

Abstract
Objective
The aim was to evaluate antitumor activity of the combination of ixabepilone and sunitinib in preclinical models of chemotherapy naïve and refractory epithelial ovarian tumors, and to investigate the mechanism of synergy of such drug combination.
Research Design
HOVTAX2 cell line was derived from a metastatic serous papillary epithelial ovarian tumor (EOC) and a paclitaxel-resistant derivative was established. Dose response curves for ixabepilone and sunitinib were generated and synergy was determined using combination indexes. The molecular mechanism of antitumor synergy was examined using shRNA silencing.
Results
The combination of ixabepilone and sunitinib demonstrated robust antitumor synergy in naïve and paclitaxel-resistant HOVTAX2 cell lines due to increased apoptosis. The GTPase, RhoB, was synergistically upregulated in cells treated with ixabepilone and sunitinib. Using shRNA, RhoB was demonstrated to mediate antitumor synergy. These results were validated in two other EOC cell lines.
Conclusions
Ixabepilone plus sunitinib demonstrated antitumor synergy via RhoB in naïve and paclitaxel-resistant cells resulting in apoptosis. This study demonstrates a novel mechanism of action leading to antitumor synergy and provides ‘proof-of-principle’ for combining molecular targeted agents with cytotoxic chemotherapy to improve antitumor efficacy. RhoB could be envisioned as an early biomarker of response to therapy in a planned Phase II clinical trial to assess the efficacy of ixabepilone combined with a receptor tyrosine kinase inhibitor such as sunitinib. To the best of our knowledge, this is the first demonstration of antitumor synergy between these two classes of drugs in EOC and the pivotal role of RhoB in this synergy.
Keywords: Ovarian cancer, receptor-tyrosine kinase, ixabepilone, sunitinib, RhoB
Introduction
Epithelial ovarian cancer (EOC) is the leading cause of gynecologic cancer death among developed nations with the majority of women presenting with advanced-stage disease and 5-year survival rates of about 25%.1, 2 Approximately 80–85% of all ovarian carcinomas in Western countries are of serous subtype with a papillary or glandular pattern.3 At the molecular level, high-grade ovarian serous carcinomas are genetically unstable and frequently have TP53 mutations, loss of BRCA1 or BRCA2 function, and/or amplification of oncogenes such as PRKCI, NOTCH3, EGFR, HER2, c-MYC and WT1 along with activation of several signaling pathways including ras/raf/MAP kinase and PI3 kinase/Akt pathways.4–7 Low levels or altered expression of apoptosis-regulating proteins such as Bcl-2, BAX, survivin and RhoB are also reported in these tumors.4, 8, 9 Reports suggest that in ovarian cancer, there is enhanced angiogenic-signaling pathway and a higher expression of PDGFRα, PDGFRβ and c-kit with autocrine or paracrine stimulation of the receptors by their respective ligands.10, 11 Dysregulation of these key cell-signaling and biochemical pathways, which ultimately leads to increased cellular proliferation and promotes cell survival, has been implicated in development and maintenance of resistance to chemotherapy and poor prognosis with shortened survival.12–15 RhoB, a small GTPase that regulates actin organization and vesicle transport, is downstream of many of these dysregulated receptors. RhoB is not mutated in cancer, but its altered expression and activity seem crucial to cancer progression and therapeutic responses.16 It is also required for apoptosis in cells exposed to microtubule stabilizing agents. Ras has also been shown to downregulate RhoB promoter transcriptional activity17, 18 while activated Akt is well known to phosphorylate and inactivate RhoB.18
Ovarian cancer is highly sensitive to chemotherapy drugs, particularly the platinum agents, and hence the current preferred treatment regimen for advanced EOC is platinum-based combination chemotherapy, usually coupled with paclitaxel.19 While this regimen has promising clinical response rates, the majority of patients will relapse with a median time-to-recurrence of about one year. A significant number of tumor recurrences become resistant to platinum agents, which significantly hinders successful treatment outcomes. Several clinical studies of chemotherapy agents in patients with platinum resistant ovarian tumors, either singly or in combination, have demonstrated moderate response rates.20 Ixabepilone, a novel epothilone B analog has demonstrated significant clinical benefit with an acceptable safety profile in patients with platinum- and taxane-resistant relapsed or refractory ovarian carcinoma.21 Ixabepilone acts in a similar way to taxanes by stabilizing microtubules resulting in arrested cell division and apoptosis; however it is structurally different from taxanes and has distinct tubulin-binding sites with preferential suppression of the dynamic instability of α/β-III-microtubules.22 It has demonstrated broad anti-tumor activity in several human pre-clinical tumor models.23 In combination with capecitabine, ixabepilone has shown improved response rates and progression-free survival in a phase III clinical trial for patients with metastatic breast cancer pre-treated or resistant to anthracyclines or taxanes.24 Combining such novel cytotoxic chemotherapeutic drugs along with agents that block specific oncogenic pathways may improve anti-tumor efficacy. It is also well recognized that such combinations with non-overlapping mechanisms of action and toxicity profiles can produce synergistic anti-tumor effects. Targeting specific and/or multiple oncogenic pathways using receptor-blocking monoclonal antibodies or tyrosine kinase inhibitors (TKI), thereby preventing activation of the signal transduction cascades, has been harnessed with some success in improving clinical outcomes in several cancers. In ovarian cancer, clinical trials with agents that block angiogenic pathways, mediated by several receptor tyrosine kinases such as VEGFR and PDGFR and c-kit, have shown efficacy. Thus, there is a strong biological rationale for further investigation in combining these agents with microtubule-targeting chemotherapy drugs such as ixabepilone, which can have direct anti-angiogenic effects in addition to antitumor activity.25, 26 In this study, we examine the anti-tumor efficacy of ixabepilone in combination with sunitinib, a multi-targeted receptor TKI in a newly developed paclitaxel naïve and resistant HOVTAX2, a pre-clinical model of high grade serous ovarian carcinoma, established from a surgically resected human ovarian tumor. We also found that activation of RhoB is critical for the antitumor synergy between these two classes of drugs.
Materials and Methods
Reagents
Ixabepilone was kindly provided by Bristol-Myers Squibb (Plainsboro, NJ). Sunitinib (Sutent®Pfizer) was kindly provided by Pfizer (New York, NY) and paclitaxel was purchased from Sigma-Aldrich (St. Louis, MO). All drugs were diluted in DMSO at various concentrations and stored at −20°C.
Tissue, cell lines and in vivo model
This study was approved by the Mayo Institutional Review Board Committee. HOVTAX2 cell line was established in the Copland laboratory from surgically removed human ovarian carcinoma. A portion of the tissue was frozen and processed for pathology review which showed a high grade serous papillary carcinoma. The rest was minced, washed in PBS (Cellgro, Herndon, VA) and cultured in Dulbecco’s modified eagle medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum (Hyclone, Logan, UT), non-essential amino acids (Cellgro), and penicillin-streptomycin-amphotericin B (Cellgro) at 37°C in a humidified atmosphere with 5% CO2. Cells were cultured for 6 months and were designated stable cell lines once they reached passage 50 and were homogeneous. OVCA420 was a gift from Dr. Robert C. Bast Jr. (University of Texas MD Anderson Cancer Center) and UWB1.289 was purchased from ATCC (Manassas, VA). Cells were maintained in the above media that was mixed with equal volume MEGM (Lonza, Walkersville, MD). Our STR analysis matched that of the ATCC profile for UWB1.289. The OVCA420 cells demonstrated a unique profile when examined by the comprehensive Deutsche Sammlung von Mikroorganismen und Zellkulturen data base (http://www.dsmz.de/human_and_animal_cell_lines/main.php?contentleft_id=101). The STR profile for OVCA420 is as follows: AMEL XX; CSF1PO 8; D13S317 12,13; D16S539 9; D18S51 14,17; D21S11 30.2; D3S1358 16,17; D5S818 13; D7S820 11; D8S1179 14,15; FGA 22; TPOX 8; VWA 16,18. For morphological studies, cells were plated in 100 mm plates and grown to ~90% confluence. Phase images were obtained on an inverted Olympus microscope (C Squared Corporation, Pittsburgh, PA). For creating paclitaxel resistant HOVTAX2, cells were treated in vitro with increasing doses of paclitaxel ranging from 0.1 nM to 100 nM every four weeks. For in vivo studies, 2 million HOVTAX2 cells in 100 µl media were injected intraperitoneal (i.p.) into 6 week old female athymic nude mice (n=5, Harlan, Tampa, FL). After 12 weeks, the mice were euthanized and tumors were identified on the diaphragm and mesentery fat of the bowel. Tumor number was counted and tumor volume (mm3) was calculated by using the formula: (0.5236)(a x b x c), whereas a=length, b=width, c=height.
DNA isolation and STR analysis
Genomic DNA from primary tissue and its matching cell line was isolated using the Purelink Genomic DNA Isolation kit (Invitrogen, Carlsbad, CA). Thirteen STR markers using fluorescently labeled primers from ABI (Applied Biosystems, Foster City, CA) as performed by the Genotyping Core facility at Mayo Clinic (Rochester, MN).
Cell proliferation assays and growth curves
For proliferation analysis, cells were plated in 12-well plates (BD Biosciences, Bedford, MA) in triplicate at a concentration of 2 × 104 cells / well. Cells were treated with either paclitaxel, ixabepilone, sunitinib or a combination of various concentrations as indicated in figure legends for 72 hours. DMSO alone was used as control. After 72 hours, cells were washed with PBS (Cellgro), trypsinized and 100 µl was counted by Coulter Particle Counter (Beckman, Brea, CA). For single dose outs of sunitinib and ixabepilone, IC50 was calculated via extrapolation of 50% growth on a log scale to determine corresponding drug concentration.
Analysis of synergy by Combination index analysis
Using the ratio of the IC50, the proportion of each compound needed in a combination dose was calculated. Experiments were then carried out using ixabepilone, sunitinib and a fixed ratio combination of both at a variety of different doses. Drug interactions were analyzed using CalcuSyn®.27 Determination of synergy, additivity or antagonism was based on the multiple drug effect equation of Chou and Talalay and was quantified by the combination index (CI). CI=1 indicates an additive effect, <1 is synergy and >1 is antagonism.28
Cell lysis and western blot analysis
Cells were plated in 10 cm plates at an initial concentration of 3×105 cells/plate for each group and grown to ~50% confluence prior to treatment for 24 hrs and then were treated with ixabepilone, sunitinib and combination of both for 72 hours. DMSO was used as control and all cells received identical volumes of DMSO (1:1000). For each condition, cells were collected including any floating cells and lysed. Cell lysis was done in RIPA buffer containing 50 mM Tris, 5 mM EDTA, 150 mM NaCl, 0.1% SDS, 0.5 % deoxycholate, 1% NP40, protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor (Pierce, Rockford, IL) followed by centrifugation. Supernatant protein concentrations were measured by BCA assay (Pierce). Twenty-five micrograms of protein was loaded on Bis-Tris / MES gels (Invitrogen) and then transferred to 0.2 µm Immobilon-P membranes (Millipore; Billerica, MA). The membranes were hybridized overnight at 4°C with the following antibodies: PDGFR, VEGFR1, PARP, p-ERK, ERK, p-AKT, p53, Rb (Cell Signaling, Beverly, MA); α-tubulin, β-actin (Sigma-Aldrich); βIII-tubulin (Abcam, Cambridge, MA); RhoB (Santa Cruz Biotechnology, Santa Cruz, CA). Secondary species-specific horseradish peroxidase-labeled antibodies (Jackson Immunoresearch, West Grove, PA) were applied in 3% milk/TBS for 45 min at room temperature. Detection was performed using Supersignal chemiluminescence kit (Pierce).
Flow cytometry
For cell death analysis, cells were plated at 7.5 × 104 cells/plate in 6 cm plates (Midwest Scientific) and treated with either 50 nM sunitinib, 10 nM ixabepilone or both for 72 hrs. Media was collected for floating cells and adhered cells were collected using 0.05% trypsin (Cellgro). Both floating and adhered cells were washed with cold PBS (Cellgro)and resuspended in cold binding buffer (BD Pharmingen, San Jose, CA) followed by staining with propidium iodide (BD Pharmingen) for 10 minutes. For cell cycle analysis, cells were plated in 10 cm plates and grown to ~50% confluence prior to serum starvation for 24 hrs. Cells were released with regular media and treatment for 48 hours. Adhered cells were collected using 0.05% trypsin (Cellgro, Manassas, VA). Cells were then fixed and stained with propidium iodide (BD Pharmingen, San Jose, CA) per the manufacturer’s protocol. FACS analysis was performed on Accuri C6 flow cytometer (Accuri, Ann Arbor, MI). Unstained cells were used as controls for setting the population parameters and overlay of histograms show no deviation or drift of channels.
Microtubule stabilization assay
Exponentially growing cells were treated with either DMSO control or 10 nM ixabepilone for 6 hours and then harvested, pelleted and washed in cold PBS (Cellgro). Pelleted cells were lysed in 50 µl hypotonic lysis buffer (1 mM MgCl22 mM EGTA, 0.5% Nonidet P-40, 20 mM Tris-HCl (pH 6.8), and 2 mM PMSF) for 5 min at 37°C. Samples were centrifuged at ~ 15,000 × g for 10 min at 22°C in a temperature-controlled centrifuge to separate the pellet (polymerized cytoskeletal fraction) from the supernatant (soluble cytosolic fraction). The pellet was resuspended in a volume of lysis buffer equal to the supernatant, and both had an equal volume of 4X LDS sample buffer (Invitrogen) added. Equal volumes of samples were used for western analysis.
Lentiviral transduction
Self-inactivating shRNA lentiviruses were generated using MISSION shRNA pLKO.1 constructs (Sigma-Aldrich, St. Louis, MO). The nontarget control was a random scrambled sequence (SHC002) and the sequences for RhoB were from clones NM_004040.2-452s1c1, NM_004040.2-461s1c1, NM_004040.2-498s1c1, NM_004040.2-623s1c1, NM_004040.2-839s1c1) (Sigma-Aldrich). Lentiviruses were packaged using HEK293FT cells by transient transfection of the pLKO.1 constructs along with ViraPower (Invitrogen) using Lipofectamine 2000 (Invitrogen). Supernatants were collected 72 hours post-transfection, passed through a 0.45 µm PVDF syringe filter (Millipore, Bedford, MA) and applied to cells for infection along with 5 µg/ml polybrene (American Bioanalytical, Natick, MA) for 24 hrs. Cells were then selected with 2 µg/ml puromycin (Fisher Scientific, Houston TX).
RNA isolation and Quantitative PCR (QPCR)
Total mRNA was isolated from cells using Purelink RNA isolation kit (Invitrogen) with DNase treatment per the manufacturer’s protocol and the O.D. 260/280 ratio of the mRNA was at least 1.8. Two-step quantitative reverse transcriptase-mediated real-time PCR (QPCR) was used to measure changes in mRNA in response to changes in RhoB expression. The RT step was achieved by synthesizing cDNA from 3 µg RNA using the High Capacity Reverse Transcription kit as per the manufacturer’s protocol (Applied Biosystems Foster City, CA). The PCR step was done using TaqMan® Fast Universal PCR Master Mix (Applied Biosystems) and TaqMan® FAMTM dye-labeled probes for RhoB (Hs00269660_s1) and GAPDH (Hs99999905_m1). Data was normalized to GAPDH for each sample. Fold change values between nontargets, control samples and treated samples were calculated using the ΔΔCt method.29
Immunostaining
IHC was performed on ovarian tissues that were mounted on slides from frozen sections. ICC was done with cell lines plated in chamber slides (Lab-Tek, Rochester, NY) that were fixed with 2% paraformaldehyde followed by permalization with methanol. Slides were blocked with diluent that contained background reducing components (Dakocytomation, Denmark) for 30 minutes and probed for the following antibodies: ER, PR, CK7, c-kit, CA125, BRCA1, BRCA2, PDGFR and VEGFR1. Images were obtained on an inverted Olympus microscope (C Squared Corporation, Tamarac, FL).
Statistical analysis
Data are presented as either percentage, actual cell number, fold change ± s.d. unless otherwise specified. Comparisons of treatment groups were analyzed by two-tailed paired Student's t-test. p<0.05 was considered statistically significant. For synergy statistics, CalcuSyn® was used based on the multiple drug effect equation of Chou and Talalay as previously described.
Results
Cell-line characterization
The patient’s EOC tumor histology demonstrated serous papillary morphology and the derived cell line HOVTAX2 exhibited epithelial morphology (Fig 1A). STR analysis at passage number 65 validated that the derived cell line matched its originating tumor tissue with stable allele sizes for the markers within 0- to 3-bp accuracy of the well-calibrated ABI sequencer. (Fig. 1B) Both tumor and cell line were negative for estrogen and progesterone receptor expression with positive expression for the following: BRCA1, BRCA2, cytokeratin, c-kit, PDGFRα and VEGFR1 (Fig s1A and B). Even though the tumor tissue was positive for Ca125, the derived cell line appeared to have lost Ca125 expression in vitro (Fig s1A and B). This newly established HOVTAX2 cell line demonstrated a wild-type p53 phenotype and a mutant retinoblastoma (Rb) phenotype, which was determined by protein accumulation seen by Western blot analysis (Fig 1B). When HOVTAX2 cells were injected i.p. into immune compromised mice, tumor growth occurred on the diaphragm and mesentery (Fig 1C) similar to that found in patients diagnosed with metastatic ovarian cancer. To model paclitaxel refractory tumors that develop in many patients, a paclitaxel-resistant cell line of HOVTAX2 was established through treatment with incremental increase of dose of paclitaxel over time. This cell line demonstrated resistance to paclitaxel treatment up to 100 nM, a dose that kills 100% of cells in the naïve cell line (Fig 1D).
Figure 1. HOVTAX2, a new serous ovarian carcinoma cell line.

A. Images of a frozen section of the patient tissue and the derivative cell line, HOVTAX2, in live culture. B. Panel showing that the STR profile for HOVTAX2 matches the STR profile of the patient tissue it was derived from. HOVTAX2 is also shown to be p53 wild-type and Rb mutant based upon protein accumulation. C. In vivo images of the diaphragm and mesentery fat of the bowel after HOVTAX2 cells were injected i.p. into 6 week old female athymic nude mice (n=5). After 12 weeks, the mice were euthanized and tumors were identified to grow on the diaphragm and mesentery fat of the bowel. Tumor number was counted and volume measured as described in the Materials and Methods. D. Cell proliferation assay illustrating HOVTAX2 naïve cells are susceptible to 72 hr treatment of 100 nM paclitaxel while HOVTAX2 paclitaxel resistant cells had no significant response. Proliferation is graphed as percentage of control average ± S.D. *p<0.05
Naïve and paclitaxel-resistant serous papillary ovarian cancer cells’ growth is inhibited by sunitinib and ixabepilone
Growth of both naïve and paclitaxel-resistant cell lines was inhibited by sunitinib (Fig 2A) and ixabepilone (Fig 3A) in nM concentrations in a dose dependent manner, after 72 hours of treatment. Western blot analysis demonstrated that VEGFR1, PDGFRα and c-kit (molecular targets of sunitinib) are expressed in both cell lines, with increased VEGFR1 and decreased PDGFRα in the paclitaxel-resistant cells (Fig 2B). Cells that are refractory to chemotherapeutics such as paclitaxel have been previously shown to have altered expression of these receptors.29–31 When the downstream targets of sunitinib were examined, there was only significant down-regulation of p-ERK in naïve and paclitaxel resistant cells (Fig 2B). A dose out of sunitinib from 50 nM to 5 µM for 30 minutes was also examined for p-AKT and total AKT with similar results to that of the 24 hour time point (data not shown). Flow cytometry showed that after treatment with sunitinib, cells became stacked in the G1 phase with a ~15% increase in naïve cells and to a lesser degree in paclitaxel-resistant cells by ~5% (Fig 2C). In order to verify that ixabepilone was stabilizing the microtubules, a microtubule stabilization assay was performed as shown by western blot, whereby β-III tubulin and α-tubulin accumulated in the polymerized fraction (P) (Fig 3B). Flow cytometry illustrated that upon ixabepilone treatment, cells became stacked in the G2/M phase with a ~36% increase in G2/M phase cells in the paclitaxel-naïve cells and an 18% increase in G2/M phase cells in paclitaxel-resistant cells (Fig 3C).
Figure 2. Effects of sunitinib in HOVTAX2.

A. Proliferation of both naïve and paclitaxel resistant cells show dose responsiveness to sunitinib at nM concentrations after 72 hrs. Data is plotted as cell number ± S.D. B. Western blot panel of cells treated with 50 nM sunitinib for 24 hrs showing that p-ERK is downregulated with no changes in p-AKT expression. β-actin is a loading control. C. Cell cycle analysis of cells starved for 24 hrs followed by 72 hr treatment of sunitinib in regular media. Data is plotted as cell phase percentage with stacking in G1 phase in response to sunitinib; more so, in naïve cells.
Figure 3. Effects of ixabepilone in HOVTAX2.

A. Proliferation of both naïve and paclitaxel resistant cells show dose responsiveness to ixabepilone at nM concentrations after 72 hrs. Data is plotted as cell number ± S.D. B. Western blot analysis of cells treated for 6 hours with 10 nM ixabepilone for microtubule stabilization assay shows that β-III tubulin and α-tubulin become polymerized in response to treatment. S=soluble, P=polymerized. C. Cell cycle analysis of cells starved for 24 hrs followed by 72 hr treatment of ixabepilone in regular media. Data is plotted as cell phase percentage with stacking in G2 phase in response to ixabepilone. Naïve cells show greater G2 stacking and decrease in G1 phase when compared to paclitaxel resistant cells.
Sunitinib synergizes with ixabepilone
In HOVTAX2 and its paclitaxel-resistant derivative, the combination of sunitinib and ixabepilone at a fixed ratio display strong antitumor synergism, as evidenced by a significant leftward shift in the synergy curves (Figs. 4A & B). Further analysis with Calcusyn software confirms the strong synergy, with CI values ranging between 0.1 and 0.4 for both cell lines (CI< 1 indicates synergy). This synergy was confirmed in two other serous papillary ovarian carcinomas, OVCA420 and UWB1.289 with CI values of 0.3 and 0.7, respectively (Fig. s2A). Flow cytometry established that the synergy was due to cell death. In the naïve cell line, sunitinib and ixabepilone induced cell death in 15.3% and 33.1% of the populations, respectively (Fig. 4C). In combination, cell death was induced by 60.3%. In the resistant analog, the combination induced cell death by 21.5%, which was significantly less than its naïve counterpart (Fig. 4C). This cell death was due to apoptosis as indicated by PARP cleavage (Fig. 4D).
Figure 4. Combinatorial effects of sunitinib and ixabepilone.

A. Synergy curves using various concentrations of sunitinib and ixabepilone used alone and in combination at a fixed ratio in HOVTAX2 naïve cells show a leftward shift. Data was analyzed by Calcusyn software and Fa-CI plot is as shown. CI=1 indicates an additive effect, <1 is synergy and >1 is antagonism. CI values are between 0.1 and 0.4 indicating strong synergy. B. Synergy curves of HOVTAX2 paclitaxel resistant cells also show a leftward shift with CI values below 1.0 on the Fa-CI plot indicating mild to moderate synergy of sunitinib and ixabepilone. C. Cell death analysis of cells treated for 72 hours alone and in combination. Naïve cells had much higher cell death in combination (60.3%) verses paclitaxel resistant cells (21.5%). D. Western blot analysis confirming that cell death was due to apoptosis by the presence of cleaved PARP.
Synergism of sunitinib and ixabepilone is mediated through upregulated RhoB
Quantitative PCR analysis indicated that sunitinib exposure of the HOVTAX2 cells resulted in increased RhoB mRNA expression (Fig. 5A, s2B). This is consistent with previous published observations that Ras, a downstream signaling partner of tyrosine kinase receptors, downregulates RhoB promoter transcriptional activity.18 In the HOVTAX2 cell line and its resistant derivative cell line, combinatorial exposure to sunitinib and ixabepilone significantly upregulated RhoB to a greater extent than exposure to sunitinib alone, with fold changes of 5.5- and 4-fold, respectively. Western blot analysis confirmed a similar pattern in RhoB protein expression (Fig. 5B). After screening several lentiviruses containing RhoB shRNA (Fig. 6A), RhoB 839 shRNA was used to silence RhoB in all cell lines (Figs. 6B & s2C). RhoB remained silenced following treatment with sunitinib and ixabepilone. HOVTAX2 cells infected with RhoB 839 shRNA lentivirus displayed no induction of apoptosis following exposure to sunitinib and ixabepilone (only a 4.5% increase in apoptosis in the treated cells compared with control), whereas cells infected with nontarget virus maintain the apoptotic response to exposure to ixabepilone and sunitinib (a 56.8% increase in apoptosis in the treated cells compared with untreated cells) (Fig. 6C). OVCA420 and UWB1.289 also showed RhoB dependence, but to a lesser degree (Fig. s2D). Thus, when RhoB is silenced, cells are less susceptible to cell death upon combination treatment indicating RhoB-dependence for antitumor synergy (Figs 6C and s2D).
Figure 5. RhoB induction in response to combinatorial treatment in HOVTAX2.
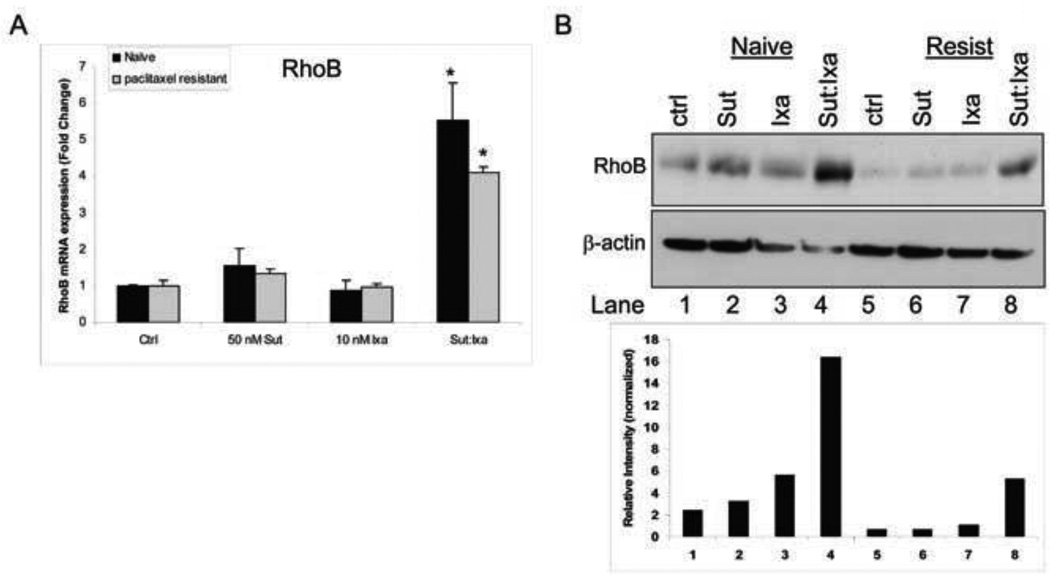
A. Quantitative PCR (QPCR) of RhoB mRNA levels after treatment with 50 nM sunitinib (Sut) and 10 nM ixabepilone (Ixa) show that in combination, RhoB levels increase ~6-fold in naïve cells and ~4-fold in paclitaxel resistant cells. Data is plotted as fold change as compared to DMSO control ± S.D. *p<0.05 B. Western blot analysis of RhoB confirms RhoB is only induced when sunitinib and ixabepilone are used in combination. β-actin is a loading control
Figure 6. Combinatorial effects of sunitinib and ixabepilone are dependent upon RhoB in HOVTAX2.

A. QPCR of RhoB mRNA levels examining lentiviral shRNA silencing of RhoB. RhoB 839 was the only shRNA that has significant silencing in naïve cells. Data is plotted as fold change as compared to nontarget control ± S.D. *p<0.05 B. QPCR of RhoB mRNA levels using nontarget and RhoB 839 shRNA with 24 hr treatment of sunitinib and ixabepilone in combination shows RhoB remains silenced. C. Cell death analysis of cells treated for 72 hours in combination with nontarget and RhoB 839 silenced cells. Cell death was not significantly induced when treated as compared to DMSO control in RhoB silenced cells. Naïve cells have a 56.8% difference with a 4.5% difference in RhoB silenced cells. Paclitaxel resistant cells have a 17.7% difference with only a 0.2% difference in RhoB silenced cells.
Discussion
Combination chemotherapy is the mainstay of most treatments against epithelial malignancies. Recently, several molecularly targeted drugs that block the growth and spread of different malignant tumors by interfering with specific signaling-pathways involved in tumor growth have shown great promise either as single agents or in combination with cytotoxic chemotherapy drugs.32–35 The current combinatorial anti-cancer therapies consist of empirically designed combinations to achieve a synergistic enhancement of the therapeutic efficacy, while overcoming, delaying and/or preventing drug resistance. Understanding the mechanistic pathways contributing to such synergy will improve development of these drug combinations, particularly the molecularly targeted agents along with cytotoxic chemotherapy drugs. Sunitinib and several other TKI drugs have been proposed as possibly effective in reversing chemotherapy resistance.36, 37 The goals of our study was to test antitumor activity of sunitinib alone in paclitaxel-naïve and paclitaxel-resistant serous ovarian cancer cell lines as well as determine if antitumor synergy occurred when combined with ixabepilone. We demonstrated a robust synergistic antitumor activity of this combination in both paclitaxel-naïve and paclitaxel-resistant ovarian tumor models. The potential utility of this combination is particularly relevant in patients with drug resistant disease as ixabepilone demonstrates efficacy in such tumors.22
Induction of apoptosis can generate reactive oxygen species, which in turn can enhance expression of VEGF. VEGF over-expression, an important survival factor for cancer cells, has been postulated as a potential mechanism of chemotherapy resistance.38 As seen in our data, the VEGF receptor 1 was upregulated whereas PDGFRα was attenuated in paclitaxel-resistant cells compared to paclitaxel-naïve cells. Autocrine activation of PDGFR has also been shown to promote the progression of ovarian cancer.39 Hence, the ability of sunitinib to inhibit the signaling through these receptor tyrosine kinases may contribute to the synergy that was observed between ixabepilone and sunitinib. We were surprised in that our findings indicate that p-ERK is inhibited by sunitinib while p-AKT appears not to be suppressed. We examined early time points up to 5 µM and found no differences between p-AKT levels in control and sunitinib treated cells. While sunitinib is known to suppress both of these growth and survival promoting pathways, there is one report indicating selective suppression of p-ERK but not p-AKT in thyroid cancer cells by sunitinib.40, 41
The reduced susceptibility of ixabepilone to drug efflux pumps such as glycoprotein (P-gp) may allow higher accumulation and concentration of ixabepilone within the tumor cells, which may explain the greater anti-tumor efficacy of this agent in resistant tumors. Our observation of cell cycle arrest induced at different phases (G1 phase arrest by sunitinib and G2/M phase by ixabepilone) may also contribute to the synergy observed between the two agents. We also have shown in this study, by using the newly established ovarian cancer cell line and other previously established ovarian cancer models, that the synergistic anti-tumor efficacy of sunitinib and ixabepilone is mediated by up-regulation of RhoB. RhoB, a small GTPase, is now identified as a critical link to antitumor signaling pathway that regulates actin organization and vesicle trafficking.17 It is not mutated in cancer, but its expression is commonly attenuated in cancers.17 In our laboratory, we have previously demonstrated that reactivation of suppressed RhoB is a critical step in inhibition of anaplastic thyroid cancer growth.16 Herein, we show that treatment of cells with sunitinib leads to upregulated RhoB mRNA expression by as yet an unknown mechanism42 and that sunitinib and ixabepilone synergistic effects on apoptosis are abrogated by silencing RhoB.
The observation of synergistic anti-tumor effects of ixabepilone and sunitinib in EOC is a novel observation. Mouse xenograft data has demonstrated single agent antitumor activity of sunitinib in naïve EOC.43 As well, a small phase II Canadian clinical trial has examined single agent sunitinib in recurrent ovarian cancer concluding that sunitinib has modest activity in recurrent platinum-sensitive ovarian cancer.44 A phase II Gynecologic Oncology Group (GOG) clinical trial has also examined single agent ixabepilone with recurrent or persistent platinum- and taxane-resistant primary ovarian or peritoneal carcinoma concluding that ixabepilone demonstrated antitumor activity and acceptable safety in patients with platinum- and taxane resistant recurrent or persistent ovarian or primary peritoneal carcinoma.21 More recently, a Phase 1 clinical trial is enrolling patients to test the combination of sunitinib and ixabepilone against progressive advanced solid tumors (http://clinicaltrials.gov/ct2/show/NCT00884676). To the best of our knowledge, this is the first demonstration of antitumor synergy between these two classes of drugs, a microtubule stabilizer and tyrosine kinase inhibitor in paclitaxel-naïve and paclitaxel-resistant EOC. Furthermore, these results provide ‘proof-of-principle’ for combining specific targeted agents (TKI’s and/or antibody directed against TK) against oncogenic pathways along with cytotoxic chemotherapy and strongly support to test these combinations in clinical trials.
Supplementary Material
A. Immunohistochemistry of frozen sections from patient 2 with serous papillary histology. The sections were positive for all stains except for estrogen receptor (ER) and progesterone receptor (PR). B. Immunocytochemistry of the derived HOVTAX2 cell line showed similar staining patterns with the exception of the loss of CA125 staining.
A. Synergy curves using various concentrations of sunitinib and ixabepilone used alone and in combination at a fixed ratio in ovarian carcinoma cell lines OVCA4 2 0 and UWB1.2 8 9. Data was analyzed by Calcusyn software and Fa-CI plot is as shown. CI=1 indicates an additive effect, <1 is synergy and >1 is antagonism. Synergy curves of both cell lines show a leftward shift with CI values between 0.3 and 0.7 on the Fa-CI plot indicating synergy of sunitinib and ixabepilone. B. Quantitative PCR (QPCR) of RhoB mRNA levels after treatment with 2 µM sunitinib (Sut) and 10 nM ixabepilone (Ixa) show that in response to sunitinib, RhoB levels increase ~6-fold in OVCA420 cells and ~16-fold in UWB1.289 cells. The combination showed no significant combinatorial effect. Western blot analysis confirms similar patters at the protein level (Data not shown). Data is plotted as fold change as compared to DMSO control ± S.D. C. QPCR of RhoB mRNA levels using nontarget and RhoB 839 shRNA with 24 hr treatment of sunitinib and ixabepilone in combination shows RhoB remains silenced. D. Cell death analysis of cells treated for 72 hours in combination with nontarget and RhoB 839 silenced cells. Cell death was not significantly induced when treated as compared to DMSO control in RhoB silenced cells. OVCA420 cells have a 94.6% difference with a 71.9% difference in RhoB silenced cells. UWB1.289 cells have a 94.4% difference with a 52.9% difference in RhoB silenced cells.
Highlights.
Cell-line characterization of serous papillary ovarian cancer cells
Sunitinib synergizes with ixabepilone
Synergism of sunitinib and ixabepilone is mediated through upregulated RhoB
Acknowledgements
We thank the Mayo Clinic Cancer Center for the use of the Genotyping Core, which provided short tandem repeat analysis services. Mayo Clinic Cancer Center is supported in part by an NCI Cancer Center Support Grant 5P30 CA15083-37. This work was funded in part from Mayo Clinic Research Committee (GC-O), NIH/NCI grant R01CA136665 and Mayo Clinic base budget (JAC).
Abbreviations
- STR
short tandem repeat
- EOC
epithelial ovarian cancer
- GOG
Gynecologic Oncology Group
- PDGFR
Platelet-derived growth factor receptor
- VEGFR
Vascular endothelial growth factor receptor
- TKI
Tyrosine kinase inhibitor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1. [Accessed 03/31/2011];SEER Stat Fact Sheets: Ovary. National Cancer Institute. 2010 http://seer.cancer.gov/statfacts/html/urinb.html.
- 2.Heintz AP, Odicino F, Maisonneuve P, et al. Carcinoma of the ovary. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006 Nov;95(Suppl 1):S161–S192. doi: 10.1016/S0020-7292(06)60033-7. [DOI] [PubMed] [Google Scholar]
- 3.Soslow RA. Histologic subtypes of ovarian carcinoma: an overview. Int J Gynecol Pathol. 2008 Apr;27(2):161–174. doi: 10.1097/PGP.0b013e31815ea812. [DOI] [PubMed] [Google Scholar]
- 4.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009 Jun;9(6):415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell DA. Origins and molecular pathology of ovarian cancer. Mod Pathol. 2005 Feb;18(Suppl 2):S19–S32. doi: 10.1038/modpathol.3800306. [DOI] [PubMed] [Google Scholar]
- 6.Despierre E, Lambrechts D, Neven P, Amant F, Lambrechts S, Vergote I. The molecular genetic basis of ovarian cancer and its roadmap towards a better treatment. Gynecol Oncol. 2010 May;117(2):358–365. doi: 10.1016/j.ygyno.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto S, Tsuda H, Kita T, et al. Clinicopathological significance of WT1 expression in ovarian cancer: a possible accelerator of tumor progression in serous adenocarcinoma. Virchows Arch. 2007 Jul;451(1):27–35. doi: 10.1007/s00428-007-0433-4. [DOI] [PubMed] [Google Scholar]
- 8.Couderc B, Pradines A, Rafii A, et al. In vivo restoration of RhoB expression leads to ovarian tumor regression. Cancer Gene Ther. 2008 Jul;15(7):456–464. doi: 10.1038/cgt.2008.12. [DOI] [PubMed] [Google Scholar]
- 9.de la Torre FJ, Garcia A, Gil-Moreno A, et al. Apoptosis in epithelial ovarian tumours Prognostic significance of clinical and histopathologic factors and its association with the immunohistochemical expression of apoptotic regulatory proteins (p53, bcl-2 and bax) Eur J Obstet Gynecol Reprod Biol. 2007 Jan;130(1):121–128. doi: 10.1016/j.ejogrb.2005.11.048. [DOI] [PubMed] [Google Scholar]
- 10.Berchuck A, Olt GJ, Everitt L, Soisson AP, Bast RC, Jr, Boyer CM. The role of peptide growth factors in epithelial ovarian cancer. Obstet Gynecol. 1990 Feb;75(2):255–262. [PubMed] [Google Scholar]
- 11.Wilczynski SP, Chen YY, Chen W, Howell SB, Shively JE, Alberts DS. Expression and mutational analysis of tyrosine kinase receptors c-kit, PDGFRalpha, and PDGFRbeta in ovarian cancers. Hum Pathol. 2005 Mar;36(3):242–249. doi: 10.1016/j.humpath.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Berchuck A, Kamel A, Whitaker R, et al. Overexpression of HER-2/neu is associated with poor survival in advanced epithelial ovarian cancer. Cancer Res. 1990 Jul 1;50(13):4087–4091. [PubMed] [Google Scholar]
- 13.Dabrow MB, Francesco MR, McBrearty FX, Caradonna S. The effects of platelet-derived growth factor and receptor on normal and neoplastic human ovarian surface epithelium. Gynecol Oncol. 1998 Oct;71(1):29–37. doi: 10.1006/gyno.1998.5121. [DOI] [PubMed] [Google Scholar]
- 14.Fraser M, Leung B, Jahani-Asl A, Yan X, Thompson WE, Tsang BK. Chemoresistance in human ovarian cancer: the role of apoptotic regulators. Reprod Biol Endocrinol. 2003 Oct 7;1:66. doi: 10.1186/1477-7827-1-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubin SC, Finstad CL, Wong GY, Almadrones L, Plante M, Lloyd KO. Prognostic significance of HER-2/neu expression in advanced epithelial ovarian cancer: a multivariate analysis. Am J Obstet Gynecol. 1993 Jan;168(1 Pt 1):162–169. doi: 10.1016/s0002-9378(12)90907-2. [DOI] [PubMed] [Google Scholar]
- 16.Marlow LA, Reynolds LA, Cleland AS, et al. Reactivation of suppressed RhoB is a critical step for the inhibition of anaplastic thyroid cancer growth. Cancer Res. 2009 Feb 15;69(4):1536–1544. doi: 10.1158/0008-5472.CAN-08-3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prendergast GC. Actin' up: RhoB in cancer and apoptosis. Nat Rev Cancer. 2001 Nov;1(2):162–168. doi: 10.1038/35101096. [DOI] [PubMed] [Google Scholar]
- 18.Jiang K, Sun J, Cheng J, Djeu JY, Wei S, Sebti S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Molecular and cellular biology. 2004 Jun;24(12):5565–5576. doi: 10.1128/MCB.24.12.5565-5576.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. [Accessed 05/03/2010];NCCN Clinical Practice Guidelines in Oncology / Ovarian Cancer v 2.2011. 2011 http://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf.
- 20.Ledermann JA, Kristeleit RS. Optimal treatment for relapsing ovarian cancer. Ann Oncol. 2010 Oct;21(Suppl 7):vii218–vii222. doi: 10.1093/annonc/mdq377. [DOI] [PubMed] [Google Scholar]
- 21.De Geest K, Blessing JA, Morris RT, et al. Phase II clinical trial of ixabepilone in patients with recurrent or persistent platinum- and taxane-resistant ovarian or primary peritoneal cancer: a gynecologic oncology group study. J Clin Oncol. 2010 Jan 1;28(1):149–153. doi: 10.1200/JCO.2009.24.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dumontet C, Jordan MA, Lee FF. Ixabepilone: targeting betaIII-tubulin expression in taxane-resistant malignancies. Mol Cancer Ther. 2009 Jan;8(1):17–25. doi: 10.1158/1535-7163.MCT-08-0986. [DOI] [PubMed] [Google Scholar]
- 23.Lee FY, Borzilleri R, Fairchild CR, et al. BMS-247550: a novel epothilone analog with a mode of action similar to paclitaxel but possessing superior antitumor efficacy. Clin Cancer Res. 2001 May;7(5):1429–1437. [PubMed] [Google Scholar]
- 24.Thomas ES, Gomez HL, Li RK, et al. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J Clin Oncol. 2007 Nov 20;25(33):5210–5217. doi: 10.1200/JCO.2007.12.6557. [DOI] [PubMed] [Google Scholar]
- 25.Bijman MN, van Nieuw Amerongen GP, Laurens N, van Hinsbergh VW, Boven E. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther. 2006 Sep;5(9):2348–2357. doi: 10.1158/1535-7163.MCT-06-0242. [DOI] [PubMed] [Google Scholar]
- 26.Jelovac D, Armstrong DK. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J Clin. 2011 Apr 26; doi: 10.3322/caac.20113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calcusyn Windows Software for Dose Effect Analysis [computer program] Cambridge, MA: Biosoft; 1996. [Google Scholar]
- 28.Chou T-C, Talalay P. Analysis of combined drug effects: a new look at a very old problem. Trends in Pharmacological Sciences. 1983;4:450–454. [Google Scholar]
- 29.Montgomery RB, Guzman J, O'Rourke DM, Stahl WL. Expression of oncogenic epidermal growth factor receptor family kinases induces paclitaxel resistance and alters beta-tubulin isotype expression. The Journal of biological chemistry. 2000 Jun 9;275(23):17358–17363. doi: 10.1074/jbc.M000966200. [DOI] [PubMed] [Google Scholar]
- 30.Xu JW, Li QQ, Tao LL, et al. Involvement of EGFR in the promotion of malignant properties in multidrug resistant breast cancer cells. International journal of oncology. 2011 Jul 26; doi: 10.3892/ijo.2011.1143. [DOI] [PubMed] [Google Scholar]
- 31.Busby JE, Kim SJ, Yazici S, et al. Therapy of multidrug resistant human prostate tumors in the prostate of nude mice by simultaneous targeting of the epidermal growth factor receptor and vascular endothelial growth factor receptor on tumor-associated endothelial cells. The Prostate. 2006 Dec 1;66(16):1788–1798. doi: 10.1002/pros.20519. [DOI] [PubMed] [Google Scholar]
- 32. [Accessed 8/29/2011];Targeted Cancer Therapies. 2011 http://www.cancer.gov/cancertopics/factsheet/Therapy/targeted. 2011.
- 33.Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Molecular cancer research : MCR. 2007 Mar;5(3):203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 34.Press MF, Lenz HJ. EGFR, HER2 and VEGF pathways: validated targets for cancer treatment. Drugs. 2007;67(14):2045–2075. doi: 10.2165/00003495-200767140-00006. [DOI] [PubMed] [Google Scholar]
- 35.Tkaczuk KH. Review of the contemporary cytotoxic and biologic combinations available for the treatment of metastatic breast cancer. Clinical therapeutics. 2009;31(Pt 2):2273–2289. doi: 10.1016/j.clinthera.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 36.Dai CL, Liang YJ, Wang YS, et al. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009 Jun 28;279(1):74–83. doi: 10.1016/j.canlet.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 37.Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug metabolism and disposition: the biological fate of chemicals. 2009 Feb;37(2):359–365. doi: 10.1124/dmd.108.024612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HS, Oh JM, Jin DH, Yang KH, Moon EY. Paclitaxel induces vascular endothelial growth factor expression through reactive oxygen species production. Pharmacology. 2008;81(4):317–324. doi: 10.1159/000119756. [DOI] [PubMed] [Google Scholar]
- 39.Matei D, Emerson RE, Lai YC, et al. Autocrine activation of PDGFRalpha promotes the progression of ovarian cancer. Oncogene. 2006 Mar 30;25(14):2060–2069. doi: 10.1038/sj.onc.1209232. [DOI] [PubMed] [Google Scholar]
- 40.Yang F, Jove V, Xin H, Hedvat M, Van Meter TE, Yu H. Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways. Molecular cancer research : MCR. 2010 Jan;8(1):35–45. doi: 10.1158/1541-7786.MCR-09-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenton MS, Marion KM, Salem AK, Hogen R, Naeim F, Hershman JM. Sunitinib inhibits MEK/ERK and SAPK/JNK pathways and increases sodium/iodide symporter expression in papillary thyroid cancer. Thyroid : official journal of the American Thyroid Association. 2010 Sep;20(9):965–974. doi: 10.1089/thy.2010.0008. [DOI] [PubMed] [Google Scholar]
- 42.Vasilaki E, Papadimitriou E, Tajadura V, Ridley AJ, Stournaras C, Kardassis D. Transcriptional regulation of the small GTPase RhoB gene by TGF{beta}-induced signaling pathways. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010 Mar;24(3):891–905. doi: 10.1096/fj.09-134742. [DOI] [PubMed] [Google Scholar]
- 43.Bauerschlag DO, Schem C, Tiwari S, et al. Sunitinib (SU11248) inhibits growth of human ovarian cancer in xenografted mice. Anticancer Res. 2010 Sep;30(9):3355–3360. [PubMed] [Google Scholar]
- 44.Biagi JJ, Oza AM, Chalchal HI, et al. A phase II study of sunitinib in patients with recurrent epithelial ovarian and primary peritoneal carcinoma: an NCIC Clinical Trials Group Study. Ann Oncol. 2011 Feb;22(2):335–340. doi: 10.1093/annonc/mdq357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Immunohistochemistry of frozen sections from patient 2 with serous papillary histology. The sections were positive for all stains except for estrogen receptor (ER) and progesterone receptor (PR). B. Immunocytochemistry of the derived HOVTAX2 cell line showed similar staining patterns with the exception of the loss of CA125 staining.
A. Synergy curves using various concentrations of sunitinib and ixabepilone used alone and in combination at a fixed ratio in ovarian carcinoma cell lines OVCA4 2 0 and UWB1.2 8 9. Data was analyzed by Calcusyn software and Fa-CI plot is as shown. CI=1 indicates an additive effect, <1 is synergy and >1 is antagonism. Synergy curves of both cell lines show a leftward shift with CI values between 0.3 and 0.7 on the Fa-CI plot indicating synergy of sunitinib and ixabepilone. B. Quantitative PCR (QPCR) of RhoB mRNA levels after treatment with 2 µM sunitinib (Sut) and 10 nM ixabepilone (Ixa) show that in response to sunitinib, RhoB levels increase ~6-fold in OVCA420 cells and ~16-fold in UWB1.289 cells. The combination showed no significant combinatorial effect. Western blot analysis confirms similar patters at the protein level (Data not shown). Data is plotted as fold change as compared to DMSO control ± S.D. C. QPCR of RhoB mRNA levels using nontarget and RhoB 839 shRNA with 24 hr treatment of sunitinib and ixabepilone in combination shows RhoB remains silenced. D. Cell death analysis of cells treated for 72 hours in combination with nontarget and RhoB 839 silenced cells. Cell death was not significantly induced when treated as compared to DMSO control in RhoB silenced cells. OVCA420 cells have a 94.6% difference with a 71.9% difference in RhoB silenced cells. UWB1.289 cells have a 94.4% difference with a 52.9% difference in RhoB silenced cells.