

Amyotrophic lateral sclerosis (ALS) is a neurologic disorder, characterized by progressive degeneration of both upper and lower motor neurons in the brain and spinal cord. Previous genetic studies have identified mutations in Cu/Zn superoxide dismutase (SOD1), transactive response binding protein 43 (TARDBP), fused in sarcoma (FUS), and valosin containing protein (VCP) genes as being causative of disease.1 Recently, an expansion of the noncoding GGGGCC hexanucleotide repeat in chromosome 9 open reading frame 72 (C9ORF72) was identified as an important novel genetic defect in patients with ALS without or with frontotemporal dementia (FTD-ALS).2,3 Here we report the frequency of this new mutation and its associated clinical features in a cohort of patients obtained from the Coriell Cell Repository.
Methods.
We studied 617 patients with a diagnosis of ALS (n = 568), FTD-ALS (n = 20), progressive muscular atrophy (PMA; n = 26), primary lateral sclerosis (PLS; n = 2), and progressive bulbar palsy (PBP; n = 1). DNA samples from patients were obtained from the Coriell Institute for Medical Research. Table 1 summarizes detailed demographic and clinical information.
Table 1.
Clinical characteristics in patients with and without C9ORF72 hexanucleotide repeat expansions
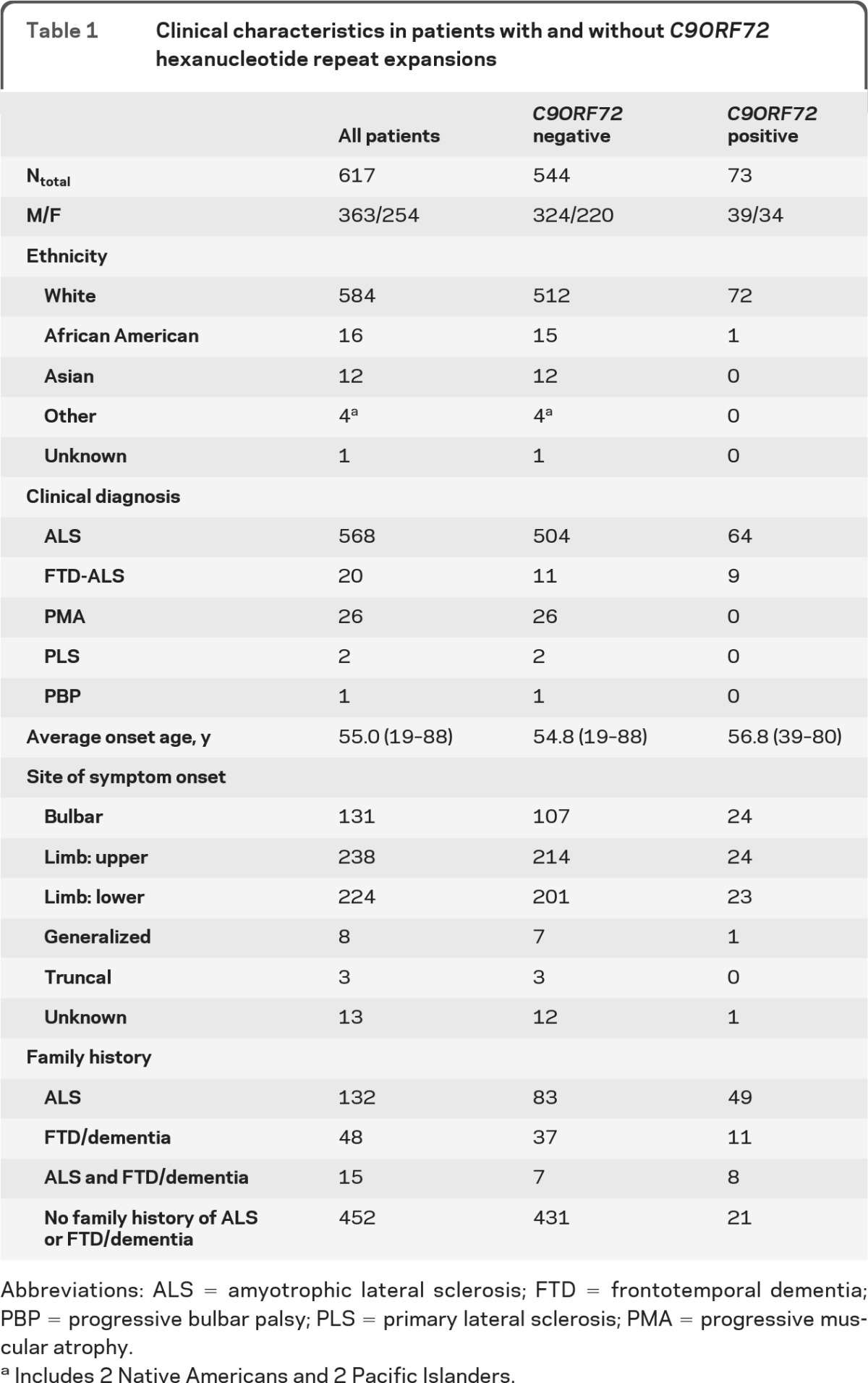
Abbreviations: ALS = amyotrophic lateral sclerosis; FTD = frontotemporal dementia; PBP = progressive bulbar palsy; PLS = primary lateral sclerosis; PMA = progressive muscular atrophy.
Includes 2 Native Americans and 2 Pacific Islanders.
The presence or absence of an expanded hexanucleotide repeat was determined using our 2-step protocol.2 First, the hexanucleotide repeat was PCR amplified in all samples using 1 fluorescently labeled primer followed by fragment-length analysis on an ABI3730 DNA analyzer. Patients showing only a single peak on the electropherogram, suggesting homozygosity in this assay, were further analyzed using the repeat-primed PCR method. A characteristic stutter amplification pattern on the electropherogram was considered evidence of a pathogenic repeat expansion.
Fisher exact tests were used to compare frequencies of demographic and clinical features among groups.
Results.
Of the 617 samples that were analyzed, 73 (11.8%) were found to carry pathogenic GGGGCC repeat expansions in C9ORF72 (table 1 and table e-1 on the Neurology® Web site at www.neurology.org). Interestingly, a significantly higher mutation frequency was observed within the FTD-ALS patient group (9/20; 45.0%), compared to the group of patients with pure ALS (64/568; 11.3%) (p = 0.0002). Among familial cases (fALS), 37.1% (49/132) showed a repeat expansion, while 4.9% (24/485) of the sporadic cases (sALS) were positive. In the remainder of our patient series, repeat-units ranged from 2 to 28, which we considered normal in this study.
Average age at onset for expansion carriers was 56.8 ± 8.0 years (range 39–80) and males accounted for 53.4% (n = 39). Ethnicities of positive cases were white (n = 72; 98.6%) and African American (n = 1; 1.4%). The mutation cases with known site of onset were equally distributed among bulbar, limb-upper, and limb-lower for the pure ALS cases (20/20/23 respectively, 1 unknown), whereas the FTD-ALS cases presented with bulbar, limb-upper, or generalized onset (4/4/1, respectively). Overall, bulbar presentation was somewhat more common in C9ORF72 mutation carriers (24/73; 32.9%) compared to non-C9ORF72 mutation carriers (107/544; 19.7%) (p = 0.014).
Discussion.
The frequency of repeat expansion carriers in fALS (37.1%) reported in this study was highly similar to that previously reported in a selected series of European patients with ALS (38.1%) and slightly higher than our published mutation frequency from a US fALS series (23.5%). This may be due to the ALS cases in our previous publication being collected at 1 location (Mayo Clinic Florida), primarily from incident patients, whereas Coriell samples were collected at multiple centers throughout the United States. The frequency of repeat expansion carriers in sALS (4.9%) in this study was highly comparable to our previously reported frequency of 4.1%.
Only limited clinical features of C9ORF72 mutation carriers have thus far been described.2–5 Clinical data available on the patients we studied indicate considerable clinical heterogeneity among mutation carriers with onset ages ranging from 39 to 80 years, with limb, bulbar, and generalized presentations at disease onset. All mutation carriers presented with upper and lower motor neuron features, with the highest mutation frequency among FTD-ALS patients. Although most mutation carriers were white, we also report the first African American patient with a C9ORF72 repeat expansion.
In our study a characteristic stutter amplification pattern by repeat-primed PCR was considered evidence for pathogenicity; however, future Southern blot analyses will be required to determine the mutant allele length in each individual patient. Future studies should also clarify the minimal length of a pathogenic repeat expansion.
The reporting of C9ORF72 mutation status of this large cohort of patients with ALS from the Coriell Institute provides an essential resource for the scientific community. Acquisition of available lymphoblast cell lines derived from this cohort of mutated patients will allow accurate repeat sizing, genotype–phenotype correlations, and a source of mutant cells for in vitro studies.
Our findings confirm that C9ORF72 GGGGCC hexanucleotide repeat expansions are a major cause of ALS and FTD-ALS.
Supplementary Material
Acknowledgment:
This study used ALS samples from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds).
Footnotes
Supplemental data at www.neurology.org
Author contributions: Nicola Rutherford: drafting/revising the manuscript for content, analysis or interpretation of data, Acquisition of data. Mariely DeJesus-Hernandez, Matt Baker, Thomas Kryston, and Patricia Brown: analysis or interpretation of data, acquisition of data. Drs. Boylan, Wszolek, and Lomen-Hoerth: drafting/revising the manuscript for content, including medical writing for content, contribution of vital reagents/tools/patents, obtaining funding. Dr. Rademakers: drafting/revising the manuscript for content, including medical writing for content, study concept and design, analysis or interpretation of data, study supervision, obtaining funding.
N. Rutherford, M. DeJesus-Hernandez, M. Baker, T. Kryston, P. Brown, and C. Lomen-Hoerth report no disclosures. K. Boylan receives research support from the ALS Association, Biogen Idec, Neuraltus Pharmaceuticals, Cytokinetics Inc, and Mayo Foundation, and has received research support from Avanir Pharmaceuticals and Synapse Biomedical. Z. Wszolek is supported by the NIH/NINDS NS057567, 1RC2NS070276, P50NS072187-01S2, Mayo Clinic Florida Research Committee CR program, and the Dystonia Medical Research Foundation. R. Rademakers receives research support from the NIH (R01 NS065782, R01AG02651, and P50 AG16574), the ALS association, the ALS Therapy Alliance, CurePSP, and the Consortium for Frontotemporal Degeneration Research. Dr. Rademakers further received honoraria for lectures or educational activities not funded by industry and serves on the medical advisory board of the Association for Frontotemporal Degeneration. Go to Neurology.org for full disclosures.
References
- 1. Ticozzi N, Tiloca C, Morelli C, et al. Genetics of familial amyotrophic lateral sclerosis. Arch Ital Biol 2011; 149: 65– 82 [DOI] [PubMed] [Google Scholar]
- 2. Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245– 256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72: 257– 268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murray ME, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 2011; 122: 673– 690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ince PG, Highley JR, Kirby J, et al. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 2011; 122: 657– 671 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.