

Abstract
The paper consists of three parts. Part 1: Definition of Syndromes. Focus is given to craniofacial syndromes in which abnormal traits in the dentition are associated symptoms. In the last decade, research has concentrated on phenotype, genotype, growth, development, function, and treatment. Part 2: Syndromes before Birth. How can the initial malformation sites in these syndromes be studied and what can we learn from it? In this section, deviations observed in syndromes prenatally will be highlighted and compared to the normal human embryological craniofacial development. Specific focus will be given to developmental fields studied on animal tissue and transferred to human cranial development. Part 3: Developmental Fields Affected in Two Craniofacial Syndromes. Analysis of primary and permanent dentitions can determine whether a syndrome affects a single craniofacial field or several fields. This distinction is essential for insight into craniofacial syndromes. The dentition, thus, becomes central in diagnostics and evaluation of the pathogenesis. Developmental fields can explore and advance the concept of dental approaches to craniofacial syndromes. Discussion. As deviations in teeth persist and do not reorganize during growth and development, the dentition is considered useful for distinguishing between syndrome pathogenesis manifested in a single developmental field and in several fields.
1. Introduction
Syndrome research reveals detailed insight into different aspects of congenital malformation of several body components. These malformations affect physical and psychological postnatal development and treatment. Such a wide-spread field of interest requires multidisciplinary teamwork, including clinicians and researchers with different backgrounds. In the last decade, focus has specifically been given to pathogenesis and genotype. A complete overview of craniofacial syndromes would involve not only the dental approach, but also several medical, psychological, and neurological clinical and theoretical approaches. The present paper focuses on 3 key aspects in craniofacial syndromes. These are definition and classification of dental deviations (Part 1); appearance of malformations before birth (Part 2) and a specific new dental approach to craniofacial syndromes (Part 3).
2. Part 1: Definition of Syndromes
Spranger et al. [1] defined syndromes accordingly: “A syndrome is a pattern of multiple anomalies thought to be pathogenetically related and not known to represent a single sequence or a polytopic field defect”. Furthermore, Spranger et al. defines “sequence” as “A pattern of multiple anomalies derived from a single known or presumed prior anomaly or mechanical factor”. A polytopic field defect is defined as “A pattern of anomalies derived from the disturbance of a single developmental field” [1].
Dorland's Illustrated Medical Dictionary defines syndrome as “A set of symptoms that occur together; the sum of signs of any morbid state; a symptom complex. In genetics, a pattern of multiple malformations thought to be pathogenetically related” [2].
A craniofacial syndrome is characterized by morphological and developmental deviations in the cranial tissue components, including teeth. It is difficult to determine whether the deviation has developed at a primary location that subsequently causes secondary alterations or whether there are general deviations in many structures and at many locations.
In some craniofacial syndromes, the genotype is known and in some it is unknown. Even though the genotype is known in a craniofacial syndrome, it is not understood why that particular genotype causes the malformations observed. A known genotype deviation can manifest itself in the cranium by many different phenotypic expressions, from severe to minor affection. How and why the genotype affects the cranial development and why it affects the cranial components differently are far from explained in full.
In the foreword to the textbook Syndromes of the Head and Neck by Gorlin et al. [3] F. Clarke Fraser stated that the symptoms in craniofacial syndromes may not be limited to the head and neck. It can therefore be questioned whether there are craniofacial syndromes that are limited to the head and neck. This is a challenge for future research.
In this section, focus is given to studies on craniofacial syndromes in which abnormal traits in the dentition are associated symptoms. Such abnormal traits could be deviations in tooth number (agenesis, supernumerarity), tooth morphology (size, dimensions, crown invaginations, and abnormal shapes of crowns and roots), tooth eruption (delayed eruption, ankylosis), and resorption (crowns and roots). Occurrence of these dental deviations is classified and exemplified in the following.
2.1. Agenesis
Agenesis has been reported in several syndromes. Schalk-van der Weide [4] reported that specific patterns of agenesis was associated with specific syndromes. Thus, absence of the following teeth was observed in these syndromes: Böök's syndrome: premolars; Rieger syndrome: maxillary incisors (constant), mandibular incisors, and premolars (occasional); Ellis-van Creveld syndrome: mandibular incisors and canines; Gorlin-Chaudry-Moss syndrome: deciduous molars, secondary premolars, and molars; Lipoid proteinosis: maxillary lateral incisors, canines, and premolars; Coffin-Lowry syndrome: maxillary lateral incisors, mandibular incisors; Orofaciodigital syndrome Type I: mandibular lateral incisors and canines; Down syndrome: third molars, mandibular second premolars, and maxillary lateral incisors; Holoprosencephaly: maxillary incisors; Hypoglossia-hypodactylia syndrome: mandibular incisors and canines; Glossopalatine ankylosis syndrome: incisors.
2.2. Supernumerary Teeth
Supernumerary teeth have been reported in Cleidocranial dysplasia [9].
2.3. Tooth Morphology
With regards to tooth morphology, large teeth have been reported in KBG syndrome where macrodontic incisors have been described [10]. Microdontic teeth have been reported in Down syndrome [11]. Deviations in tooth dimensions such as short roots and malformations of crowns and roots have been observed in Turner syndrome [12], in Klinefelter syndrome [13], and in Rothmund-Thomson syndrome [14].
2.4. Eruption
Delay in eruption has been reported in Down syndrome [15]. Complete failure of eruption has been observed in Gapo syndrome [16]. Other eruption deficiencies have also been observed.
2.5. Resorption
Delayed resorption of primary teeth has been observed in Cleidocranial dysplasia [9] and in Hyper IgE syndrome [17].
These symptoms of deviations in the dentition are all phenotypic traits of the specific syndromes mentioned. Why it is so is still not known.
Craniofacial research is currently focused especially on determination of genotypes of different syndromes [6, 18–21].
Research is also devoted to phenotypic descriptions of craniofacial syndromes [22–24].
From a treatment point of view, interest has specifically been given to the use of dental implants. Yap and Klineberg [25] stated that the success rate is lower for implants in patients with ectodermal dysplasia than it is in patients with tooth agenesis but without ectodermal dysplasia.
Optimal treatment in multidisciplinary teams is also given some focus (e.g., [26]).
In the central textbook on craniofacial syndromes [3], Gorlin et al. stated that one-third of children born with birth defects display anomalies in the head and neck. He also reported that 72 different syndromes with orofacial cleft were registered in 1971 and that this number had increased to 250 syndromes in 1990. This development involves all craniofacial phenotypes and new findings document that craniofacial phenotypes are gradually associated with symptoms outside the craniofacial area. In 2010, Trainor [27] reported that more than 700 craniofacial malformations have been described.
The goal for future research on craniofacial syndromes must be to understand the connection between symptoms in the dentition and other symptoms in the craniofacial area.
3. Part 2: Syndromes before Birth
How can the initial malformation sites in these syndromes be studied and what can we learn from it? Prenatal syndromes can be studied by ultrasound or by autopsy after spontaneous or provoked abortion.
Research on human prenatal autopsy material for demonstration of craniofacial development has a morphological character and is predominantly based on histological, histochemical, and radiographic studies. These studies depend entirely on autopsy legislation in different countries and represent primarily the embryonic period and the foetal period before the 16th week of gestation.
In contrast to human studies, experimental studies on the craniofacial development in animals allow for a variety of advanced procedures, such as cell cultures and labelling of migrating cells, and may cover the complete foetal period [28, 29].
In a series of human autopsy studies, the initial sites of malformation in craniofacial syndromes have been investigated. Of specific interest are studies on Holoprosencephaly [30, 31], Anencephaly [32–35], amniotic band rupture [36], Down syndrome (Trisomy 21) [37–40], other trisomies [41–45], Meckel syndrome [46, 47], orofacial clefts [48–50], and cranial encephalocele [51].
In order to evaluate these pathologies, it is necessary to know the initial sites of normal craniofacial development before birth. Normal development has been described in several studies and comprised in the textbook The Prenatal Human Cranium [52].
When normal development is compared with the pathologic conditions mentioned above, the following is observed.
3.1. Holoprosencephaly
The deviation is located in a midsagittal field between the eyes and spans backwards to the sella turcica. This field is called the frontonasal field.
3.2. Anencephaly
The cranial deviation corresponds to the desmal-formed theka crania, but the cartilaginous originated part of the occipital squama is not malformed. Other cranial structures may be deformed due to the absent cerebrum, but the structures are present. It is interesting that the neural hypophysis is absent.
3.3. Amniotic Band Rupture
Though amniotic band rupture in the cranium appears as a craniofacial syndrome, it is not a congenital malformation and not a syndrome. Analyses of the condition reveal that rupture of a normal developmental course has occurred and caused extensive disruptions, not limited to developmental regions (fields).
3.4. Down Syndrome
The cranial phenotype in foetuses with Down syndrome is characterized by short or absent nasal bone, short and abnormal, often cleft, palate, deviations in size and dimensions of the occipital area (the occipital or cerebellar field), and malformations in the sella turcica. Cervical spine malformation is a constant finding. Thus, deviations occur in many regions of the cranium.
3.5. Other Trisomies
The craniofacial skeleton in Trisomy 18 and 13 and Triploidy has been studied and it was shown that the developmental deviations are completely different in these trisomies. This indicates an early and specific interrelationship between genotype and phenotype.
3.6. Meckel Syndrome
In Meckel syndrome, deviations are seen in the occipital region and sella turcica.
3.7. Orofacial Clefts
The nasal bone is short in cleft lip, but not in cleft palate and combined cleft lip and palate. The sella turcica deviates especially in combined cleft lip and palate. Abnormal findings were seen in the cranial base/sella turcica and the nasal bone/maxilla.
3.8. Cranial Encephalocele
Different malformations are observed in bones in different types of cranial encephaloceles. Interestingly, the same type of malformation occurs in the sella turcica/pituitary gland region in different types of encephalocele.
3.9. Preliminary Conclusion
A general conclusion from craniofacial studies in the foetal period is that an association exists between the development of the cranium and the body axis/cervical spine and between the cranium and the central and peripheral nervous systems (CNS/PNS). This is not surprising as studies on the early gastrulation in the embryonic period have shown an axial midline structure in the body, the primitive streak [53], which will later involve the notochord and the head formation. The rostral extent of the notochord appears in the posterior wall of the sella turcica [54].
Concerning cranial development, the main conclusion is that different regions of the cranium are malformed in different genotypes. These cranial regions are called fields. In order to understand these fields, we need to turn to experimental studies on animals.
3.10. Developmental Fields
Experimental studies performed by Le Douarin et al. [28, 55] on animal tissue have revolutionized the embryological understanding of the craniofacial development. In these studies the neural crest cells from different specific regions were labelled and the authors discovered that cells from different regions on the neural tube migrate to different specific parts of the cranial face and dentition, thus forming different developmental fields.
In order to understand the human craniofacial development, it is necessary to combine morphological human studies with knowledge acquired through experimental studies regarding cell migration. Such combination studies have led to insight into developmental fields in humans [5, 7]. The developmental fields in the human cranium are illustrated in Figures 1 and 2 and in the dentition in Figures 3 and 4.
Figure 1.
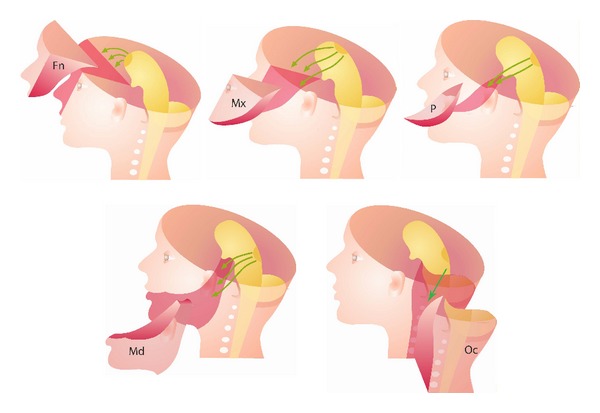
Schematic illustrations of craniofacial fields. Fn: frontonasal field, Mx: maxillary field, P: palatal field, Md: the complete mandibular field, and Oc: occipital and cervical spine field. Green arrows indicate migration paths of neural crest cells from different regions at the neural tube to different developmental fields in the cranium.
Figure 2.
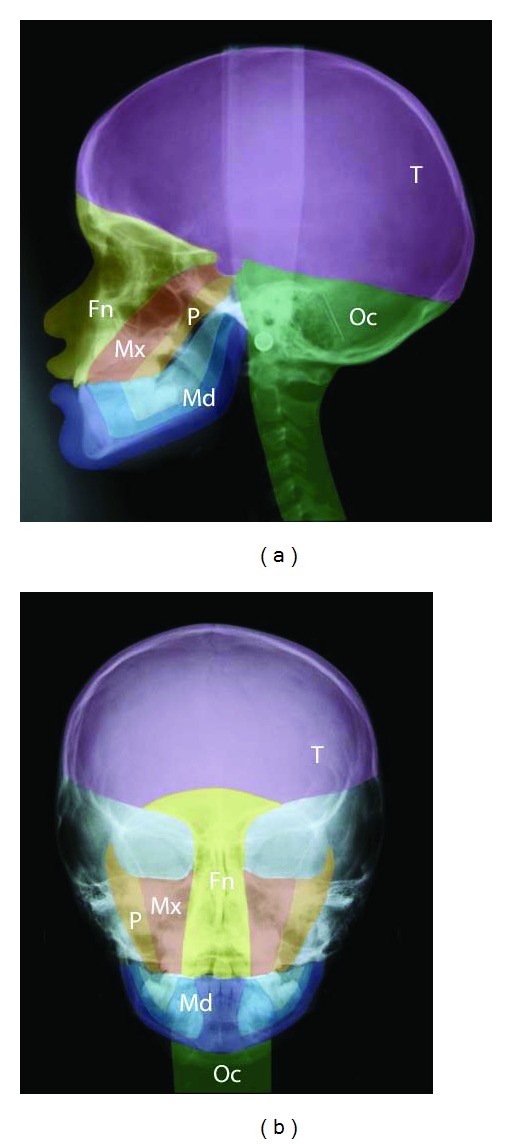
Profile (a) and frontal (b) radiograph of a girl aged 9 years with a single median maxillary central incisor. Different developmental fields are marked on the radiographs. Green: occipital and cervical spine field (Oc). Purple: theka field (T). Light and dark blue: the complete mandibular field (Md). Yellow: frontonasal field (Fn). Red: maxillary field (Mx). Orange: palatal field (P). Note that the sella turcica is a borderline region between fields. This figure is reprinted with permission from European Journal of Orthodontics 2010:32:140-147 [5].
Figure 3.
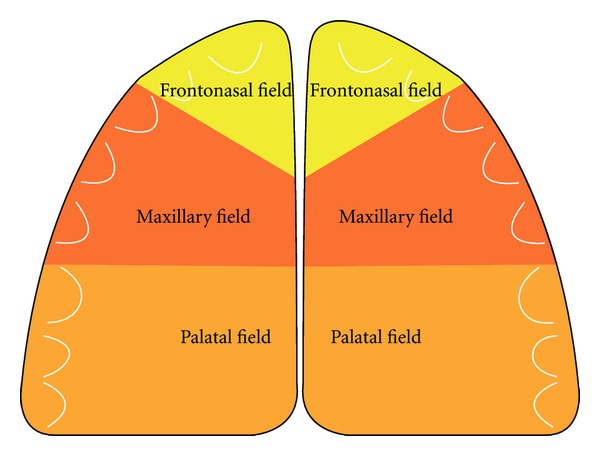
Schematic drawing of the maxilla, demonstrating three different fields in the left and right side of the maxilla.
Figure 4.
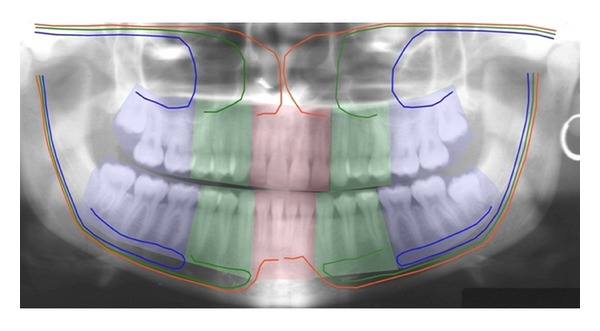
Panoramic radiograph illustrating the different fields in the maxilla and mandible with different innervation, surrounded by different ectomesenchyme. In the maxilla: red colour demonstrates the bilateral frontonasal field, innervated by the nervus nasopalatinus. Green colour demonstrates the bilateral maxillary field, innervated by the nervus maxillaris. Blue colour demonstrates the bilateral palatal fields, innervated by the nervus palatinus. In the mandible, similar fields are illustrated, innervated by different nerve branches from the nervus alveolaris inferior [6]. This figure is reprinted with permission from Orthodontic Waves 2012;71:1-16 [7].
Spranger et al. [1] defined a developmental field as “A region or part of an embryo which responds as a coordinated unit to embryonic interaction and results in complex or multiple anatomical structures. An instrinsic, nondisruptive disturbance of a developmental field will lead to a field defect” [1]. The developmental fields in the maxilla described in the present paper are the frontonasal field, the maxillary field, and the palatal field. These three bilateral fields arise from different regions on the neural crest and have three different main nerve supplies: nervus nasopalatinus, nervus maxillaris, and nervus palatines, respectively [56]. Three similar developmental fields exist in the mandible, innervated by different nerve branches, which are connected during development and mandibular growth in the bundle of peripheral nerves named the inferior alveolar nerve [57]. The interrelationship between the central and peripheral nervous systems has been studied intensively in human foetal pathological studies [58].
Developmental fields in the cranium extend in a triangular shape from the pituitary gland/sella turcica region to an outer region of the face. The sella turcica/pituitary gland region is the end region for the rostral extension of the notochord. It is therefore necessary, when describing a craniofacial developmental field, to describe the complete field in its 3-dimensional extent. The field that has been described most in detail based on foetal pathological observations is the frontonasal field [30, 34] (Figure 5). This field includes the anterior wall of the sella turcica, the anterior cranial base, the interocular region, nose, and philtrum (Figure 5). Within this field, the involvement of the vomeronasal organs producing luteinizing hormone-releasing hormone, LHRH [59], and the sella turcica/pituitary gland [60, 61] can often explain hormone-related deviations in body height and maturity associated with deviations in the frontonasal field [5].
Figure 5.
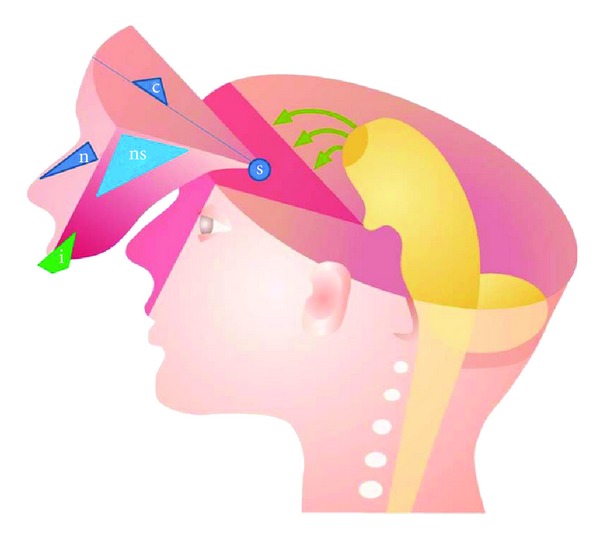
Schematic illustration of hard tissue structures within the frontonasal field in the human cranium. s: sella turcica; c: crista galli; ns: nasal septum; n: nasal bone; i: central incisor.
It has been shown that syndromes, for example, Velocardiofacial syndrome located in the palatal field in the cranium, also involve organ structures such as the brain, thymus, thyroid, and heart septum [62]. It is not obvious that malformations of these organs are caused by the same field defect, but it may be explained from the neural crest cell migration in this region [62].
Another example of craniofacial malformations interrelated with organ malformations can be seen in Cri du Chat syndrome where the cerebellum and larynx, located far apart, are developmentally associated with the posterior cranial base [63].
4. Part 3: Developmental Fields Affected in Craniofacial Syndromes
Analysis of the primary and permanent dentitions can determine whether a syndrome affects a single craniofacial field or several fields. The previous section showed how prenatal studies are useful for analysis of the initial site of organ and osseous malformations, but due to the early period of development, less useful for analysis of the dentition. Only early traces in the development of the primary teeth can be analyzed prenatally. In contrast to the prenatal tissue analysis, postnatal analysis on radiographic material is highly useful for analysis of primary and permanent teeth, but not useful at all for analysis and limitation of developmental fields. A combination of prenatal and postnatal insight is therefore necessary in order to understand how developmental fields are affected in different craniofacial syndromes.
Opitz [64] has defined developmental fields as the morphogenetic units of the embryo. He also states that processes in developmental fields are self-organizing spatially coordinated and ordered, epimorphically hierarchical, temporarily synchronized, epigenetically interactive, developmentally constrained, and phylogenetically conserved [64].
4.1. Single Field, Exemplified by SMMCI
In a craniofacial syndrome where deviations occur in a specific field, it is expected that all structures, including teeth, within the field can be deviant. Structures outside the field are not necessarily deviant. In this section, focus will be on the SMMCI syndrome (single median maxillary central incisor) with gene location 7q36.3 [65]. In this syndrome, the frontonasal field is affected. The craniofacial examination of this condition reveals a highly significantly shorter anterior cranial base [66].
In SMMCI, only one maxillary central incisor exists in both the primary and permanent dentitions [67, 68]. The malformation has been associated with sonic hedgehog gene, which normally defines the midaxial part of the frontonasal field [31, 69]. As a result, the midaxial structures of the face, maxilla, nasal cavity, nasal bone, anterior cranial fossa, and sella turcica are malformed. The sella turcica is often tiny and malformed and the growth hormone production in the pituitary gland deviates [8]. Children with SMMCI are accordingly often short in stature.
Recently, also a fusion or nonseparation of the frontal hemispheres has been registered [8]. This is a new finding that expands the craniofacial diagnostics to include brain diagnostics as well. The SMMCI condition is demonstrated by clinical and radiographic images (Figures 6, 7, 8, and 9). The illustrations show that the postnatal findings are in complete concurrence with the prenatal findings stating that only a single field (frontonasal field) is involved in the syndrome.
Figure 6.
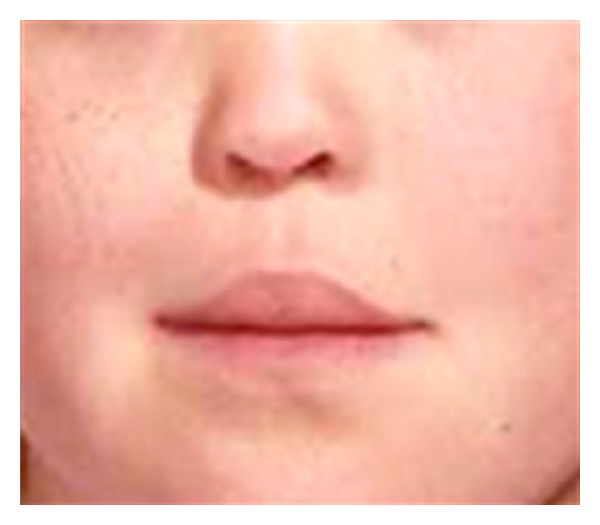
Section of photograph of a girl, aged 12 years 1 month, with SMMCI (single median maxillary central incisor). The face is characterized by tubular-shaped nose and philtrum blurred towards the prolabium without the normal s-shape. Deviations all occur within the frontonasal field, illustrated schematically in Figure 5.
Figure 7.
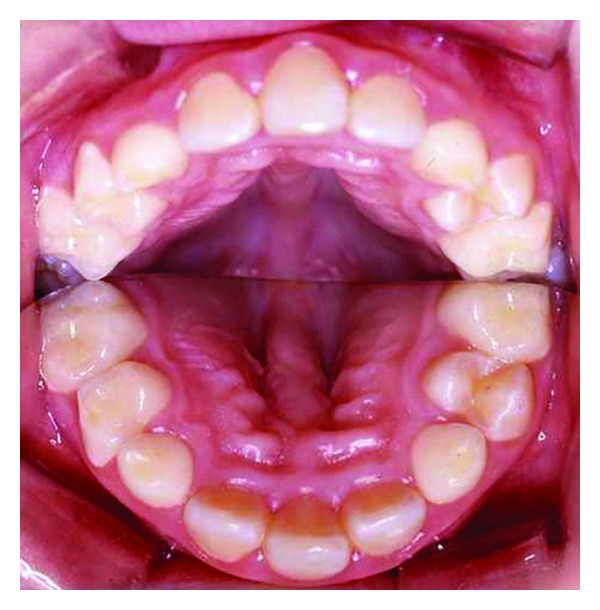
Intraoral photograph with a mirror placed between the dental arches, demonstrating the palate. The figure demonstrates a single central incisor, absence of papilla incisive, and a vault midaxially in the palate. Deviations all occur within the frontonasal field, illustrated schematically in Figure 5. The figure is reprinted with permission from Neuropediatrics 2009;40:280-283 [8].
Figure 8.
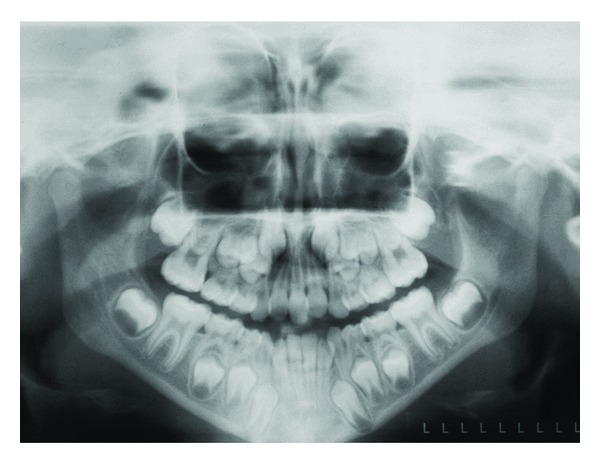
Panoramic radiograph of the dentition in a girl with SMMCI, aged 12 years 1 month, shown in Figure 6. Note the maxillary central incisor, the narrow nasal cavity, and the close-set eyes. Apart from the maxillary front the dentition looks normal. Deviations all occur within the frontonasal field, illustrated schematically in Figure 5.
Figure 9.
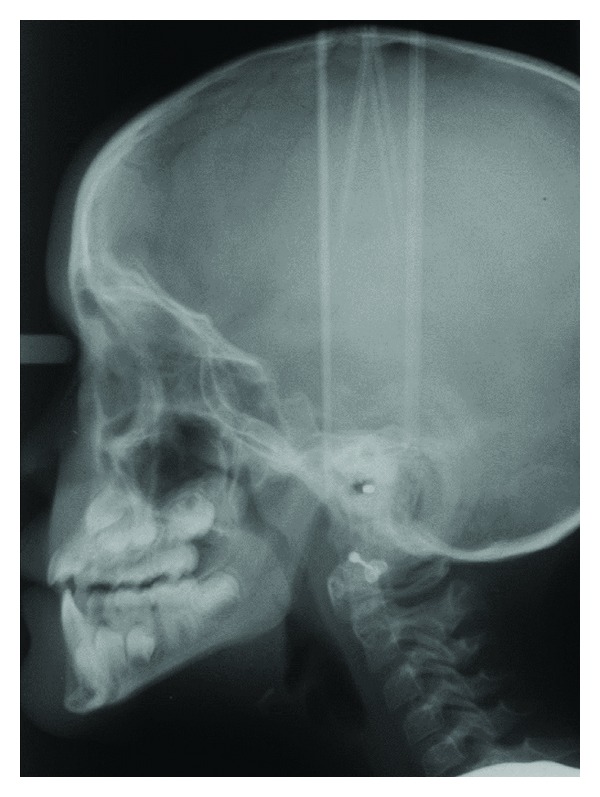
Profile radiograph of girl with SMMCI, aged 12 years 1 month, shown in Figure 6. Note the short anterior cranial fossa, the undeveloped sella turcica, and the maxillary retrognathia. Deviations all occur within the frontonasal field, illustrated schematically in Figure 5.
4.2. Several Fields, Exemplified by Trisomy 21
In several craniofacial syndromes, many of the symptoms cannot be related to fields. One of these syndromes is Trisomy 21/Down syndrome. In Down syndrome, ageneses are registered in all craniofacial fields [70, 71] in locations where ageneses are normally registered [70, 72], that is, lateral incisors in the frontonasal fields; second premolars in the maxillary field; third molars in the palatal field. In the mandible, ageneses are seen most often in the central incisor region, the second premolar region, and third molar region. Compared to normal conditions, the occurrence of agenesis is about 10 times higher in patients with Down syndrome, and especially high in the mandibular incisor region [70]. Also narrow crowns and short roots are observed in the dentition in general [3]. The craniofacial morphology reveals different malocclusions and different malformation signs in the cranium, such as absent or short nasal bone, enlarged thickness of the theka crania, and malformations in the cervical column. The Down condition is demonstrated by clinical and radiographic images (Figures 10 and 11). The illustrations show that the postnatal findings are in complete concurrence with the prenatal findings stating that symptoms occur in several fields in Down syndrome.
Figure 10.
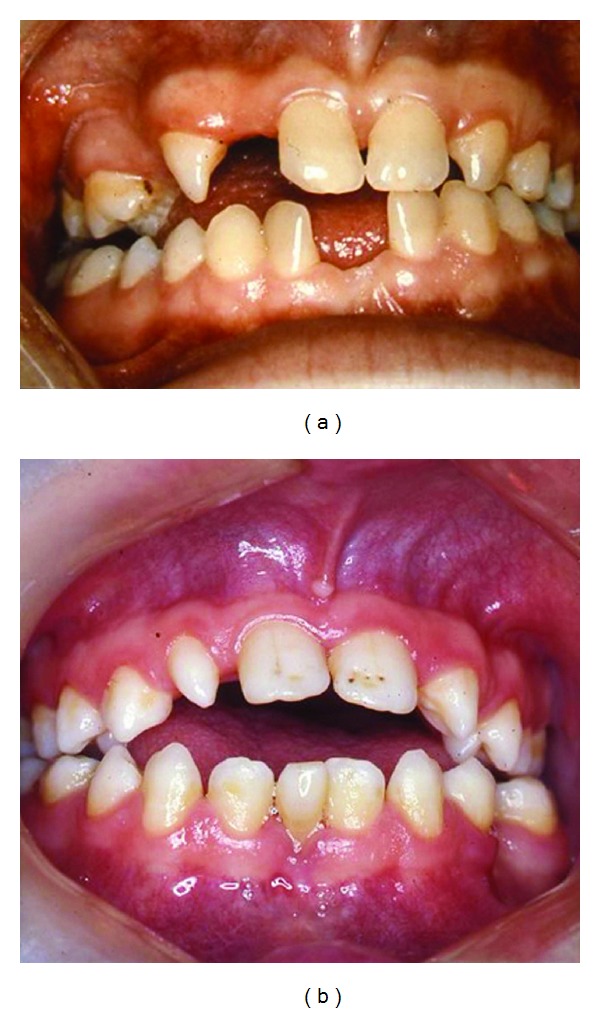
Intraoral photographs of two adult individuals with Trisomy 21/Down syndrome. In the left photograph, agenesis of the maxillary right lateral incisor and premolars is registered. Agenesis is also registered in the mandibular central incisor region. In the right photograph, an anterior vertical open bite is registered. In the maxilla a malformed right lateral incisor, agenesis of the left lateral incisor, and enamel pits at the central incisors are observed. In the mandible, agenesis of one central incisor is observed.
Figure 11.
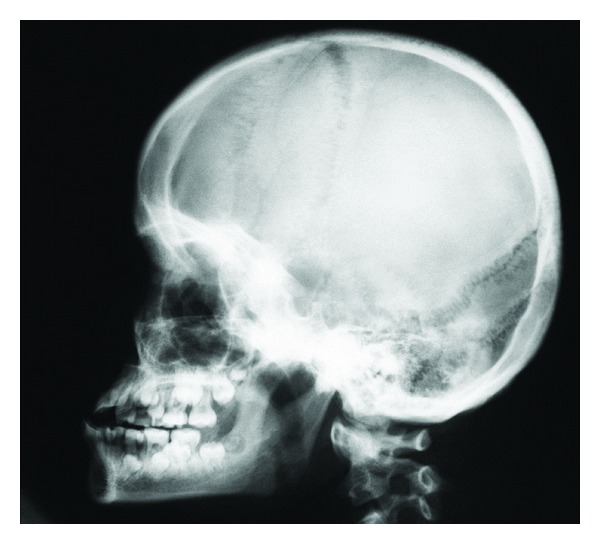
Profile radiograph of a girl with Down syndrome, aged 8 years 6 months. Note the enlarged thickness of the skull, absence of the nasal bone, deviations in the upper contour of the anterior wall of the sella turcica, malformations of the cervical spine, and maxillary retrognathia. Deviations of the skeleton occur in several craniofacial fields.
4.3. Single Field and Several Fields
This distinction between the location of dental deviations in a single field and in several fields is essential for our insight into craniofacial syndromes. It is believed that the dentition will become central in diagnostics and evaluation of the pathogenesis behind craniofacial syndromes. In a recent paper, Trainor has highlighted the role of neural crest cells in the aetiology and pathogenesis of Treacher Collins syndrome and furthermore for the potential for prevention [27]. We still need to know the genes responsible for neural crest cell migration.
The pathogenesis of SMMCI is associated with the sonic hedgehog gene that is expressed in the midcranial region anterior to the pituitary gland [31, 69].
In Down syndrome/Trisomy 21, a possible gene dosage effect associated with the extra chromosome may influence the phenotype [73]. The extra chromosome may influence the mitotic activity of cells [74].
Future research in craniofacial syndromology should include extended studies on developmental fields in order to establish a more sufficient background for elucidating the pathogenesis. In the frontonasal segment it would be interesting to focus on fields with dental deviations in the maxillary incisor region such as Kallmann syndrome [75], Rieger syndrome [3], KGB syndrome [3, 76], and cleft lip syndrome [77, 78]. In the anterior incisor field in the mandible, it would be interesting to focus on Ellis-van Creveld syndrome described by Gorlin et al. [3]. Also Williams syndrome [79] and Turner syndrome [12] may contribute to new knowledge on the pathogenesis.
It is important to realize that a syndrome has different phenotypic appearances, ranging from mild to severe. It is also obvious that syndromes that are normally confined to one field may have symptoms outside that specific field, as observed in Rieger syndrome [3]. This may be due to different genotypes. There remains still a need for a systematic analysis of the interrelationship between dental deviations, skeletal deviations, associated organ deviations, body growth, and genotypes.
In recent years, extensive experimental animal studies have focused in particular on the neural crest cell migration in the craniofacial region. These studies are highly important for understanding human development as well [80]. A study such as the previously described study on Velocardiofacial syndrome, also known as Chromosome 22q 11.2 deletion syndrome [62], with widespread symptoms in the same neural crest developmental field, may influence the classification by Spranger et al. from 1982 [1].
Part 3 shows how developmental fields can explore and advance the concept of dental approaches to craniofacial syndromes.
5. Discussion
The great advantage of dental analyses compared to all other analyses performed on human tissue is that deviations in the hard tissue persist and remain stable during the developmental course. As dental tissues do not reorganize, they are easy to analyse and use in analyses of fields. Therefore, a dental approach to craniofacial syndromes by analysis of developmental fields contributes to an understanding of the pathogenesis of craniofacial syndromes.
A problem raised by Hennekam in 2007 is which clinical condition can be called a syndrome and which cannot. There are arguments in favour for using aetiology and pathogenesis as the core issue, but Hennekam [81] also states that there are arguments to make the patient's phenotype decide the syndrome definition.
Another problem that should be solved in the craniofacial analysis is the genetics behind the craniofacial fields. Is there a signalling gradient involved in the cranial pattern formation, such as suggested in limb development? [82].
From an embryological and pathological point of view, it can be presumed that the notochord activates the neural crest cells to migration and that different genes are responsible for the different locations of neural crest cells at the neural tube. This adds another aspect to craniofacial development and syndromology and calls for scientific attention in the future.
Acknowledgment
Maria Kvetny, MA, is acknowledged for linguistic support and for manuscript and figure preparation.
References
- 1.Spranger J, Benirschke K, Hall JG. Errors of morphogenesis: concepts and terms. Recommendations of an international working group. Journal of Pediatrics. 1982;100(1):160–165. doi: 10.1016/s0022-3476(82)80261-8. [DOI] [PubMed] [Google Scholar]
- 2.Dorland. Dorland's Illustrated Medical Dictionary. 30th edition. Philadelphia, Pa, USA: Saunders; 2003. [Google Scholar]
- 3.Gorlin RJ, Cohen MM, Jr., Levin LS. Syndrome of the Head and Neck. New York, NY, USA: Oxford University Press; 1990. (Oxford Monographs on Medical Genetics No. 19). [Google Scholar]
- 4.Schalk-van der Weide Y. Oligodontia: a clinical, radiographic and genetic evaluation [thesis] Rijksuniversitet Utrecht; 1992. [Google Scholar]
- 5.Kjær I. Orthodontics and foetal pathology: a personal view on craniofacial patterning. European Journal of Orthodontics. 2010;32(2):140–147. doi: 10.1093/ejo/cjp059. [DOI] [PubMed] [Google Scholar]
- 6.De Coster PJ, Mortier G, Marks LA, Martens LC. Cranial suture biology and dental development: genetic and clinical perspectives. Journal of Oral Pathology and Medicine. 2007;36(8):447–455. doi: 10.1111/j.1600-0714.2007.00553.x. [DOI] [PubMed] [Google Scholar]
- 7.Kjær I. New diagnostics of the dentition on panoramic radiographs—focusing on the peripheral nervous system as an important aetiological factor behind dental anomalies. Orthodontic Waves. 2012;71(1):1–16. [Google Scholar]
- 8.Kjær I, Wagner A, Thomsen LL, Holm K. Brain malformation in single median maxillary central incisor. Neuropediatrics. 2009;40(6):280–283. doi: 10.1055/s-0030-1248245. [DOI] [PubMed] [Google Scholar]
- 9.Jensen BL, Kreiborg S. Development of the dentition in cleidocranial dysplasia. Journal of Oral Pathology and Medicine. 1990;19(2):89–93. doi: 10.1111/j.1600-0714.1990.tb00803.x. [DOI] [PubMed] [Google Scholar]
- 10.Herrmann J, Pallister PD, Tiddy W, Opitz JM. The KBG syndrome: a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects. 1975;11(5):7–18. [PubMed] [Google Scholar]
- 11.Geciauskas MA, Cohen MM. Mesiodistal crown diameters of permanent teeth in Down’s syndrome (mongolism) American Journal of Mental Deficiency. 1970;74(4):563–567. [PubMed] [Google Scholar]
- 12.Townsend G, Jensen BL, Alvesalo L. Reduced tooth size in 45,X (Turner syndrome) females. American Journal of Physical Anthropology. 1984;65(4):367–371. doi: 10.1002/ajpa.1330650405. [DOI] [PubMed] [Google Scholar]
- 13.Feichtinger C, Rossiwall B. Taurodontism in human sex chromosome aneuploidy. Archives of Oral Biology. 1977;22(5):327–329. doi: 10.1016/0003-9969(77)90031-0. [DOI] [PubMed] [Google Scholar]
- 14.Roinioti TD, Stefanopoulos PK. Short root anomaly associated with Rothmund-Thomson syndrome. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 2007;103(1):e19–e22. doi: 10.1016/j.tripleo.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 15.Roche AF, Barkla DH. The eruption of deciduous teeth in Mongols. Journal of Mental Deficiency Research. 1964;42:54–64. doi: 10.1111/j.1365-2788.1964.tb00800.x. [DOI] [PubMed] [Google Scholar]
- 16.Gagliardi ART, Gonzalez CH, Pratesi R. GAPO syndrome: report of three affected brothers. American Journal of Medical Genetics. 1984;19(2):217–223. doi: 10.1002/ajmg.1320190203. [DOI] [PubMed] [Google Scholar]
- 17.Becktor KB, Kjær I, Koch C. Tooth eruption, epithelial root sheath and craniofacial profile in hyper IgE syndrome: report of two cases. European Journal of Paediatric Dentistry. 2001;2(4):185–190. [Google Scholar]
- 18.Cunningham ML, Seto ML, Hing AV, Bull MJ, Hopkin RJ, Leppig KA. Cleidocranial dysplasia with severe parietal bone dysplasia: C-terminal RUNX2 mutations. Birth Defects Research Part A. 2006;76(2):78–85. doi: 10.1002/bdra.20231. [DOI] [PubMed] [Google Scholar]
- 19.Lana-Elola E, Tylzanowski P, Takatalo M, et al. Noggin null allele mice exhibit a microform of holoprosencephaly. Human Molecular Genetics. 2011;20(20):4005–4012. doi: 10.1093/hmg/ddr329. [DOI] [PubMed] [Google Scholar]
- 20.Clauss F, Manière MC, Obry F, et al. Dento-craniofacial phenotypes and underlying molecular mechanisms hypohidrotic ectodermal dysplasia (HED): a review. Journal of Dental Research. 2008;87(12):1089–1099. doi: 10.1177/154405910808701205. [DOI] [PubMed] [Google Scholar]
- 21.Kavitha B, Priyadharshini V, Sivapathasundharam B, Saraswathi TR. Role of genes in oro-dental diseases. Indian Journal of Dental Research. 2010;21(2):270–274. doi: 10.4103/0970-9290.66646. [DOI] [PubMed] [Google Scholar]
- 22.Honda E, Inoue T, Domon M, Sasaki T, Uchida T. Dental radiographic signs characteristic to Hallermann-Streiff syndrome. Oral Surgery Oral Medicine and Oral Pathology. 1990;70(1):121–125. doi: 10.1016/0030-4220(90)90189-y. [DOI] [PubMed] [Google Scholar]
- 23.Becktor KB, Becktor JP, Karnes PS, Keller EE. Craniofacial and dental manifestations of Proteus syndrome: a case report. Cleft Palate-Craniofacial Journal. 2002;39(2):233–245. doi: 10.1597/1545-1569_2002_039_0233_cadmop_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 24.Axelsson S, Kjær I, Heiberg A, Bjørnland T, Storhaug K. Neurocranial morphology and growth in Williams syndrome. European Journal of Orthodontics. 2005;27(1):32–47. doi: 10.1093/ejo/cjh065. [DOI] [PubMed] [Google Scholar]
- 25.Yap AK, Klineberg I. Dental implants in patients with ectodermal dysplasia and tooth agenesis: a critical review of the literature. The International Journal of Prosthodontics. 2009;22(3):268–276. [PubMed] [Google Scholar]
- 26.Susami T, Kuroda T, Yoshimasu H, Suzuki R. Ellis-van Creveld syndrome: craniofacial morphology and multidisciplinary treatment. Cleft Palate-Craniofacial Journal. 1999;36(4):345–352. doi: 10.1597/1545-1569_1999_036_0345_evcscm_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 27.Trainor PA. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. American Journal of Medical Genetics, Part A. 2010;152(12):2984–2994. doi: 10.1002/ajmg.a.33454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Douarin NM. The neural crest in the neck and other parts of the body. Birth Defects. 1975;11(7):19–50. [PubMed] [Google Scholar]
- 29.Slavkin HC. Molecular biology experimental strategies for craniofacial-oral-dental dysmorphology. Connective Tissue Research. 1995;32(1–4):233–239. doi: 10.3109/03008209509013728. [DOI] [PubMed] [Google Scholar]
- 30.Kjær I, Keeling JW, Graem N. The midline craniofacial skeleton in holoprosencephalic fetuses. Journal of Medical Genetics. 1991;28(12):846–855. doi: 10.1136/jmg.28.12.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kjær I, Keeling JW, Hansen BF, Becktor KB. Midline skeletodental morphology in holoprosencephaly. Cleft Palate-Craniofacial Journal. 2002;39(3):357–363. doi: 10.1597/1545-1569_2002_039_0357_msmih_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 32.Kjær I, Keeling JW, Græm N. Midline maxillofacial skeleton in human anencephalic fetuses. Cleft Palate-Craniofacial Journal. 1994;31(4):250–256. doi: 10.1597/1545-1569_1994_031_0250_mmsiha_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 33.Kjær I, Keeling JW, Graem N. Cranial base and vertebral column in human anencephalic fetuses. Journal of Craniofacial Genetics and Developmental Biology. 1994;14(4):235–244. [PubMed] [Google Scholar]
- 34.Kjær I, Fischer-Hansen B. Human fetal pituitary gland in holoprosencephaly and anencephaly. Journal of Craniofacial Genetics and Developmental Biology. 1995;15(4):222–229. [PubMed] [Google Scholar]
- 35.Lomholt JF, Fischer-Hansen B, Keeling JW, Reintoft I, Kjær I. Subclassification of anencephalic human fetuses according to morphology of the posterior cranial fossa. Pediatric and Developmental Pathology. 2004;7(6):601–606. doi: 10.1007/s10024-004-9098-z. [DOI] [PubMed] [Google Scholar]
- 36.Keeling JW, Kjær I. Diagnostic distinction between anencephaly and amnion rupture sequence based on skeletal analysis. Journal of Medical Genetics. 1994;31(11):823–829. doi: 10.1136/jmg.31.11.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kjær I, Keeling JW, Reintoft I, Nolting D, Hansen BF. Pituitary gland and sella turcica in human trisomy 21 fetuses related to axial skeletal development. American Journal of Medical Genetics. 1998;80(5):494–500. doi: 10.1002/(sici)1096-8628(19981228)80:5<494::aid-ajmg11>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 38.Lauridsen H, Hansen BF, Reintoft I, Keeling JW, Kjær I. Histological investigation of the palatine bone in prenatal trisomy 21. Cleft Palate-Craniofacial Journal. 2001;38(5):492–497. doi: 10.1597/1545-1569_2001_038_0492_hiotpb_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 39.Tuxen A, Keeling JW, Reintoft I, Fischer Hansen B, Nolting D, Kjær I. A histological and radiological investigation of the nasal bone in fetuses with Down syndrome. Ultrasound in Obstetrics and Gynecology. 2003;22(1):22–26. doi: 10.1002/uog.152. [DOI] [PubMed] [Google Scholar]
- 40.Lomholt JF, Keeling JW, Hansen BF, Ono T, Stoltze K, Kjær I. The prenatal development of the human cerebellar field in Down syndrome. Orthodontics and Craniofacial Research. 2003;6(4):220–226. doi: 10.1046/j.1397-5927.2003.00265.x. [DOI] [PubMed] [Google Scholar]
- 41.Kjær I, Keeling JW, Hansen BF. Pattern of malformations in the axial skeleton in human trisomy 21 fetuses. American Journal of Medical Genetics. 1996;65(4):332–336. doi: 10.1002/(SICI)1096-8628(19961111)65:4<332::AID-AJMG16>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 42.Kjær I, Keeling JW, Hansen BF. Pattern of malformations in the axial skeleton in human trisomy 13 fetuses. American Journal of Medical Genetics. 1997;70(4):421–426. doi: 10.1002/(sici)1096-8628(19970627)70:4<421::aid-ajmg17>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 43.Kjær I, Keeling JW, Reintoft I, Hjalgrim H, Nolting D, Hansen BF. Pituitary gland and sella turcica in human trisomy 18 fetuses. American Journal of Medical Genetics. 1998;76(1):87–92. [PubMed] [Google Scholar]
- 44.Nolting D, Hansen BF, Keeling JW, Reintoft I, Kjær I. Prenatal malformed lumbar vertebral corpora in trisomies 21, 18 and 13, evaluated radiographically and histologically. Acta Pathologica, Microbiologica et Immunologica Scandinavica. 2000;108(6):422–428. doi: 10.1034/j.1600-0463.2000.d01-78.x. [DOI] [PubMed] [Google Scholar]
- 45.Mentz RG, Engel U, Kjær I. Nasal bone length in trisomy 18, triploidy and Turner syndrome analyzed on postmortem radiographs. Ultrasound in Obstetrics and Gynecology. 2009;34(5):607–608. doi: 10.1002/uog.7436. [DOI] [PubMed] [Google Scholar]
- 46.Kjær KW, Hansen BF, Keeling JW, Kjær I. Skeletal malformations in fetuses with Meckel syndrome. American Journal of Medical Genetics. 1999;84(5):469–475. doi: 10.1002/(sici)1096-8628(19990611)84:5<469::aid-ajmg11>3.3.co;2-j. [DOI] [PubMed] [Google Scholar]
- 47.Kjær KW, Hansen BF, Keeling JW, Nolting D, Kjær I. Malformations of cranial base structures and pituitary gland in prenatal Meckel syndrome. Acta Pathologica, Microbiologica et Immunologica Scandinavica. 1999;107(10):937–944. doi: 10.1111/j.1699-0463.1999.tb01494.x. [DOI] [PubMed] [Google Scholar]
- 48.Kjær I, Reintoft I, Poulsen H, et al. A new craniofacial disorder involving hypertelorism and malformations of external nose, palate and pituitary gland. Journal of Craniofacial Genetics and Developmental Biology. 1997;17(1):23–34. [PubMed] [Google Scholar]
- 49.Lisson JA, Kjær I. Location of alveolar clefts relative to the incisive fissure. Cleft Palate-Craniofacial Journal. 1997;34(4):292–296. doi: 10.1597/1545-1569_1997_034_0292_loacrt_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 50.Hansen L, Skovgaard LT, Nolting D, Hansen BF, Kjær I. Human prenatal nasal bone lengths: normal standards and length values in fetuses with cleft lip and cleft palate. Cleft Palate-Craniofacial Journal. 2005;42(2):165–170. doi: 10.1597/03-094.1. [DOI] [PubMed] [Google Scholar]
- 51.Kjær I, Hansen BF, Keeling JW. Axial skeleton and pituitary gland in human fetuses with spina bifida and cranial encephalocele. Pediatric Pathology and Laboratory Medicine. 1996;16(6):909–926. doi: 10.1080/15513819609168714. [DOI] [PubMed] [Google Scholar]
- 52.Kjær I, Keeling JW, Hansen BF. The Prenatal Human Cranium. Normal and Pathologic Development. Copenhagen, Denmark: Munksgaard; 1999. [Google Scholar]
- 53.Voiculescu O, Bertocchini F, Wolpert L, Keller RE, Stern CD. The amniote primitive streak is defined by epithelial cell intercalation before gastrulation. Nature. 2007;449(7165):1049–1052. doi: 10.1038/nature06211. [DOI] [PubMed] [Google Scholar]
- 54.Kjær I, Becktor KB, Nolting D, Fischer Hansen B. The association between prenatal sella turcica morphology and notochordal remnants in the dorsum sellae. Journal of Craniofacial Genetics and Developmental Biology. 1997;17(3):105–111. [PubMed] [Google Scholar]
- 55.Le Douarin NM, Ziller C, Couly GF. Patterning of neural crest derivatives in the avian embryo: in vivo and in vitro studies. Developmental Biology. 1993;159(1):24–49. doi: 10.1006/dbio.1993.1219. [DOI] [PubMed] [Google Scholar]
- 56.Kjær I. Prenatal traces of aberrant neurofacial growth. Acta Odontologica Scandinavica. 1998;56(6):326–330. doi: 10.1080/000163598428257. [DOI] [PubMed] [Google Scholar]
- 57.Chávez-Lomelí ME, Mansilla Lory J, Pompa JA, Kjær I. The human mandibular canal arises from three separate canals innervating different tooth groups. Journal of Dental Research. 1996;75(8):1540–1544. doi: 10.1177/00220345960750080401. [DOI] [PubMed] [Google Scholar]
- 58.Kjær I. Neuro-osteology. Critical Reviews in Oral Biology and Medicine. 1998;9(2):224–244. doi: 10.1177/10454411980090020501. [DOI] [PubMed] [Google Scholar]
- 59.Kjær I, Hansen BF. Luteinizing hormone-releasing hormone and innervation pathways in human prenatal nasal submucosa: factors of importance in evaluating Kallmann’s syndrome. Acta Pathologica, Microbiologica et Immunologica Scandinavica. 1996;104(9):680–688. doi: 10.1111/j.1699-0463.1996.tb04929.x. [DOI] [PubMed] [Google Scholar]
- 60.Kjær I. Human prenatal craniofacial development related to brain development under normal and pathologic conditions. Acta Odontologica Scandinavica. 1995;53(3):135–143. doi: 10.3109/00016359509005963. [DOI] [PubMed] [Google Scholar]
- 61.Kjær I, Fischer-Hansen B. The adenohypophysis and the cranial base in early human development. Journal of Craniofacial Genetics and Developmental Biology. 1995;15(3):157–161. [PubMed] [Google Scholar]
- 62.Mølsted K, Boers M, Kjær I. The morphology of the sella turcica in velocardiofacial syndrome suggests involvement of a neural crest developmental field. American Journal of Medical Genetics, Part A. 2010;152(6):1450–1457. doi: 10.1002/ajmg.a.33381. [DOI] [PubMed] [Google Scholar]
- 63.Kjær I, Niebuhr E. Studies of the cranial base in 23 patients with Cri-du-Chat syndrome suggest a developmental field involved in the condition. American Journal of Medical Genetics. 1999;82(1):6–14. doi: 10.1002/(sici)1096-8628(19990101)82:1<6::aid-ajmg2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 64.Opitz JM. Development: clinical and evolutionary considerations. American Journal of Medical Genetics, Part A. 2007;143(24):2853–2861. doi: 10.1002/ajmg.a.32089. [DOI] [PubMed] [Google Scholar]
- 65.Masuno M, Fukushima Y, Sugio Y, Ikeda M, Kuroki Y. Two unrelated cases of single maxillary central incisor with 7q terminal deletion. Japanese Journal of Human Genetics. 1990;35(4):311–317. doi: 10.1007/BF01883753. [DOI] [PubMed] [Google Scholar]
- 66.Tabatabaie F, Sonnesen L, Kjær I. The neurocranial and craniofacial morphology in children with solitary median maxillary central incisor (SMMCI) Orthodontics and Craniofacial Research. 2008;11(2):96–104. doi: 10.1111/j.1601-6343.2007.00419.x. [DOI] [PubMed] [Google Scholar]
- 67.Kjær I, Becktor KB, Lisson J, Gormsen C, Russell BG. Face, palate, and craniofacial morphology in patients with a solitary median maxillary central incisor. European Journal of Orthodontics. 2001;23(1):63–73. doi: 10.1093/ejo/23.1.63. [DOI] [PubMed] [Google Scholar]
- 68.Kjær I, Balslev-Olesen M. The primary maxillary central incisor in the Solitary Median Maxillary Central Incisor syndrome. European Journal of Orthodontics. 2012;13(1):73–75. [PubMed] [Google Scholar]
- 69.Nanni L, Ming JE, Du Y, et al. SHH mutation is associated with solitary median maxillary central incisor: a study of 13 patients and review of the literature. American Journal of Medical Genetics. 2001;102(1):1–10. doi: 10.1002/1096-8628(20010722)102:1<1::aid-ajmg1336>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 70.Russell BG, Kjær I. Tooth agenesis in Down syndrome. American Journal of Medical Genetics. 1995;55(4):466–471. doi: 10.1002/ajmg.1320550415. [DOI] [PubMed] [Google Scholar]
- 71.Kieser J, Townsend G, Quick A. The Down syndrome patient in dental practice—part I: pathogenesis and general and dental features. New Zealand Dental Journal. 2003;99(1):5–9. [PubMed] [Google Scholar]
- 72.Lomholt JF, Russell BG, Stoltze K, Kjær I. Third molar agenesis in Down syndrome. Acta Odontologica Scandinavica. 2002;60(3):151–154. doi: 10.1080/000163502753740160. [DOI] [PubMed] [Google Scholar]
- 73.Roubertoux PL, Carlier M. Mouse models of cognitive disabilities in trisomy 21 (Down syndrome) American Journal of Medical Genetics Part C. 2010;154(4):400–416. doi: 10.1002/ajmg.c.30280. [DOI] [PubMed] [Google Scholar]
- 74.Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down’s syndrome mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(5):1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mølsted K, Kjær I, Giwercmand A, Vesterhauge S, Skakkebæk NE. Craniofacial morphology in patients with Kallmann’s syndrome with and without cleft lip and palate. Cleft Palate-Craniofacial Journal. 1997;34(5):417–424. doi: 10.1597/1545-1569_1997_034_0417_cmipwk_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 76.Kumar H, Prabhu N, Cameron A. KBG syndrome: review of the literature and findings of 5 affected patients. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and Endodontology. 2009;108(3):e72–e79. doi: 10.1016/j.tripleo.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 77.Nielsen BW, Mølsted K, Skovgaard LT, Kjær I. Cross-sectional study of the length of the nasal bone in cleft lip and palate subjects. Cleft Palate-Craniofacial Journal. 2005;42(4):417–422. doi: 10.1597/04-001.1. [DOI] [PubMed] [Google Scholar]
- 78.Gorlin RJ, Cervenka J, Pruzansky S. Facial clefting and its syndromes. Birth Defects. 1971;7(7):3–49. [PubMed] [Google Scholar]
- 79.Axelsson S, Bjørnland T, Kjær I, Heiberg A, Storhaug K. Dental characteristics in Williams syndrome: a clinical and radiographic evaluation. Acta Odontologica Scandinavica. 2003;61(3):129–136. doi: 10.1080/00016350310001451. [DOI] [PubMed] [Google Scholar]
- 80.Cobourne MT, Mitsiadis T. Neural crest cells and patterning of the mammalian dentition. Journal of Experimental Zoology Part B. 2006;306(3):251–260. doi: 10.1002/jez.b.21084. [DOI] [PubMed] [Google Scholar]
- 81.Hennekam RCM. What to call a syndrome. American Journal of Medical Genetics, Part A. 2007;143(10):1021–1024. doi: 10.1002/ajmg.a.31674. [DOI] [PubMed] [Google Scholar]
- 82.Towers M, Wolpert L, Tickle C. Gradients of signalling in the developing limb. Current Opinions in Cell Biology. 2012;24(2):181–187. doi: 10.1016/j.ceb.2011.11.005. [DOI] [PubMed] [Google Scholar]