

Abstract
mir-548 is a larger, poorly conserved primate-specific miRNA gene family. 69 human mir-548 genes located in almost all human chromosomes whose widespread distribution pattern implicates the evolutionary origin from transposable elements. Higher level of nucleotide divergence was detected between these human miRNA genes, which mainly derived from divergence of multicopy pre-miRNAs and homologous miRNA genes. Products of mir-548, miR-548-5p, and miR-548-3p showed inconsistent evolutionary patterns, which partly contributed to larger genetic distances between pre-miRNAs. “Seed shifting” events could be detected among miR-548 sequences due to various 5′ ends. The events led to shift of seed sequences and target mRNAs, even generated to new target mRNAs. Additionally, the phenomenon of miRNA:miRNA interaction in the miRNA gene family was found. The potential interaction between miRNAs may be contributed to dynamic miRNA expression profiles by complementarily binding events to form miRNA:miRNA duplex with 5′-/3′-overhangs. The miRNA gene family had important roles in multiple biological processes, including signaling pathways and some cancers. The potential abundant roles and functional implication further led to the larger and poorly conserved gene family with genetic variation based on transposable elements. The evolutionary pattern of the primate-specific gene family might contribute to dynamic expression profiles and regulatory network.
1. Introduction
MicroRNAs (miRNAs), a distinct class of ~22 nt single-stranded noncoding endogenous RNAs, play pivotal roles in negatively regulating gene expression by targeting mRNAs with an influence on multiple biological processes in plants and animals, including cell growth, differentiation, and apoptosis [1–3]. The primary transcript (pri-miRNA) encoding multiple miRNAs is generated by polymerase II in the nucleus. For example, miRNA members in a gene cluster are cotranscribed as a single polycistronic transcript to coregulate several categories of genes simultaneously [4–6]. Subsequently, the pri-miRNA is converted into pre-miRNA with a hairpin structure by Drosha [7–9]. Pre-miRNA is then translocated to the cytoplasm via exportin-5 [7] where the miRNA:miRNA* duplex is released from the hairpin structure by Dicer [10, 11]. Mature miRNA is loaded into RISC (RNA induced silencing complex) to mediate mRNA targeting [12, 13]. Although miRNA* is typically degraded as an inactive sequence, accumulating reports indicate that miRNA* also contributes to the gene network regulation as a potential active miRNA [14–17]. Posttranscriptional silencing of target genes by miRNAs occurs either by targeting specific cleavage of homologous mRNAs, or by targeting specific inhibition of protein synthesis [18]. Some miRNAs have higher level of sequence similarity and form the miRNA gene family, and even coregulate complex biological processes.
The small noncoding RNAs are well evolutionary conserved across large phylogenetic distances [19], and they have been subjected to the evolutionary patterns, genetic and phylogenetic analysis [20–22]. Compared with other components in the complex gene regulatory networks, the small RNA sequences show a different evolutionary pattern [20, 23]. Despite well-conserved sequences, miRNAs show evolutionary stable “seed shifting” events across different animal species, especially toward the 3′ ends [22]. The diverse biological roles of the small noncoding regulatory RNAs have been investigated recently, but the origin and evolution of miRNAs still remain largely obscure. Some literatures show that miRNAs may originate from repetitive elements, especially for transposable elements (TEs) [24–32]. For example, a larger human gene family, hsa-mir-548, are derived from Made1 transposable elements and have important roles in multiple biological processes [28]. However, with more members in the gene family have been identified, it is quite necessary to further study its evolutionary and functional relationship by systematic analysis, especially based on their potential biological roles in complex regulatory network. Here, according to known members in mir-548 gene family, we sought to indicate the evolutionary pattern and functional implication of the larger gene family.
2. Results
According to the annotation in the miRBase database (Release 18.0), mir-548 was a larger primate-specific gene family. There were 100 miRNA members that were distributed in primates, including Homo sapiens (hsa, 69), Pongo pygmaeus (ppy, 2), Pan troglodytes (ptr, 20), and Macaca mulatta (mml, 9). Except for hsa-mir-548, the miRNA genes in other primates were mainly predicted by softwares based on sequence similarity. Therefore, we herein mainly analyzed and discussed human mir-548.
As a larger human gene family, hsa-mir-548 was located on almost all of the human chromosomes, especially for chromosomes 6, 8, and X (Figure 1). There were 69 members in the gene family, and higher level of nucleotide divergence could be detected between these homologous miRNA genes. The main reasons were mainly derived from a larger number of miRNA gene members and multicopy pre-miRNAs. Some miRNAs, such as hsa-miR-548f and hsa-miR-548 h, were found 5 multicopy pre-miRNAs that were located on different chromosomes. Although these multicopy miRNA precursors could yield the same miRNA sequence, other regions, including adjacent nucleotides of miRNA sequence, showed inconsistent nucleotide substitution patterns (see Figure S1 in Supplementary Materials available online at doi:10.1155/2012/679563). Similarly, mature miRNAs also showed higher level of nucleotide divergence, including nucleotide substitution and insertion/deletion (Figures 2 and 3). Compared to other larger miRNA gene families, such as hsa-let-7 gene family [16], hsa-mir-548 gene family showed higher level of nucleotide divergence and was a poorly conserved miRNA gene family (Figures 2 and 3). The phenomenon of “seed shifting” events could be detected among miR-548 due to involved in various 5′ ends (Figure 2). The events led to variety of “seed sequences,” which also influenced the prediction of target mRNAs.
Figure 1.
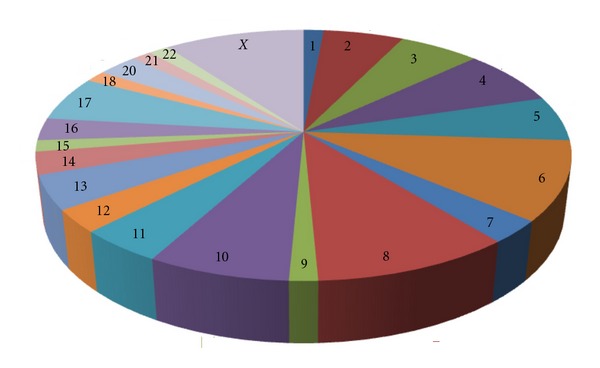
Pie distribution of mir-548 gene family in human chromosomes. The location of mir-548 gene family members is detected in human chromosomes except chromosomes 19 and Y. Note that 30.43% of the members are located in chromosomes 6, 8, and X.
Figure 2.

An example of nucleotide divergence patterns of mature miRNAs. Substitution (transition/transversion) and insertion/deletion can be detected among different mature miRNAs. Simultaneously, their seed sequences can be found shifting events because different 5′ ends.
Figure 3.
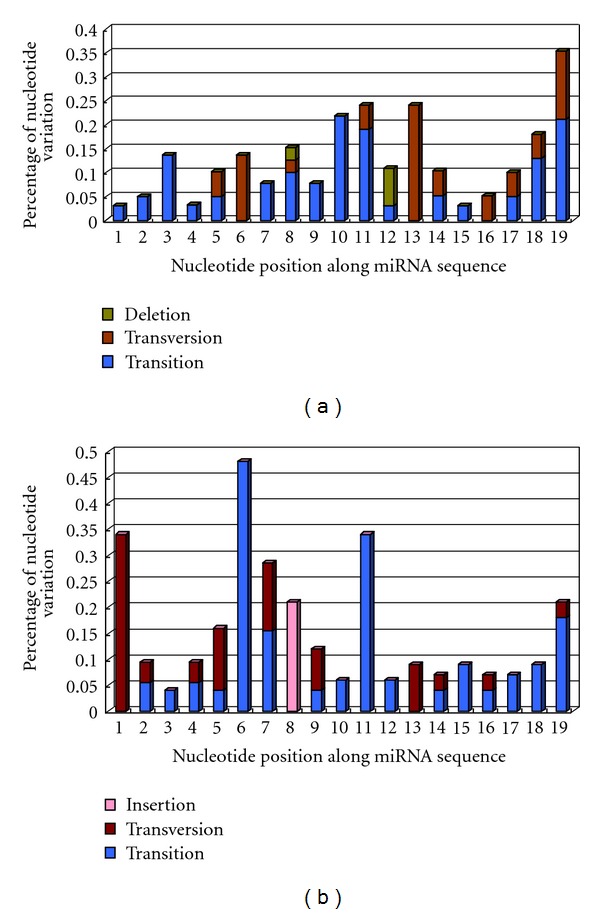
Mutational profiles of hsa-miR-548-5p (a) and hsa-miR-548-3p (b) populations. Products of hsa-mir-548 gene family are estimated their mutational profiles according to each nucleotide position along miRNA sequence. Although these miRNA sequences have higher sequence similarity, higher level of nucleotide variation can be detected between miR-548-5p and miR-548-3p populations, especially the latter.
Based on all hsa-mir-548 sequences, NJ tree showed that some of miRNA genes had larger genetic distances with other members, such as hsa-mir-548e, 548ao, 548i-4 and 548 m (Figure S2). Multicopy pre-miRNAs for a specific miRNA might be reconstructed in different clusters based on phylogenetic tree (Figure S2) and phylogenetic network by using neighbor-net method (data not shown here). Although both arms of hsa-mir-548 could yield mature miRNAs (miR-548-5p and miR-548-3p), hsa-miR-548-5p was more conservation than hsa-miR-548-3p. Nucleotide diversity of miR-548-5p was 0.19 ± 0.01, while nucleotide diversity of miR-548-3p was 0.20 ± 0.02. Average number of nucleotide differences of miR-548-5p and miR-548-3p were 3.81 and 4.30, respectively. Higher frequency of nucleotide substitution and insertion/deletion could be detected between miR-548-3p sequences (Figure 3). Their phylogenetic networks showed different evolutionary patterns. Hsa-miR-548-3p indicated a complex network with more median vectors (Figure 4). According to the predicted target mRNAs of hsa-miR-548, functional enrichment analysis showed that the miRNA gene family played important roles in multiple biological processes, including various human diseases (Table 1). For example, they were involved in regulation of actin cytoskeleton, MAPK signaling pathway, ubiquitin mediated proteolysis, colorectal cancer, glioma, and nonsmall cell lung cancer (Table 1).
Figure 4.
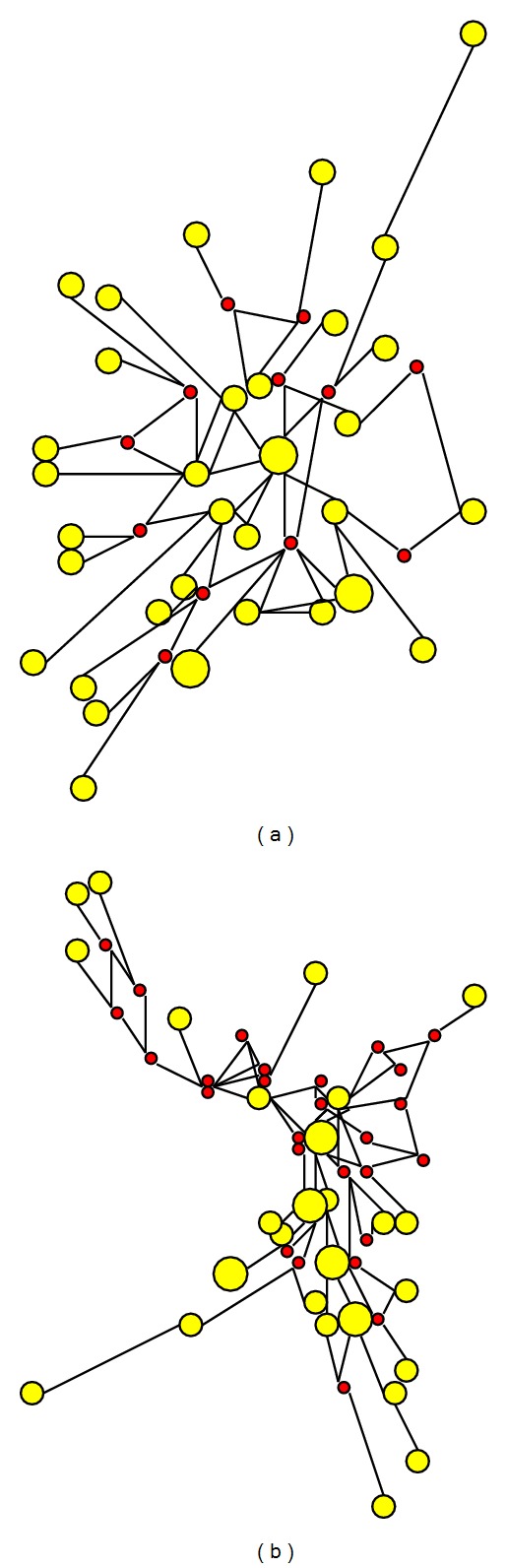
Phylogenetic networks of hsa-miR-548-5p (a) and hsa-miR-548-3p (b). 37 sequences of miR-548-5p and 33 sequences of miR-548-3p are reconstructed, respectively. More median vector (red circles) are found in network of miR-548-3p.
Table 1.
Target genes of hsa-miR-548 gene family enrichment pathway analysis.
| Pathway | Count (gene no.) | Enrichment P value | Target genes |
|---|---|---|---|
| Regulation of actin cytoskeleton | 14 | 2.10E − 07 | VAV2;IQGAP2;RRAS2;ITGB1;PAK7;SOS1;DOCK1;PPP1CB;SSH2;BAIAP2;ABI2;APC;ITGB3;SSH1 |
| MAPK signaling pathway | 14 | 3.33E − 06 | DUSP4;PRKCB;RPS6KA3;RRAS2;BDNF;MAP3K2;SOS1;ELK4;TRAF6;STK4;CACNB4;MAP3K13;FOS;PRKCA |
| Ubiquitin mediated proteolysis | 13 | 6.77E − 09 | UBE2E3;CDC27;RCHY1;UBA2;FBXW7;HERC3;UBR5;UBE2D1;TRAF6;HERC4;UBE2B;VHL;MID1 |
| Focal adhesion | 12 | 4.04E − 06 | VAV2;PRKCB;COL1A1;VASP;ITGB1;PAK7;SOS1;DOCK1;PPP1CB;PTEN;ITGB3;PRKCA |
| Melanogenesis | 11 | 2.73E − 08 | CREBBP;PRKCB;FZD3;FZD9;CALM2;KITLG;WNT5A;CREB1;CALM1;FZD7;\linebreak PRKCA |
| Phosphatidylinositol signaling system | 10 | 1.40E − 08 | PRKCB;CDS2;CALM2;ITPKB;DGKB;PTEN;CDS1;INPP5E;CALM1;PRKCA |
| Tight junction | 10 | 3.43E − 06 | LLGL2;PRKCB;RRAS2;PPP2R2B;VAPA;PARD6B;PTEN;TJP1;PPP2R2A;PRKCA |
| Wnt signaling pathway | 10 | 9.24E − 06 | NFAT5;CREBBP;PRKCB;TBL1X;FZD3;FZD9;WNT5A;FZD7;APC;PRKCA |
| Calcium signaling pathway | 10 | 4.79E − 05 | ERBB3;PRKCB;TRPC1;CALM2;ITPKB;ATP2B4;ERBB4;CALM1;SLC8A1;PRKCA |
| Cell cycle | 9 | 8.41E − 06 | CCNA2;CREBBP;YWHAZ;CDC27;YWHAE;CDK6;CCNB1;CHEK2;MCM6 |
| Axon guidance | 9 | 1.72E − 05 | EPHA3;NFAT5;EPHB1;EFNB2;ITGB1;PAK7;DCC;SEMA6D;ABLIM3 |
| Insulin signaling pathway | 9 | 2.76E − 05 | PCK1;IRS2;CALM2;SOS1;PPARGC1A;PPP1CB;RHOQ;PRKAR2B;CALM1 |
| p53 signaling pathway | 8 | 1.08E − 06 | CCNG2;RCHY1;CDK6;PPM1D;PTEN;CCNB1;CHEK2;APAF1 |
| Long-term potentiation | 8 | 2.05E − 06 | CREBBP;PRKCB;RPS6KA3;CALM2;PPP1CB;CALM1;GRIA1;PRKCA |
| Colorectal cancer | 8 | 4.85E − 06 | APPL1;FZD3;FZD9;SOS1;DCC;FZD7;APC;FOS |
| Glioma | 7 | 8.43E − 06 | PRKCB;CALM2;SOS1;CDK6;PTEN;CALM1;PRKCA |
| ErbB signaling pathway | 7 | 5.73E − 05 | ERBB3;EREG;PRKCB;PAK7;SOS1;ERBB4;PRKCA |
| Glycosphingolipid biosynthesis-lactoseries | 6 | 3.65E − 07 | B4GALT4;B3GALT2;B4GALT1;FUT9;ST8SIA1;GCNT2 |
| Nonsmall cell lung cancer | 6 | 3.14E − 05 | RASSF5;PRKCB;SOS1;CDK6;STK4;PRKCA |
| B cell receptor signaling pathway | 6 | 2.01E − 04 | VAV2;NFAT5;PRKCB;SOS1;DAPP1;FOS |
| Adherens junction | 6 | 2.67E − 04 | CREBBP;LMO7;SSX2IP;TJP1;BAIAP2;PTPRB |
| TGF-beta signaling pathway | 6 | 4.50E − 04 | ACVR2B;BMPR1A;CREBBP;SMAD7;BMPR2;BMPR1B |
| GnRH signaling pathway | 6 | 0.001 | PRKCB;MAP3K2;CALM2;SOS1;CALM1;PRKCA |
| Leukocyte transendothelial migration | 6 | 0.002 | RASSF5;VAV2;PRKCB;VASP;ITGB1;PRKCA |
| Natural killer cell mediated cytotoxicity | 6 | 0.005 | VAV2;NFAT5;PRKCB;SOS1;SH2D1A;SH2D1A;PRKCA |
| Cytokine-cytokine receptor interaction | 6 | 0.076 | ACVR2B;BMPR1A;TNFSF13B;KITLG;BMPR2;BMPR1B |
| Basal cell carcinoma | 5 | 3.79E − 04 | FZD3;FZD9;WNT5A;FZD7;APC |
| Cholera-infection | 5 | 5.26E − 04 | PRKCB;TJP1;ATP6V0A2;KDELR2;PRKCA |
| Adipocytokine signaling pathway | 5 | 9.43E − 04 | ACSL6;PCK1;IRS2;PPARGC1A;ACSL5 |
| PPAR signaling pathway | 5 | 0.001 | ACSL6;PCK1;FABP3;ME1;ACSL5 |
| ECM-receptor interaction | 5 | 0.003 | CD47;COL1A1;DAG1;ITGB1;ITGB3 |
| Gap junction | 5 | 0.004 | PRKCB;MAP3K2;SOS1;TJP1;PRKCA |
| T cell receptor signaling pathway | 5 | 0.008 | VAV2;NFAT5;PAK7;SOS1;FOS |
| Renal cell carcinoma | 5 | 0.001 | CREBBP;EPAS1;PAK7;SOS1;EPAS1;VHL |
| Small cell lung cancer | 5 | 0.003 | ITGB1;CDK6;TRAF6;PTEN;APAF1 |
| Alzheimer's disease | 5 | 0.049 | CALM2;UQCRB;CALM1;APAF1;NDUFA4 |
Here, we only list important pathways that involve at least 5 target mRNAs of hsa-miR-548 gene family.
Interestingly, we found that some miRNA members were natural or endogenous sense/antisense miRNA genes (Table 2). Hsa-mir-548aa and hsa-mir-548d were located on the sense and antisense strands of the same genomic region, and their products could complementarily bind to each other and formed miRNA:miRNA duplex with 5′-/3′-overhangs. The duplex between miRNAs was similar to typical miRNA:miRNA* duplex, although miRNA-miRNA interaction was not involving a loop structure (Figure S3). Strikingly, due to multicopy pre-miRNAs, the miRNA pairs were detected on chromosomes 8 and 17, respectively (Table 2). The miRNA-miRNA interaction not only existed in sense/antisense miRNAs, and also was detected between other miRNAs, such as hsa-miR-548 h-4-3p, and hsa-miR-548c-5p/hsa-miR-548o-2-5p/hsa-miR-548am-5p, although these miRNA pairs were located on different genomic regions or even different chromosomes (Table 2). The functional interaction networks of these miRNA pairs were reconstructed based on the top 20 predicted target mRNAs. Although miRNA:miRNA interaction could be found between miRNA pairs, they might coregulate biological processes (Figure S4).
Table 2.
miRNA:miRNA interaction in hsa-mir-548 gene family.
| Sense/antisense miRNA | miRNAs | Pre-miRNA on chromosome |
|---|---|---|
| mul-pre-1: miR-548aa/548d-5p | miR-548aa | 8(+): 124360274-124360370 |
| miR-548d-5p | 8(−): 124360274-124360370 | |
| mul-pre-2: miR-548aa/548d-5p | 17(+): 65467605-65467701 | |
| 17(−): 65467605-65467701 | ||
|
| ||
| miR-548c/548z | miR-548c | 12(+): 65016289-65016385 |
| miR-548z | 12(−): 65016289-65016385 | |
|
| ||
| miR-548h-4-3p/548c-5p | miR-548h-4-3p | 8(−): 26906370-26906480 |
| miR-548c-5p | 12(+): 65016289-65016385 | |
| miR-548h-4-3p/548o-2-5p | miR-548o-2-5p | 20(+): 37145206-37145275 |
| miR-548h-4-3p/548am-5p | miR-548am-5p | X(−): 16645135-16645208 |
|
| ||
| miR-548t-3p/548d-1-3p | miR-548t-3p | 4(+): 174189311-174189384 |
| miR-548d-1-3p | 8(−): 124360274-124360370 | |
| miR-548t-3p/548d-2-3p | miR-548d-2-3p | 17(−): 65467605-65467701 |
| miR-548t-3p/548ad | miR-548ad | 2(+): 35696471-35696552 |
|
| ||
| miR-548z/548o-2-5p |
miR-548z | 12(−): 65016289-65016385 |
| miR-548o-2-5p | 20(+): 37145206-37145275 | |
| miR-548z/548am | miR-548z | 12(−): 65016289-65016385 |
| miR-548am | X(−): 16645135-16645208 | |
Except for hsa-miR-548aa/548d-5p and hsa-miR-548c/548z are endogenous sense/antisense miRNAs, other pairs are located on different chromosomes.
3. Discussion
According to the known data, mir-548 gene family mainly expresses in primates. The members of this family are poorly conserved in their sequences, and higher levels of nucleotide divergence exist in some of the members, or even in multicopy pre-miRNAs (Figure S1). Among 69 hsa-mir-548 genes, 1–5 multicopy pre-miRNAs for a specific miRNA can be detected, although adjacent nucleotides are inconsistent and consist of nucleotide variations (Figure S1). The phylogenetic tree based on hsa-mir-548 population shows that some of the members have larger genetic distances than the others (Figure S2). Although this gene family is widely located in almost all the human chromosomes, distribution bias can be found, especially for chromosomes 6, 8, and X (Figure 1). Given that the larger and poorly conserved gene family is originated from the transposable elements (TEs) [28, 32], such universe distribution is probably derived from the evolutionary origin. The evolutionary process also leads to the diversity of miRNA sequences with nucleotide substitution, insertion, or deletion (Figures 2 and 3). Simultaneously, the dynamic evolution further strengthens the functions of the poorly conserved gene family, including their versatile biological roles in complex regulatory network.
Although homologous miRNA sequences are found among different members, “seed shifting” events can be detected (Figure 2). The alternation of 5′ ends of miRNA sequences leads to the variety of seed sequences, and even generates the new target mRNAs. The variety of seed sequences implicates their diverse roles in multiple biological processes, including regulating important signaling pathways and human tumorigenesis (Table 1). Indeed, accumulated evidence show that multiple isomiRs, as well as various sequences, can be generated from a given miRNA locus due to the alternative and imprecise Drosha and Dicer cleavage during pre-miRNA processing [33–38]. This fact also partly contributes to the complexity of the regulatory network, especially when mentioning those isomiRs with novel 5′ ends and seed sequences. Taken together, the “seed shifting” events occurring among different members of miR-548 gene family and among multiple isomiRs in a given miR-548 locus are, respectively, derived from the dynamic evolutionary process and miRNAs processing mechanism, and strengthen the biological roles of miRNA in gene regulatory network. Furthermore, the functional variety may be the adaption to the complex biological processes, and simultaneously implicates the evolutionary trend. Interestingly, except for the seed shifting events in miR-548, higher level of nucleotide divergence can also be detected, especially for the miR-548-3p sequences (Figures 3 and 4). Although both arms of hsa-mir-548 yield to mature miRNA sequences (miR-548-5p and miR-548-3p), they show inconsistent mutational profiles and evolutionary patterns (Figures 3 and 4). The nucleotide divergences in the miRNA sequences lead to the various target mRNAs and biological roles (Table 1). The evolutionary trends and patterns are mainly driven by the functional selection pressure from the complex cellular environment and are based on the repetitive elements and transposable elements.
Based on the larger gene family, we also found that some of the members have potential interaction and form miRNA:miRNA duplex (Table 2 and Figure S3). The miRNA-miRNA interaction is recently identified in the miRNA world, which restricts the transcription process [11, 39–42]. These miRNA pairs, such as hsa-mir-548aa and hsa-mir-548d, may be the sense/antisense miRNAs in the same genomic region, or have not location correlation (Table 2). Indeed, the miRNA-miRNA interaction is more prevalent and complex than we previously thought. In this sense, more miRNA members will be discovered and identified, including those active miRNAs* serving as the gene network regulators [14–17]. The abundant miRNA data will indicate more potential miRNA pairs which can complementarily bind to each other and form miRNA:miRNA duplex. In addition, even if the analysis is not so stringent and permits some mismatches and the complementarily binding of nucleotide U and G, the interaction among miRNAs still can be found as a typical structure of the miRNA:miRNA* duplex. The functional interaction of miRNA pairs can be predicted through their target mRNAs, although they have potential interactions by forming a miRNA:miRNA duplex and restricting each enrichment levels (Figure S4). The miRNA-miRNA interaction in the miR-548 gene family may be the results of the inverted repeat transposons during evolutionary history, while these miRNA pairs might have more potential and abundant regulatory roles and contribute to dynamic expression profiles and multiple biological processes.
4. Materials and Methods
All the miRNA members in the mir-548 gene family (http://www.mirbase.org/cgi-bin/mirna_summary.pl?fam=MIPF0000317), including their annotations, miRNA/miRNA* and pre-miRNA sequences from different animal species, were obtained from the miRBase database (Release 18.0, http://www.mirbase.org/) [43]. Multiple sequence alignment of miRNA and pre-miRNA sequences were aligned with Clustal X 2.0 [44]. Phylogenetic trees of pre-miRNAs based on Neighbor-Joining (NJ) method were reconstructed with MEGA 5.0 [45] by 1,000 bootstrap resampling. Nucleotide diversity and average number of nucleotide differences of miR-548-5p and miR-548-3p populations were estimated in DnaSP version 5 software [46]. Percentage of nucleotide substitution and insertion/deletion at each position was estimated for miR-548-5p and miR-548-3p without considering gaps/missing sites in the terminus regions. The most abundant nucleotide at each position was selected as the reference nucleotide, which would help estimate substitution trend more precisely.
Further, phylogenetic network of pre-miRNA members was reconstructed in SplitsTree 4.10 [47] by using the Neighbor-Net method [48] based on Jukes-Cantor model. Based on the network, we attempted to reconstruct the evolutionary history and discover potential evolutionary pattern. All the gaps/missing data were deleted in the phylogenetic tree. In order to infer ancestral miRNA members to understand origin of miRNAs, we also reconstructed evolutionary network [49] with Network 4.6.1.0 (http://www.fluxus-engineering.com/) based on the mature miRNA sequences. Due to various 5′/3′ ends, miR-548 sequences (including miR-548-5p and miR-548-3p) were dealt according to their pre-miRNAs based on the core sequences. Some miRNAs that largely deviated from the core miRNA sequences were removed from the analysis.
Based on a great amount of miRNA members in the gene family, we also searched for the phenomenon of interaction between miRNAs according to miRNA and pre-miRNA sequences. If miRNA sequence could be accurately mapped to other pre-miRNAs, potential miRNA:miRNA interaction could be detected. Generally, the miRNA pairs might be located on sense/antisense strands in the same genomic region, or located on different genomic regions. Similar to miRNA:miRNA* duplex, they could form miRNA:miRNA duplex with 5′-/3′-overhangs.
We integrated the predicted Target mRNAs of the prediction software programs Pictar [50], TargetScan [51] and miRanda programs [52]. These genes were then queried for gene ontology (GO) enrichments by using CapitalBio Molecule Annotation System V4.0 (MAS, http://bioinfo.capitalbio.com/mas3/). We also constructed functional interaction networks using Cytoscape v2.8.2 Platform [53].
Supplementary Material
Figure S1: Multiple alignment of multicopy pre-miRNAs of hsa-miR-548f and their NJ tree.
Figure S2: The phenomenon of miRNA-miRNA interaction by forming miRNA:miRNA duplex.
Figure S3: A phylogenetic tree based on all pre-miRNAs of miR-548 gene family by using NJ model.
Figure S4: A functional interaction network of miRNA pairs.
Authors' Contribution
T. Liang, L. Guo, and C. Liu are contributed equally to this work.
Acknowledgments
This work was supported by a research Grant from the National Natural Science Foundation of China (2012104GZ30055), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (12KJB360001), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nature Reviews Genetics. 2004;5(5):396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 3.Plasterk RHA. Micro RNAs in animal development. Cell. 2006;124(5):877–881. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 4.Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA. 2003;9(2):175–179. doi: 10.1261/rna.2146903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299(5612):p. 1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- 6.Kim VN, Nam JW. Genomics of microRNA. Trends in Genetics. 2006;22(3):165–173. doi: 10.1016/j.tig.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes and Development. 2003;17(24):3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303(5654):95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409(6818):363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 10.Khvorova A, Reynolds A, Jayasena SD. Erratum: Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115(4):p. 505. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 11.Guo L, Liang T, Gu W, Xu Y, Bai Y, Lu Z. Cross-mapping events in miRNAs reveal potential miRNA-Mimics and evolutionary implications. PLoS One. 2011;6(5) doi: 10.1371/journal.pone.0020517.e20517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Carmell MA, Rivas FV, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305(5689):1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 13.Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Molecular Cell. 2004;15(2):185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC. The regulatory activity of microRNA∗ species has substantial influence on microRNA and 3′ UTR evolution. Nature Structural and Molecular Biology. 2008;15(4):354–363. doi: 10.1038/nsmb.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okamura K, Ishizuka A, Siomi H, Siomi MC. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes and Development. 2004;18(14):1655–1666. doi: 10.1101/gad.1210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo L, Lu Z. The fate of miRNA∗ strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? PLoS One. 2010;5(6, article e11387) doi: 10.1371/journal.pone.0011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jagadeeswaran G, Zheng Y, Sumathipala N, et al. Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics. 2010;11(1, article no. 52) doi: 10.1186/1471-2164-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Research. 2005;65(9):3509–3512. doi: 10.1158/0008-5472.CAN-05-0298. [DOI] [PubMed] [Google Scholar]
- 19.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294(5543):858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 20.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nature Reviews Genetics. 2007;8(2):93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 21.Niwa R, Slack FJ. The evolution of animal microRNA function. Current Opinion in Genetics and Development. 2007;17(2):145–150. doi: 10.1016/j.gde.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Guo L, Sun B, Sang F, Wang W, Lu Z. Haplotype distribution and evolutionary pattern of miR-17 and miR-124 families based on population analysis. PLoS One. 2009;4(11, article e7944) doi: 10.1371/journal.pone.0007944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheeler BM, Heimberg AM, Moy VN, et al. The deep evolution of metazoan microRNAs. Evolution and Development. 2009;11(1):50–68. doi: 10.1111/j.1525-142X.2008.00302.x. [DOI] [PubMed] [Google Scholar]
- 24.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nature Structural and Molecular Biology. 2006;13(12):1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 25.Devor EJ, Peek AS, Lanier W, Samollow PB. Marsupial-specific microRNAs evolved from marsupial-specific transposable elements. Gene. 2009;448(2):187–191. doi: 10.1016/j.gene.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hertel J, Lindemeyer M, Missal K, et al. The expansion of the metazoan microRNA repertoire. BMC Genomics. 2006;7, article no. 25 doi: 10.1186/1471-2164-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piriyapongsa J, Mariño-Ramírez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176(2):1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One. 2007;2(2, article no. e203) doi: 10.1371/journal.pone.0000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piriyapongsa J, Jordan IK. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA. 2008;14(5):814–821. doi: 10.1261/rna.916708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends in Genetics. 2005;21(6):322–326. doi: 10.1016/j.tig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Yuan Z, Sun X, Jiang D, et al. Origin and evolution of a placental-specific microRNA family in the human genome. BMC Evolutionary Biology. 2010;10(1, article no. 346) doi: 10.1186/1471-2148-10-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan Z, Sun X, Liu H, Xie J. MicroRNA genes derived from repetitive elements and expanded by segmental duplication events in mammalian genomes. PLoS One. 2011;6(3) doi: 10.1371/journal.pone.0017666.e17666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, Fahlman RP. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Research. 2009;37(8):2461–2470. doi: 10.1093/nar/gkp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuchenbauer F, Morin RD, Argiropoulos B, et al. In-depth characterization of the microRNA transcriptome in a leukemia progression model. Genome Research. 2008;18(11):1787–1797. doi: 10.1101/gr.077578.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Current Biology. 2002;12(9):735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 36.Morin RD, O'Connor MD, Griffith M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Research. 2008;18(4):610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruby JG, Jan C, Player C, et al. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127(6):1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 38.Guo L, Lu Z. Global expression analysis of miRNA gene cluster and family based on isomiRs from deep sequencing data. Computational Biology and Chemistry. 2010;34(3):165–171. doi: 10.1016/j.compbiolchem.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Shearwin KE, Callen BP, Egan JB. Transcriptional interference—a crash course. Trends in Genetics. 2005;21(6):339–345. doi: 10.1016/j.tig.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hongay CF, Grisafi PL, Galitski T, Fink GR. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell. 2006;127(4):735–745. doi: 10.1016/j.cell.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 41.Stark A, Bushati N, Jan CH, et al. A single Hox locus in Drosophila produces functional microRNAs from opposite DNA strands. Genes and Development. 2008;22(1):8–13. doi: 10.1101/gad.1613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai EC, Wiel C, Rubin GM. Complementary miRNA pairs suggest a regulatory role for miRNA:miRNA duplexes. RNA. 2004;10(2):171–175. doi: 10.1261/rna.5191904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozomara A, Griffiths-Jones S. MiRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Research. 2011;39(1):D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 45.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 47.Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14(1):68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- 48.Bryant D, Moulton V. Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Molecular Biology and Evolution. 2004;21(2):255–265. doi: 10.1093/molbev/msh018. [DOI] [PubMed] [Google Scholar]
- 49.Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 50.Krek A, Grün D, Poy MN, et al. Combinatorial microRNA target predictions. Nature Genetics. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 51.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 52.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biology. 2004;2(11) doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. doi: 10.1093/bioinformatics/btq675.btq675 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Multiple alignment of multicopy pre-miRNAs of hsa-miR-548f and their NJ tree.
Figure S2: The phenomenon of miRNA-miRNA interaction by forming miRNA:miRNA duplex.
Figure S3: A phylogenetic tree based on all pre-miRNAs of miR-548 gene family by using NJ model.
Figure S4: A functional interaction network of miRNA pairs.