

Abstract
The gliding Gram-negative Lysobacter bacteria are emerging as a promising source of new bioactive natural products. These ubiquitous freshwater and soil microorganisms are fast growing, simple to use and maintain, and genetically amenable for biosynthetic engineering. This Highlight reviews a group of biologically active and structurally distinct natural products from the genus Lysobacter, with a focus on their biosyntheses. Although Lysobacter sp. are known as prolific producers of bioactive natural products, detailed molecular mechanistic studies of their enzymatic assembly have been surprisingly scarce. We hope to provide a snapshot of the important work done on the lysobacterial natural products and to provide useful information for future biosynthetic engineering of novel antibiotics in Lysobacter.
1 Introduction
The genus Lysobacter belongs to the Xanthomonadaceae family within the gamma-proteobacteria. These bacteria are gliding Gram-negative strict aerobes that are ubiquitous inhabitants of soil and freshwater habitats.1,2 Christensen and Cook proposed this genus in 1978 to include a number of bacterial species which were distinguished by their production of lytic enzymes and their resultant ability to lyse prokaryotic and eukaryotic microbes. This trait, as well as a high genomic G+C content (65–70%) and their gliding motility, is shared with myxobacteria, but lysobacters are not known to form fruiting bodies. Lysobacter species have been occasionally misidentified as members of a similar plant-associated genus and fellow members of the family Xanthomonadaceae, Stenotrophomonas, and other assignments, such as Cytophaga, Sorangium, or Polyangium, for lysobacters can be found in the older literature.2–7
While the bioactive natural products from the Gram-positive actinomycetes have been extensively studied for decades, natural products of Gram-negative bacteria have received comparatively little attention. A number of Lysobacter species are now recognized as bacterial predators with an impressive arsenal of bioactive small molecules, which gives them potential both as biocontrol agents and producers of promising drug leads. An overview of the chemistry of gliding bacteria, including Lysobacter, was published in 2007.7 Here, we focus on the bioactive natural products from the genus Lysobacter, with an emphasis on the biosynthetic mechanisms and biosynthetic genes for cyclodepsipeptides, cyclic lipodepsipeptides, cephem-type β-lactams, and polycyclic tetramate macrolactams (PTM).
2 Structure, activity and biosynthesis of Lysobacter natural products
2.1 Cyclodepsipeptides
The cyclodepsipeptide lysobactin (katanosin B, 1) was initially isolated from Lysobacter sp. ATCC53042 in 1988 (Fig. 1).8,9 Its structure was also independently determined by Tymiak et al. in 1989.10 Among the 11 amino acid residues, six are non-proteinogenic. Structural analogues, including linear lysobactin and desleucyllysobactin generated from the hydrolysis of lysobactin, almost completely abolished biological activity while a circular analogue, L-alanyl-desleucyllysobactin possessed some activity against methicillin-resistant Staphylococcus aureus (MRSA). D-alanyl-desleucyllysobactin was the most active analogue, which suggested that the rigidity of the ring structure and the presence of a D-amino acid at the N-terminus are important for the antibacterial activity of lysobactin.
Fig. 1.

The structure of lysobactin (katanosin B) and katanosin A. Non-proteinogenic amino acid residues are highlighted.
Almost at the same time as the isolation of lysobactin, two depsipeptides, katanosin A (2) and katanosin B (1) were isolated from Cytophaga strain PBJ-5356 that was discovered from a soil sample collected in Katanocity, Osaka Prefecture, Japan.11 The structural determination showed that katanosin B has the same structure as lysobactin, while katanosin A differs from B by one amino acid residue, valine in 2 and isoleucine in 1 (Fig. 1).12
Lysobactin exhibited activity against multiple pathogenic Gram-positive and Gram-negative bacteria and displayed high efficacy in treating the Gram-positive bacterial infections.9 Most notably, lysobactin was found to be effective against MRSA (MIC 0.39 μg ml−1, 2-fold lower than vancomycin) and vancomycin-resistant enterococci (VRE, MIC 0.78 μg ml−1, 50-fold lower than vancomycin). Lysobactin’s activity was proposed to result from inhibition of both the membrane biosynthesis and cell wall biosynthesis.9,13 Due to its significant antibacterial activity, extensive synthetic efforts have been made to obtain partial and total structure of lysobactin.14–23
The biosynthetic genes for lysobactin were first cloned by Bernhard et al. in 1996.24 They isolated a 4.6 kb DNA fragment encoding a partial nonribosomal peptide synthetase (NRPS). This fragment’s relevance to lysobactin biosynthesis was confirmed through gene disruption. The rest of the biosynthetic genes were not reported until 2011 when Hou et al. obtained the whole cluster for lysobactin biosynthesis by genome sequencing (Fig. 2).25 The cluster contains two giant multi-modular NRPSs (LybA and LybB), which together compose 11 modules that directly correlate with the 11 amino acids present in lysobactin. Three of the adenylation (A) domains were heterologously expressed and characterized in vitro, which provided evidence for a co-linear logic of the NRPS biosynthesis of lysobactin. Interestingly, no explicit epimerization (E) domain was found in the NRPSs responsible for the incorporation of two D-amino acids in lysobactin, and two dual function C/E domains were proposed to be responsible for the D-configuration (Fig. 2). Two unusual tandem thioesterases (TE I and II) were found in the C-terminus of LybB, and biochemical characterization showed that TE I is solely responsible for product cyclization and release, while TE II is only involved in hydrolytic cleavage of stuck products from mis-primed peptidyl carrier proteins (PCP).
Fig. 2.

The biosynthetic assembly of lysobactin.25 A, adenylation domain; C, condensation domain; E, epimerase domain; C/E, dual function (condensation/epimerase) domain, PCP, peptidyl carrier protein; TE, thioesterase domain. The function of the two unusual tandem thioesterases (TE I and II) is also included.
2.2 Cyclic lipodepsipeptides
This group of cyclodepsipeptides differs from 1 and 2 by having a β-hydroxyl fatty acid chain that forms both an amide bond and an ester bond with the peptide. Two families of cyclic lipodepsipeptides have been isolated from lysobacters. The first is the WAP-8294A family (Fig. 3, only shown are three members 3–5). In 1997, Kato et al. isolated WAP-8294A2 (3) from Lysobacter sp. 8294 grown in a rich “production medium”.26 The compound contains a total of 12 amino acids and a β-hydroxyl fatty acid (3-hydroxy-7-methyloctanoic acid) (Fig. 3). The carbonyl carbon of the β-hydroxyl fatty acid forms an amide bond with the peptide’s N-terminus, and the β-hydroxyl condenses into an ester bond with the C-terminus.26–29 Six of the 12 amino acid residues assume a D-configuration, two contain an N-methylation, and one contains a β-hydroxylation (Fig. 3).
Fig. 3.

The structure of three members of the WAP-8294A family. Non-proteinogenic amino acid residues are highlighted.
WAP-8294A2 showed activity identical to that of vancomycin (MIC = 0.78 μg ml−1) against clinical MRSA isolates, and more importantly, it was significantly more effective than vancomycin in treating mice with systematic MRSA infections. The mean ED50 values of vancomycin and WAP-8294A2 against nine MRSA strains were 5.3 mg kg−1 and 0.38 mg kg−1, respectively, indicating that WAP-8294A2 is 14 times more active in vivo than vancomycin. Additionally, WAP-8294A2 exhibited good activity (MIC = 6.3 μg ml−1) against VRE, as well as against S. aureus protoplasts, suggesting that WAP-8294A2 possesses a mechanism of action distinct from that of vancomycin. WAP-8294A2’s mode of action was further investigated in comparison with vancomycin.27 The mechanism of vancomycin’s bactericidal effects is through inhibition of transpeptidation of peptidoglycan by noncovalent binding of vancomycin to the D-Ala-D-Ala moiety in the peptidoglycan monomer.30,31 A vancomycin antagonist, diacetyl-L-lysyl-D-alanine-D-alanine, did not inhibit the activity of WAP-8294A2, but a variety of phospholipids, particularly phosphatidylglycerol and cardiolipin, reduced WAP-8294A2’s inhibition zone against a MRSA strain in a disc diffusion assay.27 This suggests that WAP-8294A2’s bactericidal effects result from selective targeting of phospholipids in the cell membrane. WAP-8294A2 is currently in phase I and II clinical trials at aRigen.32
Despite its significant activity, the molecular mechanism for WAP-8294A2 biosynthesis had not been reported until recently. Genome mining of the Lysobacter enzyomegenes OH11, which possesses potent activities against both fungi and bacteria, revealed a large number of NRPSs and polyketide synthases (PKSs).33 Among them, a gene cluster was discovered containing two large modular NRPSs that make up twelve modules. In silico analysis suggested that the domain composition and specificity of the modules matched well with the amino acid composition of WAP-8294A2, including E domains and methyltransferase (MT) domains required for D-amino acid incorporation and N-methylation, respectively. To identify the products of this gene cluster, Zhang et al. generated several gene disruption mutants, including one (WAPS1) of the two NRPSs. The mutants lost activity against Gram-positive bacteria, and at least two HPLC peaks present in the wild type extract disappeared from the mutants. The main peak was isolated from the wild type and found to have the antibacterial activity. Subsequent analysis by HR-MS and 1H and 13C NMR showed that this compound was identical to WAP-8294A2. The results confirmed L. enzymogenes OH11 as a new producer of WAP-8294A2 and revealed the gene cluster required for WAP-8294A2 biosynthesis (Fig. 4).
Fig. 4.

The biosynthetic assembly of WAP-8294A.33 A, adenylation domain; ACL, acyl-CoA ligase; C, condensation domain; CIII, type-III condensation domain; E, epimerase domain; MT, methyltransferase domain; PCP, peptidyl carrier protein; TE, thioesterase domain. Copyright © 2011, American Society for Microbiology. Antimicrobial Agents and Chemotherapy, Dec. 2011, pp. 5581–5589.
The WAP-8294A2 synthetase represents a large NRPS complex, consisting of 45 domains within 12 modules (Fig. 4). The modules make up a linear assembly line that matches the sequence of the amino acids. Interestingly, no acyl-CoA ligase was found in the gene cluster. Such an enzyme is usually necessary for activating the β-hydroxyl-fatty acid before its incorporation into the lipodepsipeptide products. The disruption of one of the acyl-CoA ligases found in the genome led to a significant reduction of WAP-8294A production, suggesting that this pathway may recruit acyl chain-activating enzymes in trans.33 The NPRS assembly starts with a condensation (C) domain, which is consistent with its likely function of condensing the activated acyl chain with the first amino acid (L-Ser1). Following a sequential incorporation of the 12 amino acids by the 12 modules, the linear lipopeptidyl chain is cyclized and released by the TE domain located at the end of assembly line. This TE is expected to have relaxed substrate specificity toward fatty acyl chain, since WAP-8294A analogs differ mainly in the acyl chain length.26–29 This could provide a way to generate new WAP-8294A analogues, through precursor feeding or over-expression of a particular acyl-CoA ligase.
The tripropeptins (6–12), isolated from Lysobacter sp. BMK333-48F3, represent the second family of cyclic lipodepsipeptides (Fig. 5).34–37 They consist of eight amino acid residues and a branched acyl group varying in length from eleven to sixteen carbons, three of which are part of the cyclodepsipeptide ring. Five of the eight residues are non-proteinogenic: D-allo-threonine, D-proline, threo-β-hydroxy-D-aspartic acid, L-trans-3-hydroxyproline, and threo-β-hydroxy-L-aspartic acid. Interestingly, a new tripropeptin was identified from strain BMK333-48F3 after isoleucine was fed to the culture.37 Rather than the characteristic iso-acyl moiety of tri-propeptins A–E and Z, the new tripropeptin featured an anteiso-acyl moiety; its methyl branch shifted from the penultimate to the antepenultimate carbon, indicating that the source of the branched acyl moieties is an amino acid (either leucine or isoleucine). The amino acid is presumably decarboxylated and deaminated to provide the branched acid, which would be activated to serve as the starter unit for a polyketide synthase, supplying the fatty acyl moiety. This novel structure was dubbed tripropeptin anteiso-C (12), and it showed antibacterial properties similar to tripropeptins C and D.
Fig. 5.

The structure of the tripropeptin family. Non-proteinogenic amino acid residues are highlighted.
The activity of the tripropeptins generally increases with the length of the acyl chain, although tripropeptin E (10) had a lower activity against much of the bioassay panel than tripropeptins C (8) and D (9).34–36 Tripropeptins C and D were shown to have excellent activity against Gram-positive bacteria, including MRSA, and also showed activity against VRE. Studies of tri-propeptin C showed that its mechanism of action involves formation of an equimolar complex with Ca2+ and undecaprenyl pyrophosphate, a central intermediate in the lipid cycle of bacterial cell wall biosynthesis.38 This complexation blocks the lipid cycle of cell wall biosynthesis, in a mode of action similar to that of bacitracin. But tripropeptin C showed strong activity (MIC = 0.25 μg ml−1) against both Bacillus subtilis and its bacitracin-efflux mutants, indicating that tripropeptin C is not exported by bacitracin efflux systems. Some enterococci, streptococci, and bacilli that possess such bacitracin efflux systems and are resistant to bacitracin are highly sensitive to tripropeptin C. Although this family of cyclic lipodepsipeptides is structurally interesting and biologically active, its biosynthetic genes and biosynthetic mechanism have not been reported.
2.3 Cephem-type β-lactams
Cephem-type β-lactam antibiotics include cephalosporins, cephabacins and cephamycins. These antibiotics share a core structure, the cephem nucleus (Fig. 6). Cephems are produced by multiple microorganisms, including fungi, Streptomyces, Xanthomonas, and Lysobacter.39 This review will focus on Lysobacter strains, which produce the cephabacins (13–18).
Fig. 6.

The structure of the cephabacin family. PK, polyketide; NRP, nonribosomal peptide.
Cephabacins were isolated from L. lactamgenus strain YK90.40,41 Their structures were determined by spectroscopic analyses involving IR, UV, CD and 1H-, 13C-NMR and were categorized into two major groups, the F group (13–15) and H group (16–18).42 The F group contains an unusual formylamino substituent at C-7 of cephem nucleus, which makes this group highly resistant to hydrolysis by various β-lactamases. Both groups inhibited the peptidoglycan synthesis of E. coli and B. subtilis and showed a protective effect in mice infection.43
The cephalosporin biosynthetic pathway has been initially studied in fungi and actinomycetes.39 The first characterized member of the corresponding Lysobacter pathway was the ORF2 gene (pcbC) encoding isopenicillin N synthase, which is a key enzyme in penicillin/cephalosporin biosynthesis (Fig. 7).44 It catalyzes isopenicillin N formation from δ-(L-α-aminoadipyl)-L-cysteinyl-D-valine (ACV). The minimal gene cluster required for the synthesis of cephabacin core structure was defined by transferring an expression vector pCP7 into Pseudomonas putida, a host with ε-lysine-aminotransferase that supplied α-aminoadipic acid.45 ORF1 (pcbAB), encoding a three-module NRPS (ACV synthetase), was also characterized by expression in Pseudomonas putida. The formation of the tripeptide ACV was observed by HPLC and confirmed using authentic standard. The remaining ORFs encode epimerase, expandase (deacetoxycephalosporin C synthetase), hydroxylase (deacetylcephalosporin C synthetase), and functions for tailoring and resistance (Fig. 7).44–46 In contrast to other β-lactam antibiotics, cephabacins have a hybrid polyketide-peptide chain attached to the methylene hydroxyl moiety at the C-3 of the cephem nucleus. Two ORFs located upstream of the pcbAB cluster were found to encode a hybrid PKS-NRPS (cpbI) and a NRPS (cpbK).47 The CpbI protein contains 3 NRPS modules and one PKS module, comprising a total of 14 domains (C-A3-PCP-C-A2-PCP-C-A1-PCP-KS-AT-KR-ACP-TE), and CpbK protein contains one NRPS module, comprising 3 domains (C-A4-PCP). The four A domains were biochemically characterized, with A1 activating L-arginine and the other three A domains all activating L-alanine.48 The in vitro results confirmed that these four NRPS modules are responsible for the oligopeptide chain incorporation in cephabacin biosynthesis. The function of the PKS in the hybrid PKS-NRPS was also probed by in vitro expression of KS, ACP and AT domains independently in E. coli.49 Using synthetic [L-Ala-L-Ala-L-Ala-L-3H-Arg]-SNAC as substrate, the formation of peptidyl-S-KS was observed when the purified KS was incubated with the 4′-phosphopantetheinyl transferase Sfp. This result showed the selective recognition of the tetrapeptide, a presumed product of the 4-module NRPS, by the PKS of the hybrid NRPS-PKS. Furthermore, the AT domain was shown to use malonyl-CoA as the substrate, which is likely the source of the 2-carbon unit linked to the oligopeptide of cephabacins (Fig. 7). Despite this progress, the pathway is not completely understood. For example, the final condensation step between the polyketide-oligopeptide and the cephem core has not been characterized. It remains unclear how the formylamino moiety in the F group of cephabacins is incorporated. In addition, the mechanism dictating the oligopeptide chain length, as seen in different cephabacin analogues, is not clear.
Fig. 7.

A proposed biosynthetic pathway to cephabacins. A, adenylation domain; ACP, acyl carrier protein; AT, acyltransferase domain; C, condensation domain; KR, ketoreductase domain; KS, ketosynthase domain; PCP, peptidyl carrier protein; TE, thioesterase domain.
2.4 Polycyclic tetramate macrolactams
In 1985, two isomeric antifungal compounds isolated from L. gummosus ATCC 39472 were reported as acyltetramates with the quasi molecular ion [M + H]+ at m/z 511.277 and empirical formulae of C29H38N2O6.50 The two isomers, named catacandins A and B, had good activity against Candida species (MIC 3–12 μg ml−1), but no activity against Gram-negative or Gram-positive bacteria. No further studies were reported on the catacandins. Fully elucidated structures of tetramate-containing macrolactams were first reported as isolates from marine sponges and/or their associated microbiota in the early 1990s.51,52 Two new tetramic acid (2,4-pyrrolidinedione)-containing macrolactams, maltophilin (19) and dihydromaltophilin (20), were isolated later in the decade from terrestrial Stenotrophomonas maltophilia and Streptomyces sp. (Fig. 8).53,54 Maltophilin and dihydromaltophilin were also found to exhibit potent antifungal activity, but no investigations were done on their biosynthetic gene clusters or mechanisms. The quasi molecular ion [M + H]+ of maltophilin is at m/z 511.3, identical to those of the catacandins, and it contains the same acyltetramic moieties which were reported in the catacandins’ partial structures. In the absence of contrary evidence, it seems likely that catacandins A and B were analogues of maltophilin. Very similar to maltophilin is xanthobaccin (21) (Fig. 8), a tetramate macrolactam isolated from Lysobacter sp. strain SB-K88 in 2005 and found to be active against fungal pathogens of sugar beet.3,4 Interestingly, xanthobaccin differs from maltophilin and dihydromaltophilin in the stereochemistry of the two carbon centers at the junctions between the 17-membered macrolactam and the adjoining 6-membered ring, as well as one of the carbons joining the 6-membered ring to the penultimate 5-membered ring.
Fig. 8.
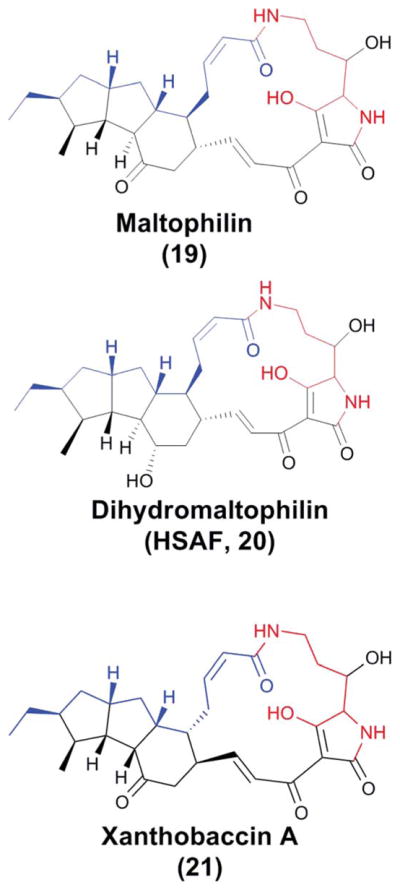
The structures of three members of the polycyclic tetramate macrolactam (PTM) family. Portions colored with blue and black represent the two separate hexaketide chains, and the red-colored region represents the ornithine-originated potion.
A “heat stable antifungal factor” (HSAF) was discovered from the biocontrol agent L. enzymogenes C3 that has powerful anti-fungal activity.55,56 HSAF exhibited inhibitory activity against a wide range of fungal species. Subsequent structural determination showed that HSAF is identical to dihydromaltophilin (20). Like the previously discovered tetramic acid-containing macrolactams, HSAF contains a 5,5,6-tricyclic system fused with a 17-membered macrolactam that includes the tetramic acid moiety (Fig. 8).
The mode of action of HSAF was studied using Aspergillus nidulans as the test organism.55 The results indicated that HSAF specifically interferes with the biosynthesis of sphingolipids by targeting a ceramide synthase unique to filamentous fungi. Further studies showed that HSAF caused thickening of the cell wall due to the accumulation of sphingolipid intermediates, leading to increased chitin deposition and irreversibly blocking elongation of the hyphal tips.57 Sphingolipids represent an attractive new target for the development of novel antifungal drugs because their structure in fungal cells is distinct from that in mammalian cells.58 This novel mode of action, together with the unusual chemical structure that is distinct from any existing antifungal drug on the market, makes HSAF a potentially promising drug lead.
Following the identification of the antifungal factor, the genetic locus responsible for HSAF biosynthesis in L. enzymogenes C3 was also identified.56 In the center of this locus is a gene encoding a hybrid PKS-NRPS, which comprises one PKS module and one NRPS module, making up a total of nine domains (KS-AT-DH-KR-ACP-C-A-PCP-TE) (Fig. 9). A disruption of this PKS-NRPS led to loss of HSAF production as well as the antifungal activity, showing that HSAF biosynthesis requires a hybrid PKS-NRPS mechanism. This was the first biosynthetic gene cluster reported for tetramate-containing macrolactams. The structure of HSAF suggests it is likely to be constructed from two hexaketides, connected by an ornithine residue which forms amide bonds with the two polyketides through both its α- and δ-amines (Fig. 10). The five-membered tetramic acid is formed by a carbon-carbon bond between ornithine’s carbonyl carbon and one hexaketide’s α-carbon. According to the general paradigm established for the biosynthesis of bacterial non-aromatic polyketides, multiple modules of PKS would be needed in order to synthesize these two separate hexaketide chains. However, no further PKS genes were found in the flanking regions of the hybrid PKS-NRPS. Later, the draft genome sequence of L. enzymogenes OH11 was completed, and the results showed that strain OH11 has all the HSAF genes found in strain C3 (Fig. 9). Within the HSAF gene cluster, the two strains have a 96% identity. Remarkably, the genome sequence data showed that the hybrid PKS-NRPS is the only one at this locus.59
Fig. 9.

The HSAF biosynthetic gene cluster.59 KS, β-ketosynthase; AT, acyltransferase; DH, dehydratase; KR, β-ketoreductase; ACP, acyl carrier protein; C, condensation; A, adenylation; PCP, peptidyl carrier protein; TE, thioesterase.
Fig. 10.

A possible biosynthetic pathway to HSAF. Note that the sequence of the OX enzymes-catalyzed cyclizations could be different from the proposed here and similar pathways involving epoxide openings (carbocation chemistry) could also be possible.
The presence of only a single PKS-NRPS in the gene cluster is striking, because bacterial modular PKS are usually non-iterative. To understand the mechanism, Lou et al. heterologously expressed the NRPS domains.59 In vitro characterization using the purified enzymes demonstrated that the A domain specifically activated L-ornithine and the four-domain NRPS was able to catalyze the reactions between acyl-S-ACP and ornithinyl-S-NRPS to form the expected tetramate product. The results showed that this NRPS is responsible for forming both α- and δ-amide bonds as well as the carbon–carbon bond between ornithine’s carbonyl carbon and one hexaketide’s α-carbon (Fig. 10). These results also support the hypothesis that HSAF biosynthesis involves an iterative PKS-NRPS, which is a previously unrecognized mechanism for bacterial polyketide-peptides. Another unusual activity was observed in the study of the TE domain. The in vitro data showed that this TE possesses the activities of both a protease and a peptide ligase, which suggests that the TE may be involved in formation of the amide bonds in the tetramate macrolactam.60
Flanking the hybrid PKS-NRPS are a cascade of redox enzymes, a putative regulator and a transporter (Fig. 9). Two of the redox enzymes have recently been characterized.61 A feature common to nearly all polycyclic tetramate macrolactam (PTM) is that they carry a 3-hydroxyl on the ornithine residue.62,63 A disruption of the putative sterol desaturase (SD) gene abolished HSAF production, but yielded a new compound. The structural determination of this compound showed that it is 3-dehydroxy HSAF (3-deOH-HSAF). This compound exhibited nearly no antifungal activity, showing the crucial role of the 3-hydroxyl moiety. The SD gene was then expressed in E. coli, and HSAF was produced in this E. coli strain upon feeding 3-deOH-HSAF. In vitro, the SD enzyme was able to convert 3-deOH-HSAF to HSAF in the presence of NADPH. The results demonstrated that the SD gene encodes a 3-hydroxylase of the HSAF carbon chain. Additionally, when the ferredoxin reductase gene, which is located next to the SD in the cluster, was co-expressed with the SD in E. coli, the hydroxylase activity was significantly enhanced.61
So far, HSAF is the only PTM whose biosynthetic mechanism is being investigated using both genetic and biochemical approaches. However, many aspects still need to be elucidated. For example, how the single PKS module dictates the chain length and structure of the two separate polyketide chains, which is probably the most intriguing aspect, is not very clear. Another very interesting aspect is how the two polyketide chains eventually lead to the 5,5,6-tricyclic system. The cooperation of the unusual PKS-NRPS with the cascade of redox enzymes is likely to play a key role, as in the proposed biosynthetic mechanism shown in Fig. 8. Further biochemical characterization of each of these enzymes should eventually unveil the secrets of PTM biosynthesis.
2.5 Other natural products isolated from Lysobacter species
In addition to the four families of compounds discussed above, a number of natural products with interesting activity and chemistry have also been reported. Here, we only highlight lactivicin (22–23) and myxin (24) (Fig. 11). Lactivicin was initially isolated from two bacterial strains, Empedobacter lactamgenus YK-258 and Lysobacter albus YK-422.64 It was found to exist as an epimeric mixture in equilibrium under aqueous conditions. The absolute configuration of the two diastereomers (1A, 1B) was assigned by circular dichroism and X-ray crystallography. Lactivicin has a unique structure containing a L-cycloserine ring linked to a γ-lactone ring. It is highly active against Gram-positive bacteria and moderately active against Gram-negative bacteria.65 It displayed a mode of action similar to most β-lactam antibiotics, although it does not contain a β-lactam ring.66–68 Chemically synthesized lactivicin analogues were used for SAR studies.69–72 The biosynthesis of lactivicin has not been reported. The chemical structure suggests an assembly from two building blocks, namely N-acetyl-L-serine and α-hydroxylglutamic acid. An alternative pathway may involve an NRPS-catalyzed condensation between L-cycloserine and α-ketoglutarate, which would lead to the nitrogen-carbon linkage between the L-cycloserine ring and the γ-lactone ring. The acylation of the amino group of L-serine could precede the condensation, thus “deactivating” the primary amine so that the attack of amide nitrogen in the cycloserine ring on the α-carbonyl of α-ketoglutarate would be facilitated.
Fig. 11.

The structures of lactivicin and myxin. The two diasteromers of lactivicin are in equilibrium under aqueous conditions.
Another interesting natural product is the phenazine N-oxide myxin, which was the first reported lysobacterial natural product.73–75 The initial report was prior to the definition of the genus “Lysobacter” by Christensen and Cook in 1978, and the producer organism was described as a “Sorangium”.1,2 Myxin in copper chelate form, cuprimyxin (Unitrop®, Hoffmann-La Roche), is an antibiotic used in treatment of animal skin infections. The mechanism of action involves inhibition of DNA synthesis.76–78 Recently, myxin was shown to cleave DNA strands under both aerobic and anaerobic conditions via a free radical mechanism, indicating that myxin may have therapeutic value in treating cancer, especially hypoxia solid tumors.79 Biosynthesis of myxin has not been reported, while the biosynthesis of phenazine-type antibiotics has been extensively studied.80–83
3 Conclusion
The gliding Gram-negative Lysobacter bacteria are emerging as a promising source of bioactive natural products. They are especially good at producing complex peptides and other amino acid derived natural products. All of the complex peptides discussed in this review, cyclodepsipeptides (lysobactin and katanosin A) and cyclic lipodepsipeptides (WAP-8294As and tripropeptins), exhibit potent antibacterial activities, including anti-MRSA. Particularly notable is WAP-8294A2, which shows exceptionally potent activities against MRSA and VRSA and is currently in Phase-I/II clinical studies.26–29,32 Although both WAP-8294A2 and daptomycin are cyclic lipodepsipeptides, there are some differences in the structures. The N-terminus of WAP-8294A2 is acylated with a β-hydroxyl fatty acid that forms a lactone with the C-terminus carboxyl of the peptide, whereas the N-terminus of daptomycin is acylated with a non-hydroxylated fatty acid and the cyclization takes place between the C-terminus carboxyl and the hydroxyl group of an internal amino acid residue (threonine). In light of the enormous success of daptomcyin (~$560 million sales in 2009), the exploitation of the biosynthesis of this group of complex cyclopeptides could lead to new anti-infectives.
The polycyclic tetramate macrolactams (HSAF) possess a distinct structure and a new mode of action.55,56,62,63 Most remarkably, the biosynthesis of these compounds involves only a single-module PKS-NRPS, although HSAF apparently requires two separate polyketide chains that are linked together by one amino acid (ornithine) via two amide bonds. Iterative PKSs are common in fungi, but no such iterative PKS-NRPS system has been characterized in bacteria. Furthermore, the PTM-type iterative system is distinct from the fungal iterative system in that it is always clustered with a cascade of redox enzymes. Several bacterial modular PKSs were also reported to act iteratively.84,85 However, no other bacterial iterative PKS has so far been reported that is capable of making two separate polyketide chains and interacting with a NRPS to make a hybrid polyketide-peptide product. Finally, PTM-type biosynthetic genes are present in phylogenetically diverse bacteria ranging from proteobacteria to actinomycetes.62,63 In the past few years, genetic and biochemical characterizations have started to reveal a previously unrecognized biosynthetic mechanism for hybrid PK-NRP in prokaryotic organisms.
Cephabacins are new β-lactam antibiotics carrying an unusual formylamino moiety on β-lactam ring, which makes this group of antibiotics highly resistant to β-lactamases. On the other hand, lactivicin does not contain a β-lactam ring but a cycloserine and a γ-lactone, yet it is the only known natural non-β-lactam antibiotic that binds to penicillin-binding proteins. Myxin has an unusual phenazine-N-oxide structure and has been in commercial production for decades.
In spite of their many interesting features in structures and activities, almost none of the lysobacterial natural products have been biosynthetically engineered to generate new analogues. This severely lags behind what has been accomplished in their Gram-positive counterparts, the actinomycetes. The main reasons include the lack of molecular mechanistic understanding of the biosynthetic pathways and insufficient tools for genetic manipulation of this group of gliding bacteria. The identification and characterization of the biosynthetic genes for WAP-8294A2, HSAF, lysobactin, and cephabacins should aid in mechanistic understanding and open a new area of antibiotic research and discovery.
Acknowledgments
The research in the Du lab has been supported by NSFC (31028019), the NIH (R01AI097260), Nebraska Research Council and Nebraska Research Initiatives. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank Dr Haoxin Wang for critical reading of the manuscript.
Biographies

Yunxuan Xie earned her B.Sc. degree (2002) from Tianjin Normal University and M.Sc. degree (2005) from Nankai University, China. She is currently pursuing a Ph.D. at the Department of Chemistry, University of Nebraska-Lincoln, under the guidance of Dr Liangcheng Du. Her research is on the biosynthetic mechanism for bioactive natural products isolated from Lysobacter species.

Stephen J. Wright earned his B.Sc. degree from Wayne State College (Nebraska) in 2009. He is pursuing a master’s degree at the Department of Chemistry, University of Nebraska-Lincoln, under the direction of Dr Liangcheng Du. He is studying the regulatory and biosynthetic mechanisms of natural products in Lysobacter species.

Dr Shen Yuemao received his B.Sc. degree in Chemistry from Anhui Normal University in 1986 and M.Sc. degree in natural products chemistry from KIB, the Chinese Academy of Sciences, in 1989. He was a visiting Scientist and a joint Ph.D. student at the Floss Lab at the University of Washington, in 1995–1999. He received his Ph.D. degree from KIB in 1999. Before joining the faculty of Shandong University in 2009, he was an Intern Investigator (1989–1991), Assistant Investigator (1991–1995), Associate Investigator (1998–1999) and Investigator (1999–2004) at KIB, and Professor at Xiamen University (2004–2009). His research interests are on isolation, structure elucidation, and biosynthesis of natural products with anti-infective or antitumor activities.

Dr Liangcheng Du received his B.Sc. degree in Biology from Yunnan University, M.Sc. degree in Biochemistry from KIB, the Chinese Academy of Sciences, and Ph.D. degree in Biochemistry from The Royal Veterinary and Agricultural University, Denmark. From 1997–2001, he was a postdoctoral associate of Prof. Ben Shen at the University of California at Davis. He was appointed as Assistant Professor at the Department of Chemistry, University of Nebraska-Lincoln, in 2001, and promoted to Associate Professor in 2007. The current research includes the biosynthesis of natural products from Lysobacter, plant pathogenic fungi and endophytic fungi.
References
- 1.Christensen P, Cook FD. Int J Syst Bacteriol. 1978;28:367–393. [Google Scholar]
- 2.Reichenbach H. In: Prokaryotes. Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E, editors. Springer; New York: 2006. pp. 939–957. [Google Scholar]
- 3.Nakayama T, Homma Y, Hashidoko Y, Mizutani J, Tahara S. Appl Environ Microbiol. 1999;65:4334–4339. doi: 10.1128/aem.65.10.4334-4339.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Islam MT, Hashidoko Y, Deora A, Ito T, Tahara S. Appl Environ Microbiol. 2005;71:3786–3796. doi: 10.1128/AEM.71.7.3786-3796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan RF, Holtman MA, Zylstra GJ, White JF, Kobayashi DY. J Appl Microbiol. 2003;94:1079–1086. doi: 10.1046/j.1365-2672.2003.01932.x. [DOI] [PubMed] [Google Scholar]
- 6.Hayward AC, Fegan N, Fegan M, Stirling GR. J Appl Microbiol. 2010;108:756–770. doi: 10.1111/j.1365-2672.2009.04471.x. [DOI] [PubMed] [Google Scholar]
- 7.Nett M, Konig GM. Nat Prod Rep. 2007;24:1245–1261. doi: 10.1039/b612668p. [DOI] [PubMed] [Google Scholar]
- 8.O’Sullivan J, McCullough JE, Tymiak AA, Kirsch DR, Trejo WH, Principe PA. J Antibiot. 1988;41:1740–1744. doi: 10.7164/antibiotics.41.1740. [DOI] [PubMed] [Google Scholar]
- 9.Bonner DP, Osullivan J, Tanaka SK, Clark JM, Whitney RR. J Antibiot. 1988;41:1745–1751. doi: 10.7164/antibiotics.41.1745. [DOI] [PubMed] [Google Scholar]
- 10.Tymiak AA, Mccormick TJ, Unger SE. J Org Chem. 1989;54:1149–1157. [Google Scholar]
- 11.Shoji J, Hinoo H, Matsumoto K, Hattori T, Yoshida T, Matsuura S, Kondo E. J Antibiot. 1988;41:713–718. doi: 10.7164/antibiotics.41.713. [DOI] [PubMed] [Google Scholar]
- 12.Kato T, Hinoo H, Terui Y, Kikuchi JK, Shoji J. J Antibiot. 1988;41:719–725. doi: 10.7164/antibiotics.41.719. [DOI] [PubMed] [Google Scholar]
- 13.Maki H, Miura K, Yamano Y. Antimicrob Agents Chemother. 2001;45:1823–1827. doi: 10.1128/AAC.45.6.1823-1827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armaroli S, Cardillo G, Gentilucci L, Gianotti M, Tolomelli A. Org Lett. 2000;2:1105–1107. doi: 10.1021/ol005659o. [DOI] [PubMed] [Google Scholar]
- 15.Cardillo G, Gentilucci L, Gianotti M, Tolomelli A. Eur J Org Chem. 2000:2489–2494. [Google Scholar]
- 16.Hafez AM, Dudding T, Wagerle TR, Shah MH, Taggi AE, Lectka T. J Org Chem. 2003;68:5819–5825. doi: 10.1021/jo034150e. [DOI] [PubMed] [Google Scholar]
- 17.Egner BJ, Bradley M. Tetrahedron. 1997;53:14021–14030. [Google Scholar]
- 18.Yadav JS, Chandrasekhar S, Reddy YR, Rao AVR. Tetrahedron. 1995;51:2749–2754. [Google Scholar]
- 19.Guzman-Martinez A, Lamer R, VanNieuwenhze MS. J Am Chem Soc. 2007;129:6017–6021. doi: 10.1021/ja067648h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Nussbaum F, Anlauf S, Benet-Buchholz J, Habich D, Kobberling J, Musza L, Telser J, Rubsamen-Waigmann H, Brunner NA. Angew Chem, Int Ed. 2007;46:2039–2042. doi: 10.1002/anie.200604232. [DOI] [PubMed] [Google Scholar]
- 21.Ooi T, Kameda M, Taniguchi M, Maruoka K. J Am Chem Soc. 2004;126:9685–9694. doi: 10.1021/ja048865q. [DOI] [PubMed] [Google Scholar]
- 22.Stawikowska R, Stawikowski M, Cudic P. Biopolymers. 2007;88:569–569. [Google Scholar]
- 23.Stawikowska R, Stawikowski M, Cudic P. Adv Exp Med Biol. 2009;611:385–386. doi: 10.1007/978-0-387-73657-0_168. [DOI] [PubMed] [Google Scholar]
- 24.Bernhard F, Demel G, Soltani K, Dohren HV, Blinov V. DNA Seq. 1996;6:319–330. doi: 10.3109/10425179609047570. [DOI] [PubMed] [Google Scholar]
- 25.Hou J, Robbel L, Marahiell MA. Chem Biol. 2011;18:655–664. doi: 10.1016/j.chembiol.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Kato A, Nakaya S, Ohashi Y, Hirata H. J Am Chem Soc. 1997;119:6680–6681. [Google Scholar]
- 27.Kato A, Nakaya S, Kokubo N, Aiba Y, Ohashi Y, Hirata H, Fujii K, Harada K. J Antibiot. 1998;51:929–935. doi: 10.7164/antibiotics.51.929. [DOI] [PubMed] [Google Scholar]
- 28.Harad KI, Suzuki M, Kato A, Fujii K, Oka H, Ito Y. J Chromatogr, A. 2001;932:75–81. doi: 10.1016/s0021-9673(01)01235-3. [DOI] [PubMed] [Google Scholar]
- 29.Kato A, Hirata H, Ohashi Y, Fujii K, Mori K, Harada K. J Antibiot. 2011;64:373–379. doi: 10.1038/ja.2011.9. [DOI] [PubMed] [Google Scholar]
- 30.Healy VL, Lessard IA, Roper DI, Knox JR, Walsh CT. Chem Biol. 2000;7:R109–119. doi: 10.1016/s1074-5521(00)00116-2. [DOI] [PubMed] [Google Scholar]
- 31.Nicolaou KC, Boddy CNC, Brase S, Winssinger N. Angew Chem, Int Ed. 1999;38:2097–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 32.Pirri G, Giuliani A, Nicoletto SF, Pizzuto L, Rinaldi AC. Cent Eur J Biol. 2009;4:258–273. [Google Scholar]
- 33.Zhang W, Li Y, Qian G, Wang Y, Chen H, Li YZ, Liu F, Shen Y, Du L. Antimicrob Agents Chemother. 2011;55:5581–5589. doi: 10.1128/AAC.05370-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hashizume H, Igarashi M, Hattori S, Hori M, Hamada M, Takeuchi T. J Antibiot. 2001;54:1054–1059. doi: 10.7164/antibiotics.54.1054. [DOI] [PubMed] [Google Scholar]
- 35.Hashizume H, Hirosawa S, Sawa R, Muraoka Y, Ikeda D, Naganawa H, Igarashi M. J Antibiot. 2004;57:52–58. doi: 10.7164/antibiotics.57.52. [DOI] [PubMed] [Google Scholar]
- 36.Hashizume H, Hattori S, Igarashi M, Akamatsu Y. J Antibiot. 2004;57:394–399. doi: 10.7164/antibiotics.57.394. [DOI] [PubMed] [Google Scholar]
- 37.Hashizume H, Igarashi M, Sawa R, Adachi H, Nishimura Y, Akamatsu Y. J Antibiot. 2008;61:577–582. doi: 10.1038/ja.2008.78. [DOI] [PubMed] [Google Scholar]
- 38.Hashizume H, Sawa R, Harada S, Igarashi M, Adachi H, Nishimura Y, Nomoto A. Antimicrob Agents Chemother. 2011;55:3821–3828. doi: 10.1128/AAC.00443-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brakhage AA, Thon M, Sprote P, Scharf DH, Al-Abdallah Q, Wolke SM, Hortschansky P. Phytochemistry. 2009;70:1801–1811. doi: 10.1016/j.phytochem.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Ono H, Nozaki Y, Katayama N, Okazaki H. J Antibiot. 1984;37:1528–1535. doi: 10.7164/antibiotics.37.1528. [DOI] [PubMed] [Google Scholar]
- 41.Harada S. J Antibiot. 1984;37:1536–1545. doi: 10.7164/antibiotics.37.1536. [DOI] [PubMed] [Google Scholar]
- 42.Tsubotani S, Hida T, Kasahara F, Wada Y, Harada S. J Antibiot. 1984;37:1546–1554. doi: 10.7164/antibiotics.37.1546. [DOI] [PubMed] [Google Scholar]
- 43.Nozaki Y, Okonogi K, Katayama N, Ono H, Harada S, Kondo M, Okazaki H. J Antibiot. 1984;37:1555–1565. doi: 10.7164/antibiotics.37.1555. [DOI] [PubMed] [Google Scholar]
- 44.Kimura H, Suzuki M, Sumino Y. J Ferment Bioeng. 1995;80:118–123. [Google Scholar]
- 45.Kimura H, Miyashita H, Sumino Y. Appl Microbiol Biotechnol. 1996;45:490–501. doi: 10.1007/BF00578461. [DOI] [PubMed] [Google Scholar]
- 46.Kimura H, Izawa M, Sumino Y. Appl Microbiol Biotechnol. 1996;44:589–596. doi: 10.1007/BF00172490. [DOI] [PubMed] [Google Scholar]
- 47.Sohn YS, Nam DH, Ryu DD. Metab Eng. 2001;3:380–392. doi: 10.1006/mben.2001.0200. [DOI] [PubMed] [Google Scholar]
- 48.Demirev AV, Lee CH, Jaishy BP, Nam DH, Ryu DD. FEMS Microbiol Lett. 2006;255:121–128. doi: 10.1111/j.1574-6968.2005.00067.x. [DOI] [PubMed] [Google Scholar]
- 49.Seon LJ, Vladimirova MC, Demirev AV, Kim BG, Lim SK, Nam DH. J Microbiol Biotechnol. 2008;18:427–433. [PubMed] [Google Scholar]
- 50.Meyers E, Cooper R, Dean L, Johnson JH, Slusarchyk DS, Trejo WH, Singh PD. J Antibiot. 1985;38:1642–1648. doi: 10.7164/antibiotics.38.1642. [DOI] [PubMed] [Google Scholar]
- 51.Gunasekera SP, Gunasekera M, Mccarthy P. J Org Chem. 1991;56:4830–4833. [Google Scholar]
- 52.Kanazawa S, Fusetani N, Matsunaga S. Tetrahedron Lett. 1993;34:1065–1068. [Google Scholar]
- 53.Jakobi M, Winkelmann G, Kaiser D, Kempter C, Jung G, Berg G, Bahl H. J Antibiot. 1996;49:1101–1104. doi: 10.7164/antibiotics.49.1101. [DOI] [PubMed] [Google Scholar]
- 54.Graupner PR, Thornburgh S, Mathieson JT, Chapin EL, Kemmitt GM, Brown JM, Snipes CE. J Antibiot. 1997;50:1014–1019. doi: 10.7164/antibiotics.50.1014. [DOI] [PubMed] [Google Scholar]
- 55.Li S, Du L, Yuen G, Harris SD. Mol Biol Cell. 2006;17:1218–1227. doi: 10.1091/mbc.E05-06-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu F, Zaleta-Rivera K, Zhu X, Huffman J, Millet JC, Harris SD, Yuen G, Li XC, Du L. Antimicrob Agents Chemother. 2007;51:64–72. doi: 10.1128/AAC.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li SJ, Calvo AM, Yuen GY, Du LC, Harris SD. J Eukaryotic Microbiol. 2009;56:182–187. doi: 10.1111/j.1550-7408.2008.00384.x. [DOI] [PubMed] [Google Scholar]
- 58.Thevissen K, Francois IE, Aerts AM, Cammue BP. Curr Drug Targets. 2005;6:923–928. doi: 10.2174/138945005774912771. [DOI] [PubMed] [Google Scholar]
- 59.Lou LL, Qian GL, Xie YX, Hang JL, Chen HT, Zaleta-Riyera K, Li YY, Shen YM, Dussault PH, Liu FQ, Du LC. J Am Chem Soc. 2011;133:643–645. doi: 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lou L, Chen H, Cerny RL, Li Y, Shen Y, Du L. Biochemistry. 2012;51:4–6. doi: 10.1021/bi2015025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Huffman J, Li Y, Du L, Shen Y. Med Chem Comm. 2012 doi: 10.1039/c2md20026k. [DOI] [Google Scholar]
- 62.Blodgett JA, Oh DC, Cao S, Currie CR, Kolter R, Clardy J. Proc Natl Acad Sci U S A. 2010;107:11692–11697. doi: 10.1073/pnas.1001513107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao S, Blodgett JA, Clardy J. Org Lett. 2010;12:4652–4654. doi: 10.1021/ol1020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harada S, Tsubotani S, Hida T, Ono H, Okazaki H. Tetrahedron Lett. 1986;27:6229–6232. [Google Scholar]
- 65.Nozaki Y, Katayama N, Harada S, Ono H, Okazaki H. J Antibiot. 1989;42:84–93. doi: 10.7164/antibiotics.42.84. [DOI] [PubMed] [Google Scholar]
- 66.Nozaki Y, Katayama N, Ono H, Tsubotani S, Harada S, Okazaki H, Nakao Y. Nature. 1987;325:179–180. doi: 10.1038/325179a0. [DOI] [PubMed] [Google Scholar]
- 67.Macheboeuf P, Fischer DS, Brown T, Zervosen A, Luxen A, Joris B, Dessen A, Schofield CJ. Nat Chem Biol. 2007;3:565–569. doi: 10.1038/nchembio.2007.21. [DOI] [PubMed] [Google Scholar]
- 68.Brown T, Charlier P, Herman R, Schofield CJ, Sauvage E. J Med Chem. 2010;53:5890–5894. doi: 10.1021/jm100437u. [DOI] [PubMed] [Google Scholar]
- 69.Tamura N, Matsushita Y, Kawano Y, Yoshioka K. Chem Pharm Bull. 1990;38:116–122. doi: 10.1248/cpb.38.116. [DOI] [PubMed] [Google Scholar]
- 70.Tamura N, Matsushita Y, Yoshioka K, Ochiai M. Tetrahedron. 1988;44:3231–3240. [Google Scholar]
- 71.Harada S, Tsubotani S, Hida T, Koyama K, Kondo M, Ono H. Tetrahedron. 1988;44:6589–6606. [Google Scholar]
- 72.Natsugari H, Kawano Y, Morimoto A, Yoshioka K, Ochiai M. J Chem Soc, Chem Commun. 1987:62–63. [Google Scholar]
- 73.Peterson EA, Gillespie DC, Cook FD. Can J Microbiol. 1966;12:221–230. doi: 10.1139/m66-031. [DOI] [PubMed] [Google Scholar]
- 74.Weigele M, Leimgruber W. Tetrahedron Lett. 1967;8:715–718. doi: 10.1016/s0040-4039(00)90579-3. [DOI] [PubMed] [Google Scholar]
- 75.Weigele M, Maestrone G, Mitrovic M, Leimgruber W. Antimicrob Agents Chemother. 1970;10:46–49. [PubMed] [Google Scholar]
- 76.Lesley SM, Behki RM. J Bacteriol. 1967;94:1837–1845. doi: 10.1128/jb.94.6.1837-1845.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hollstein U, Van Gemer RJ. Biochemistry. 1971;10:497–504. doi: 10.1021/bi00779a023. [DOI] [PubMed] [Google Scholar]
- 78.Hollstein U, Butler PL. Biochemistry. 1972;11:1345–1350. doi: 10.1021/bi00758a003. [DOI] [PubMed] [Google Scholar]
- 79.Chowdhury G, Sarkar U, Pullen S, Wilson WR, Rajapakse A, Fuchs-Knotts T, Gates KS. Chem Res Toxicol. 2012;25:197–206. doi: 10.1021/tx2004213. [DOI] [PubMed] [Google Scholar]
- 80.Laursen JB, Nielsen J. Chem Rev. 2004;104:1663–1685. doi: 10.1021/cr020473j. [DOI] [PubMed] [Google Scholar]
- 81.Mavrodi DV, Blankenfeldt W, Thomashow LS. Annu Rev Phytopathol. 2006;44:417–445. doi: 10.1146/annurev.phyto.44.013106.145710. [DOI] [PubMed] [Google Scholar]
- 82.Mentel M, Ahuja EG, Mavrodi DV, Breinbauer R, Thomashow LS, Blankenfeldt W. Chem Bio Chem. 2009;10:2295–2304. doi: 10.1002/cbic.200900323. [DOI] [PubMed] [Google Scholar]
- 83.Mavrodi DV, Peever TL, Mavrodi OV, Parejko JA, Raaijmakers JM, Lemanceau P, Mazurier S, Heide L, Blankenfeldt W, Weller DM, Thomashow LS. Appl Environ Microbiol. 2010;76:866–879. doi: 10.1128/AEM.02009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shao L, Qu XD, Jia XY, Zhao QF, Tian ZH, Wang M, Tang GL, Liu W. Biochem Biophys Res Commun. 2006;345:133–139. doi: 10.1016/j.bbrc.2006.04.069. [DOI] [PubMed] [Google Scholar]
- 85.Wenzel SC, Gross F, Zhang Y, Fu J, Stewart AF, Muller R. Chem Biol. 2005;12:349–356. doi: 10.1016/j.chembiol.2004.12.012. [DOI] [PubMed] [Google Scholar]