, Smith GE, Petersen RC. Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology. 2008b;70:1740–1752. doi: 10.1212/01.wnl.0000281688.77598.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Alzheimer's Disease Neuroimaging. Initiatives Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Gitcho A, Sun F, Kuczynski B, Mungas D, Haan M. Brain imaging evidence of preclinical Alzheimer's disease in normal aging. Ann Neurol. 2006;59:673–681. doi: 10.1002/ana.20799. [DOI] [PubMed] [Google Scholar]
- Jagust WJ, Reed B, Mungas D, Ellis W, Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–877. doi: 10.1212/01.wnl.0000269790.05105.16. [DOI] [PubMed] [Google Scholar]
- Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, Foster NL, Petersen RC, Weiner MW, Price JC, Mathis CA Alzheimer's Disease Neuroimaging Initiative. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193–1199. doi: 10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan EF, Goodglass H, Weintraub S. The Boston Naming Test. 2nd. Lea and Febiger; Philadelphia: 1982. [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Mistur R, Switalski R, Tsui W, Glodzik L, Li Y, Pirraglia E, Santi S, Reisberg B, Wisniewski T, de Leon MJ. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2009;36:811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51:336–345. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- Payton ME, Greenstone MH, Schenker N. Overlapping confidence intervals or standard error intervals: What do they mean in terms of statistical significance? J Insect Sci. 2003;3:34. doi: 10.1093/jis/3.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR, Jr, Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–209. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Uecker A, Caselli RJ, Lewis S, Bandy D, de Leon MJ, De Santi S, Convit A, Osborne D, Weaver A, Thibodeau SN. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer's disease. Ann Neurol. 1998;44:288–291. doi: 10.1002/ana.410440226. [DOI] [PubMed] [Google Scholar]
- Reitan R. Validity of the Trail-Making Test as an indication of organic brain damage. Percept Mot Skills. 1958;8:271–276. [Google Scholar]
- Rey A. l'Examen Clinique en Psychologie. Presses Universitaires de France; Paris: 1964. [Google Scholar]
- Ridha BH, Barnes J, Bartlett JW, Godbolt A, Pepple T, Rossor MN, Fox NC. Tracking atrophy progression in familial Alzheimer's disease: a serial MRI study. Lancet Neurol. 2006;5:828–834. doi: 10.1016/S1474-4422(06)70550-6. [DOI] [PubMed] [Google Scholar]
- Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. Am J Psychiatry. 1984;141:1356–1364. doi: 10.1176/ajp.141.11.1356. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O'Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Schoonenboom NS, van der Flier WM, Blankenstein MA, Bouwman FH, Van Kamp GJ, Barkhof F, Scheltens P. CSF and MRI markers independently contribute to the diagnosis of Alzheimer's disease. Neurobiol Aging. 2008;29:669–675. doi: 10.1016/j.neurobiolaging.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, Thompson PM, Jack CR, Jr, Weiner MW. Alzheimer's Disease Neuroimaging Initiative. MRI Of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain. 2009;132:1067–1077. doi: 10.1093/brain/awp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Korecka M, Clark CM, Lee VM, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov. 2007;6:295–303. doi: 10.1038/nrd2176. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ Alzheimer's Disease Neuroimaging Initiative. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbert LC, Quinn JF, Moore MM, Corbridge E, Ball MJ, Murdoch G, Sexton G, Kaye JA. Changes in premorbid brain volume predict Alzheimer's disease pathology. Neurology. 2003;61:487–492. doi: 10.1212/01.wnl.0000079053.77227.14. [DOI] [PubMed] [Google Scholar]
- Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, Kaplan A, La Rue A, Adamson CF, Chang L, Guze BH, Corder EH, Saunders AM, Haines JL, Pericak-Vance MA, Roses AD. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA. 1995;273:942–947. [PubMed] [Google Scholar]
- Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttilä T. Cerebrospinal fluid {beta}-amyloid forty-two and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66:382–389. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- Van de Pol LA, Hensel A, van der Flier WM, Visser P, Pijnenburg YA, Barkhof F, Gertz HJ, Scheltens P. Hippocampal atrophy on MRI in frontotemporal lobar degeneration and Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2006;77:439–442. doi: 10.1136/jnnp.2005.075341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR, Jr Alzheimer's Disease Neuroimaging Initiative. MRI and CSF biomarkers in normal, MCI, and AD subjects: diagnostic discrimination and cognitive correlations. Neurology. 2009;73:287–293. doi: 10.1212/WNL.0b013e3181af79e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Shaw LM, Trojanowski JQ, Aisen PS, Weiner M, Petersen RC, Jack CR., Jr Alzheimer's Disease Neuroimaging Initiative. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol. 2010;67:308–316. doi: 10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler DA. Wechsler Adult Intelligence Scale–Revised. Psychological Corporation; New York: 1987. [Google Scholar]
- Zarow C, Vinters HV, Ellis WG, Weiner MW, Mungas D, White L, Chui HC. Correlates of hippocampal neuron number in Alzheimer's disease and ischemic vascular dementia. Ann Neurol. 2005;57:896–903. doi: 10.1002/ana.20503. [DOI] [PMC free article] [PubMed] [Google Scholar]