

Abstract
Adenosine derivatives bearing an N6-(3-iodobenzyl) group, reported to enhance the affinity of adenosine-5′-uronamide analogues as agonists at A3 adenosine receptors (J. Med. Chem. 1994, 37, 636–646), were synthesized starting from methyl β-d-ribofuranoside in 10 steps. Binding affinities at A1 and A2a receptors in rat brain membranes and at cloned rat A3 receptors from stably transfected CHO cells were compared. N6-(3-Iodobenzyl)adenosine was 2-fold selective for A3 vs A1 or A2a receptors; thus it is the first monosubstituted adenosine analogue having any A3 selectivity. The effects of 2-substitution in combination with modifications at the N6- and 5′-positions were explored. 2-Chloro-N6-(3-iodobenzyl)adenosine had a Ki value of 1.4 nM and moderate selectivity for A3 receptors. 2-Chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide, which displayed a Ki value of 0.33 nM, was selective for A3 vs A1 and A2a receptors by 2500- and 1400-fold, respectively. It was 46,000-fold selective for A3 receptors vs the Na+-independent adenosine transporter, as indicated in displacement of [3H]N6-(4-nitrobenzyl)-thioinosine binding in rat brain membranes. In a functional assay in CHO cells, it inhibited adenylate cyclase via rat A3 receptors with an IC50 of 67 nM. 2-(Methylthio)-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide and 2-(methylamino)-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide were less potent, but nearly as selective for A3 receptors. Thus, 2-substitution (both small and sterically bulky) is well-tolerated at A3 receptors, and its A3 affinity-enhancing effects are additive with effects of uronamides at the 5′-position and a 3-iodobenzyl group at the N6-position.
Introduction
The novel A3 adenosine receptor1 may be important in the regulation of CNS, cardiac, inflammatory, and reproductive functions. The expression of A3 adenosine receptors in normal vs asthmatic lung tissue has been studied.2 The A3 adenosine receptor was cloned from rat brain and rat testes cDNA libraries.1,3 Its activation stimulates phosphatidylinositol metabolism in antigen-exposed mast cells4 and inhibits adenylate cyclase in transfected CHO cells.1 Activation of A3 receptors enhances the release of inflammatory mediators from mast cells,4,5 lowers blood pressure,6 and depresses locomotor activity.7. A cerebroprotective effect of chronic administration of an A3 agonist has been discovered.8 The activation of A3 receptors is also thought to be related to the cardioprotective preconditioning response following exposure to adenosine agonists.9
The structure-activity relationships of adenosine derivatives and xanthine derivatives at the rat A3 versus A1 and A2a receptors have been explored.10,11 The affinity of various ligands at sheep12 and human13 A3 receptors has been reported to be very different from rat. At rat A3 receptors, most xanthines known to bind to A1 and A2 receptors do not act as antagonists.10
We recently reported new adenosine agonist derivatives of moderate A3 selectivity.7,10;11 The 5′-methyluronamide modification of adenosine and the N6-benzyl group, either alone or in combination, increases affinity in binding to A3 receptors relative to A1 and A2a receptors.10 Optimization of substituent groups has led to the development of the highly potent A3 agonist N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (IB-MECA, 1, Figure 1) which is 50-fold selective for A3 vs either A1 or A2 receptors. A closely related, but less selective radioligand, [125I]AB-MECA, 2, was developed for characterization of A3 receptors and found to have a Kd value of 3.6 nM in binding to rat A3 receptors in the RBL-2H3 mast cell line.14
Figure 1.

Structures of high-affinity A3 receptor agonists.
In this study we have extended our previous development of A3 selective agonists. By combining the two modifications at 5′- and N6-positions, which were found earlier to result in moderate selectivity, with a third site of modification, the 2-position, we have dramatically increased selectivity. This study presents the first compounds that combine very high potency and selectivity, which should make them very useful as pharmacological tools and potential therapeutic agents.
Results
There remains a need for the development of highly selective A3 agonists. Although the new A3 radioligand [125I]AB-MECA, 2,14 is of nanomolar potency at A3 receptors and more potent than the previously used [125I]APNEA, 4,1 it is not very selective for A3 vs A1 or A2a receptors (Table 1). The presence of the 4-amino group of 2 decreases selectivity in comparison to moderately A3 selective agonist, IB-MECA, 1.7,11 The N6-derivative of adenosine, APNEA, 3, has been used recently in pharmacological studies6 to stimulate A3 receptors, although it is actually A1-selective. Until present, no monosubstituted adenosine derivatives have been reported to be selective for A3 receptors.10 In our previous study of high-affinity 5′,N6-disubstituted adenosine derivatives,11 only 50–70-fold selectivity for A3 vs A1 receptors had been achieved.
Table 1.
Affinities of 5′-Uronamide Derivatives in Radioligand Binding Assays at Rat Brain A1, A2a, and A3 Receptorsa–c
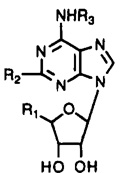 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ki (nM) | |||||||||
| compd | R1 | R2 | R3 | A1a | A2ab | A3c | A1/A3 | A2a/A3 | |
| 1d | CH3NHCO | H | 3-I-Bz | 54 | 56 | 1.1 | 49 | 51 | |
| 2d | CH3NHCO | H | 3-I-4-NH2Bz | 18 | 197 | 1.3 | 14 | 160 | |
| 3 | HOCH2 | H | 4-NH2Ph(CH2)2 | 14f | 172 ± 50 | 116 ± 18 | 0.16 | 1.5 | |
| 4 | HOCH2 | H | 3-I-4-NH2Ph(CH2)2 | 2.1f | 15.5g | 0.14 | |||
| 5e | C2H5NHCO | NH(CH2)2-p-Ph-(CH2)2COOH | H | 2600 | 15 | 584 | 4.4 | 0.026 | |
| 6 | C2H5NHCO | NH(CH2)2-p-Ph-(CH2)2CONH(CH2)2NH2 | H | 400 | 5.7 | 50 ± 24 | 8 | 0.11 | |
| 7e | HOCH2 | Cl | H | 9.3 | 63 | 1890 | 0.0049 | 0.033 | |
| 8d | HOCH2 | H | cyclopentyl | 0.59 | 462 | 240 | 0.0025 | 1.9 | |
| 9d | HOCH2 | Cl | cyclopentyl | 0.6 | 950 | 237 | 0.0025 | 4.0 | |
| 10 | HOCH2 | H | 3-I-Bz | 20.0 ± 8.5 | 17.5 ± 0.5 | 9.5 ± 1.4 | 2.1 | 1.8 | |
| 11 | HOCH2 | Cl | 3-I-Bz | 18.5 ± 4.7 | 38.5 ± 2.0 | 1.41 ± 0.17 | 13 | 27 | |
| 12 | HOCH2 | NH2 | 3-I-Bz | 63.8 ± 15.1 | 117 ± 15 | 181 ± 30 | 0.35 | 0.65 | |
| 13 | CH3NHCO | Cl | 3-I-Bz | 820 ± 570 | 470 ± 365 | 0.33 ± 0.08 | 2500 | 1400 | |
| 14 | CH3NHCO | CH3NH | 3-I-Bz | 4890 ± 2580 | 4120 ± 210 | 3.12 ± 0.64 | 1600 | 1300 | |
| 15 | CH3NHCO | CH3S | 3-I-Bz | 2140 ± 100 | 3210 ± 1360 | 2.30 ± 0.96 | 930 | 1400 | |
Displacement of specific [3H]PIA binding, unless noted, in rat brain membranes expressed as Ki ± SEM in nM (n = 3–6).
Displacement of specific [3H]CGS 21680 binding, unless noted, in rat striatal membranes, expressed as Ki ± SEM in nM (n = 3–6).
Displacement of specific binding of N6-[[125I]-4-amino-3-iodobenzyl]adenosine-5′-N-methyluronamide14 from membranes of CHO cells stably transfected with the rat A3–cDNA, expressed as Ki ± SEM in nM (n = 3–7).
Values are from Gallo-Rodriguez et al.11
Values are from van Galen et al.10 A3 affinity measured by displacement of specific binding of [125I]APNEA in membranes of CHO cells stably transfected with the rat A3–cDNA.1 Ki values at A1 receptors are vs specific binding of [3H]–N 6–cyclohexyladenosine or [3H]R-PIA. Ki values at A2a receptors are vs specific binding of [3H]NECA in the presence of 50 nM N 6-cyclopentyladenosine or vs specific binding of [3H]CGS 21680 in rat striatal membranes.
IC50 values (nM) vs displacement of specific binding of [125I]APNEA in rat brain membranes.23
Kd value (nM) from saturation of binding of [125I]APNEA in membranes of CHO cells stably transfected with the rat A3–cDNA.1
New adenosine analogues (compounds 10–15, Table 1) were synthesized according to Schemes 1 and 2 and characterized (Table 2) and tested in radioligand binding assays14–16 for affinity at rat brain A1, A2a, and A3 adenosine receptors. The compounds were assayed as follows: at A1 receptors in rat cortical membranes using [3H]-N6-[(R)-phenylisopropyl]adenosine15; at A2a receptors in rat striatal membranes using [3H]-CGS 2168016; at A3 receptors using [125I]AB-MECA, 2,14 in membranes of CHO cells stably transfected with cDNA for rat brain A3 receptors.1
Scheme 1a.

a Reagents: (a) TBDPSiCl, DMAP, DMF, room temperature; (b) Bz2O, py; (c) n-Bu4NF, THF; (d) RuO2, NaIO4, CHCl3–CH3CN–H2O (2:2:3); (e) EDAC, DMAP, MeOH; (f) MeNH2, THF, 75 °C; (g) BzCl, py–CH2Cl2; (h) Ac2O, H2SO4, AcOH.
Scheme 2.

Table 2.
Characterization of Intermediates and 2-Substituted-N 6-(3-iodobenzyl)adenosine Derivatives
| compd | mp (°C) | formula | analysis |
|---|---|---|---|
| 10 | 172 | C17H18N5O4I | C, H, N |
| 11 | foam | C17H17N5O4ClI•0.3MeOH | C, H, N |
| 12 | 152–154 | C17H19N6O4I•1.2MeOH | C, H, N |
| 13 | 206–207 | C18H18N6O4ClI•0.5MeOH | C, H, N |
| 14 | 190 | C19H23N7O4I | a |
| 15 | 179 | C19H21N6O4IS | a |
| 17 | syrup | C22H30O5Si | C, H |
| 18 | syrup | C36H38N7Si | C, H |
| 19 | syrup | C20H20N7 | C, H, N |
| 20 | syrup | C20H18N8•0.63H2O | C, H |
| 21 | 92.2–93.7 | C21H20N8 | C, H |
| 22 | syrup | C21H21NO7•0.5H2O | C, H, N |
| 23a,b | foam | C22H21NO8•0.3H2O | C, H, N |
| 24a | 222–224 | C12H19N5ClI | C, H, N |
| 25 | foam | C32H26N6O6ClI•1.0C6H14 | C, H, N |
| 26 | foam | C38H29N5O7ClI0.2C6H14 | C, H, N |
High-resolution MS (m/z) measured in FAB+ mode. 14: calcd for C19H23N7O4I 540.0856, found 540.0867. 15: calcd for C19H21N6O4I1S1 557.0468, found 557.0482.
Substitution at the 2-position is often associated with selectivity of adenosine agonists for A2a vs A1 receptors. For example, CGS 21680, 5, and APEC, 6, both having sterically bulky 2-substituents, were reported to be highly selective for A2a receptors in models of adenylate cyclase.22 Compound 6 was more potent at all three adenosine receptor subtypes than 5, and both compounds displayed a potency order of A2a > A3 > A1, consistent with the findings of van Galen et al.10 that 2-substitution of adenosine is well-tolerated in binding to A3 receptors. Among monosubstituted derivatives of adenosine, 2-(phenylamino)- and 2-chloroadenosine, 7, have Ki values for inhibition of binding of [125I]APNEA (N6-[2-(4-aminophenyl)ethyl]adenosine) at rat A3 receptors of 4.4 and 1.9 µM, respectively.10 Substitution at the 2-position is also compatible with N6-substitution for affinity at A3 receptors. For example, 2-chloro-N6-cyclopentyladenosine, 9 (Table 1), is nearly identical in its receptor binding profile to N6-cyclopentyladenosine, 8.
N6-(3-Iodobenzyl)adenosine, 10, was prepared from 6-chloropurine riboside and 3-iodobenzylamine hydrochloride in the presence of triethylamine in ethanol at 80 °C. Compound 10 was 2-fold selective for A3 vs A1 or A2a receptors, making it is the first monosubstituted adenosine analogue with any selectivity for A3 receptors. 2-Chloro-N6-(3-iodobenzyl)adenosine, 11, was 7-fold more potent than 10 at A3 receptors and of moderate selectivity. 2-Amino substitution of adenosine analogues is also compatible with N6-substitution in A3 receptor binding but is not as favorable as 2-chloro for potency and selectivity. For example, 2-amino-N6-(3-iodobenzyl)-adenosine, 12, was less potent than the 2-H analogue, 10, by factors of 3.2 (A1 receptors), 6.7 (A2a receptors), and 19 (A3 receptors).
To evaluate the effects of triple substitution of adenosine, i.e. at 5′-, 2-, and N6-positions on the affinity at A3 receptors, we developed a general synthetic strategy in which a 5′-uronamide sugar moiety (Scheme 1) was condensed with a purine moiety, such as a substituted adenosine derivative. The key sugar intermediate 23 was synthesized starting from methyl β-d-ribofuranoside (16), which was commercially available or could be synthesized17 from d-ribose, in eight steps. The primary alcohol of 16 was selectively protected with tert-butyldiphenylsilyl chloride18 to provide 17, and the remaining alcohols were followed with benzoyl protection to provide 18. Desilylation of 18 with TBAF/THF gave compound 19. The 5′-position of 19 was oxidized using ruthenium tetroxide19 to give compound 20 which was purified after methylation by silica gel column chromatography. The methylamide at 5-position was introduced by nucleophilic displacement of 21 with methylamine in THF and benzoyl reprotection of resulting 2,3-diol to give the sugar intermediate 23.
In order to synthesize N6-(3-iodobenzyl)-2-substituted-adenosine derivatives, it was necessary to prepare the corresponding adenine derivative (Schemes 2 and 3). 2,6-Dichloropurine reacted with 3-iodobenzylamine hydrochloride in the presence of triethylamine in ethanol at room temperature to provide N6-(3-iodobenzyl)-2-chloroadenine, 24a, which was silylated before coupling to give 24b. The glycosidic bond was formed upon treatment of the 1′-O-acetyl riboside derivative 23 with the 9-silylated adenine derivative 24b in the presence of TMSOTf as a Lewis acid catalyst (Scheme 2). Condensation of 22 with 24b produced ribose ring opened product. Benzoyl groups of 25 were deprotected with NH3/MeOH to produce 13, which reacted with various nucleophiles such as methylamine/THF and sodium thiomethoxide/DME to yield compounds 14 and 15. The benzoyl groups of 26 (Scheme 3) were similarly deprotected to produce the riboside derivative 11.
Scheme 3a.

a Reagents: (a) 3-iodobenzylamine–HCl, triethylamine, EtOH; (b) HMDS, (NH4)2SO4; (c) TMSOTf, ClCH2CH2Cl; (d) NH3/MeOH.
The assignments of the anomeric structure of compounds 25 and 26 were performed based on the comparison of the coupling pattern of anomeric proton of compounds 11 and 13 with compound 10, which is of known anomeric structure being derived from 6-chloropurine riboside.
The combination of 2-substitution with the substituent groups of compound 1 resulted in very high potency and selectivity for A3 receptors. The A3 affinity of the 2-chloro analogue, 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide, 13, was 3-fold greater than for IB-MECA, 1. The affinity at A1 and A2a receptors was diminshed relative to 1, by 15- and 9-fold, respectively. Thus, selectivities of approximately 2500-fold vs A1 receptors and 1400-fold vs A2a receptors were achieved. 2-(Methylamino)-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide, 14, was less potent (Ki value 3 nM), but still highly selective for A3 receptors. 2-(Methylthio)-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide, 15, was also highly selective for A3 receptors.
The selectivity of several of the adenosine derivatives vs a nucleoside transporter previously characterized in brain24 was probed. These experiments were carried out because of the structural similarity of the present adenosine derivatives to various 6-benzyl ethers or thioether derivatives of purine ribosides, known to be high affnity antagonists of adenosine uptake via this transporter.24 The simple N6-benzyl derivative of adenosine was not selective for the receptors vs the adenosine transporter. In contrast, 2-chloro-N6-3-iodobenzyl)adenosine-5′-N-methyluronamide, 13, was 46 000-fold selective for A3 receptors vs the Na+-independent adenosine transporter, as indicated in displacement of [3H]-S-(4-nitrobenzyl)-6-thioinosine binding in rat brain membranes. Thus, in this series of 2,6,5′-trisubstituted adenosine derivatives there was a high degree of selectivity for A3 receptors vs potential antagonism of adenosine uptake.
The agonist properties of the selective ligands were also examined (Figure 2). In a functional assay using membranes from CHO cells stably transfected with rat A3 receptors, compounds 1 and 13 inhibited adenylate cyclase with IC50 values of 90.0 ± 22.5 and 66.8 ± 9.0 nM (n = 4), respectively. Both derivatives were full agonists, with a maximal 41% inhibition of forskolin-stimulated adenylate cyclase. These two derivatives were considerably more potent in the A3 receptor functional assay than were either N6-benzylNECA (IC50 of 1.61 µM) or NECA (IC50 of 5.6 ± 1.9 µM, n = 3). These findings establish a rank order of potency similar to that observed in binding assays, but at higher concentrations. The Ki values for N6-benzylNECA and NECA at rat A3 receptors in CHO cells vs [125I]APNEA were 6.8 and 113 nM, respectively.10 Thus, the IC50 values for these four agonists to inhibit adenylate cyclase were 50–240-fold higher than the respective Ki values at A3 receptors.
Figure 2.

Inhibition of adenylate cyclase in membranes from CHO cell stably transfected with rat A3 receptors. The assay was carried out as described in the Experimental Procedures in the presence of 1 µM forskolin. Each data point is shown as mean ± SEM for four to seven determinations. Adenosine derivatives were (number of separate experiments in parentheses): solid triangles, 1, N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (4); open circles, 13, 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (4); solid circles, N6-benzylNECA (7); and open squares, NECA (3). IC50 values were 1, 90.0 ± 22.5 nM; 13, 66.8 ± 9.0 nM; N6-benzylNECA, 1.61 µM; NECA, 5.6 ± 1.9 µM.
Discussion
In two earlier studies10,11 we demonstrated that combined modification of adenosine at 5′- and at N6-positions with groups that enhanced A3 potency resulted in moderate A3 selectivity. We previously showed N6-benzyladenosine-5′-N-ethyluronamide (N6-benzyl-NECA) to be a full agonist in inhibiting adenylate cyclase via rat A3 receptors.10 However, that derivative was only 1 order of magnitude selective for rat A3 receptors vs either A1 or A2a receptors in binding assays. In this study we have introduced triple substitution of adenosine as a means of enhancing the degree of A3 selectivity, and selectivity in binding assays of 3 orders of magnitude has now been achieved. 2-Chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide, the most potent and selective agent in binding assays, was also shown to be a full agonist in the inhibition of adenylate cyclase, with an IC50 of 67 nM. The agonist potency was also greater than that of other agonists, indicating a parallel between binding affinities and relative potencies in this functional assay. The agonist properties in another relevant functional assay, stimulation of A3-mediated phosphoinositide metabolism, are currently being examined.
Adenosine agonists of high selectivity, such as 13, 14, and 15, are needed for defining the role of A3 receptors in vivo. We have demonstrated that selective agonists may have therapeutic potential as cerebroprotective agents.8,20 Recently Downey and colleagues9 have demonstrated the cardioprotective potential of A3 receptor activation, based on use of APNEA coadministered with a xanthine antagonist that does not act at A3 receptors. In this study we have shown that APNEA is 8-fold A1 selective, and its pharmacological use is limited to such combination with antagonists of both A1 and A2a receptors. Clearly, the availablility of ligands such as 13 could be critical in pharmacological studies of A3 receptors. A highly selective A3 ligand would be useful as a radioligand, since the currently used high affinity ligand [125I]AB-MECA, is not sufficiently selective for general application in tissue.14
It will be necessary to establish the selectivities of these novel A3 agonists in different species, due to the unusually large species dependence in ligand affinity at this subtype, although differences appear to be more pronounced for antagonists than for agonists.12,13,25 It is to be noted that 2-chloroadenosine is 17-fold less potent than NECA at rat A3 receptors,10 whereas at sheep A3 receptors 2-chloroadenosine is only 1.7-fold less potent than NECA.12 Thus, since the most selective compound in the present series, 13, contains the 2-chloro substitution, it is likely that the selectivity will not be substantially diminshed in other species, such as sheep and human. We have shown a high degree of correlation in the relative affinities of adenosine derivatives at rat vs human A3 receptors.25
The selectivity of compound 1 for adenosine receptors vs other neurotransmitter/modulator receptors was shown.11 In this study, we have shown a high degree of selectivity of the doubly-substituted derivative, 1, and the present triply-substituted adenosine derivatives for A3 receptors vs the NBTI-sensitive adenosine uptake site. We have not tested the adenosine derivatives at the normally low-affinity A2b receptor, but substitution at the 2-position of adenosine has been shown not to be well-tolerated at the mouse fibroblast A2b receptor.26
In conclusion, 2-substitution is well-tolerated at A3 receptors, whether it be with a small group (e.g., 11) or a large group (e.g., 6). The potency-enhancing effects of 2-substituents appeared to follow the order: chloro > thioether > amine. The effects of 2-substitution to enhance A3 affinity are also additive with effects of uronamides at the 5′-position and a 3-iodobenzyl group at the N6-position. The A3 affinity-enhancing effect of a 2-chloro group was not additive with an N6-cyclopentyl group. The combination of most favorable modifications at three positions has led to very potent and highly selective agonist ligands, compounds 13–15.
Experimental Procedures
Chemistry
New compounds were characterized (and resonances assigned) by 300 MHz proton nuclear magnetic resonance mass spectroscopy using a Varian GEMINI-300 FT-NMR spectrometer. Unless noted, chemical shifts are expressed as ppm downfield from tetramethylsilane. Synthetic intermediates were characterized by chemical ionization mass spectrometry (NH3) and adenosine derivatives by fast atom bombardment mass spectrometry (positive ions in a noba or m-bullet matrix) on a JEOL SX102 mass spectrometer. In the EI mode accurate mass was determined using a VG7070F mass spectrometer. C, H, and N analyses were carried out by Atlantic Microlabs (Norcross, GA), and ±0.4% was acceptable. All adenosine derivatives were judged to be homogeneous using thin-layer chromatography (silica, 0.25 mm, glass backed, Alltech Assoc., Deerfield, IL) following final purification. 2-Chloroadenosine, NECA, CGS 21680, N6-cyclopentyladenosine, and 2-chloro-N6-cyclopentyladenosine were obtained from Research Biochemicals International (Natick, MA). IB-MECA and APEC were synthesized as reported.11,27 APNEA and iodo-APNEA were the gift of Prof. Ray A. Olsson (University of South Florida, Tampa, FL).
N6-(3-Iodobenzyl)-9-β-d-ribofuranosyladenine (10)
A mixture of 6-chloropurine riboside (purchased from Aldrich Chemical Co., 100 mg, 0.35 mmol), triethylamine (0.146 mL, 1.05 mmol), and 3-iodobenzylamine hydrochloride (103 mg, 0.38 mmol) in ethanol (2 mL) was heated for 18 h at 85 °C in a sealed bottle. After the reaction mixture was concentrated to dryness, the residue was purified by silica gel column chromatography (CHCl3–MeOH, 10:1) to give compound 10 (148 mg, 88%) as a colorless solid: 1H NMR (DMSO-d6) δ 3.54 (m, 1 H, H-5′a), 3.67 (m, 1 H, H-5′b), 3.96 (d, J = 3.3 Hz, 1 H, H-4′), 4.14 (m, 1 H, H-3′), 4.60 (m, 1 H, H-2′), 4.66 (br s, 2 H, CH2), 5.16 (d, J = 4.4 Hz, 1 H, exchangeable with D2O, 3′-OH), 5.34 (br s, 1 H, exchangeable with D2O, 5′-OH), 5.43 (d, J = 6.1 Hz, 1 H, exchangeable with D2O, 2′-OH), 5.89 (d, J = 6.0 Hz, 1 H, H-1′), 7.11 (pseudo t, J = 8.0 and 7.8 Hz, 1 H, H-5″), 7.36 (d, J = 7.6 Hz, 1 H, H-4″ or -6″), 7.58 (d, J = 7.8 Hz, 1 H, H-4″ or -6″), 7.72 (s, 1 H, H-2″), 8.21 (s, 1 H, H-2 or -8), 8.40 (s, 1 H, H-2 or -8), 8.48 (br s, 1 H, exchangeable with D2O, N6-H).
2-Chloro-N6-(3-iodobenzyl)-9-β-d-ribofuranosyladenine (11)
A mixture of compound 26 (760 mg, 0.916 mmol) and NH3/MeOH (15 mL) was stirred for 66.5 h at room temperature. After the reaction mixture was concentrated to dryness, the residue was purified by silica gel column chromatography (CHCl3–MeOH, 20:1) to yield compound 11 (445 mg, 94%) as a foam: 1H NMR (DMSO-d6) δ 3.55 (m, 1 H, H-5′a), 3.65 (m, 1 H, H-5′b), 3.94 (d, J = 3.6 Hz, 1 H, H-4′), 4.12 (m, 1 H, H-3′), 4.51 (q, J = 5.5 Hz, 1 H, H-2′), 4.60 (br d, J = 5.7 Hz, 2 H, CH2), 5.04 (pseudo t, J = 5.7 and 5.5 Hz, 1 H, exchangeable with D2O, 5′-OH), 5.19 (d, J = 4.9 Hz, 1 H, exchangeable with D2O, OH), 5.47 (d, J = 6.0 Hz, 1 H, exchangeable with D2O, OH), 5.83 (d, J = 5.5 Hz, 1 H, H-1′), 7.13 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-5″), 7.36 (d, J = 7.5 Hz, 1 H, H-4″ or -6″), 7.60 (d, J = 7.9 Hz, 1 H, H-4″ or -6″), 7.74 (s, 1 H, H-2″), 8.43 (s, 1 H, H-8), 8.94 (br t, J = 6.0 Hz, 1 H, exchangeable with D2O, NH).
2-Amino-N6-(3-iodobenzyl)-9-β-d-ribofuranosyladenine (12)
A mixture of 2-amino-6-chloropurine riboside (purchased from Aldrich Chemical Co., 80 mg, 0.26 mmol), 3-iodobenzylamine hydrochloride (71.5 mg, 0.265 mmol), and triethylamine (0.11 mL, 0.79 mmol) in ethanol (1.6 mL) was heated for 24 h at 80 °C. After the reaction mixture was concentrated to dryness the residue was purified by silica gel column chromatography (CHCl3–MeOH, 20:1 → 10:1) to yield compound 12 (99 mg, 75%) as a colorless solid: 1H NMR (DMSO-d6) δ 3.52 (m, 1 H, H-5′a), 3.63 (m, 1 H, H-5′b), 3.89 (m, 1 H, H-4′), 4.10 (m, 1 H, H-3′), 4.50 (m, 1 H, H-2′), 4.60 (br s, 2 H, CH2), 5.08 (d, J = 4.6 Hz, 1 H, exchangeable with D2O, 3′-OH), 5.35 (m, 2 H, exchangeable with D2O, 5′- and 2′-OH), 5.73 (d, J = 6.2 Hz, 1 H, H-1′), 5.83 (br s, 2 H, exchangeable with D2O, NH2), 7.11 (pseudo t, J = 7.9 and 7.8 Hz, 1 H, H-5″), 7.36 (d, J = 7.8 Hz, 1 H, H-4″ or -6″), 7.58 (d, J = 7.8 Hz, 1 H, H-4″ or -6″), 7.70 (s, 1 H, H-2″), 7.94 (s, 1 H, H-8).
2-Chloro-N6-(3-iodobenzyl)-9-[5-(methylcarbmoyl)-β-d-ribofuranosyl]adenine (13)
A mixture of compound 25 (27 mg, 0.036 mmol) and NH3/MeOH (15 mL) was stirred for 16 h at room temperature. After rotary evaporation of the volatiles, the residue was purified by silica gel column chromatography (CHCl3–MeOH, 20:1 → 10:1) to give compound 13 (13.4 mg, 68.7%) as a colorless solid: 1H NMR (DMSO-d6) δ 2.72 (d, J = 4.3 Hz, 3 H, NHCH3), 4.17 (br s, 1 H, H-3′), 4.32 (s, 1 H, H-4′), 5.55 (m, 1 H, H-2′), 4.61 (br d, J = 5.5 Hz, 2 Hz, CH2), 5.56 (d, J = 6.4 Hz, 1 H, exchangeable with D2O, 2′-OH), 5.72 (d, J = 4.3 Hz, 1 H, exchangeable with D2O, 3′-OH), 5.92 (d, J = 7.2 Hz, 1 H, H-1′), 7.13 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-5″), 7.36 (d, J = 7.5 Hz, 1 H, H-4″ or -6″), 7.61 (d, J = 7.8 Hz, 1 H, H-4″ or -6″), 7.75 (s, 1 H, H-2″), 8.27 (br d, J = 4.3 Hz, 1 H, exchangeable with D2O, NH), 8.49 (s, 1 H, H-8), 9.02 (br t, J = 6.2 and 5.7 Hz, 1 H, exchangeable with D2O, N6H).
N6-(3-Iodobenzyl)-2-(methylamino)-9-[5-(methylcarbamoyl)-β-d-ribofuranosyl]adenine (14)
A solution of 13 (10 mg, 0.018 mmol) in 2 N CH3NH2/THF (1.5 mL) was heated for 3 days at 90 °C. After the reaction mixture was concentrated to dryness, the residue was purified on a preparative TLC (CHCl3–MeOH, 10:1) to give 14 (7 mg, 70%) as a colorless solid: 1H NMR (DMSO-d6) δ 2.66 (d, J = 4.7 Hz, 3 H, -NHCH3), 2.76 (d, J = 4.3 Hz, 3 H, NHCH3), 4.18 (m, 1 H, H-3′), 4.25 (s, 1 H, H-4′), 4.57 (br s, 2 H, CH2), 4.69 (m, 1 H, H-2′), 5.47 (d, J = 6.5 Hz, 1 H, exchangeable with D2O, 2′-OH), 5.59 (d, J = 4.6 Hz, 1 H, exchangeable with D2O, 3′-OH), 5.84 (d, J = 7.2 Hz, 1 H, H-1′), 6.28 (br d, J = 4.4 Hz, exchangeable with D2O, NH), 7.11 (pseudo t, J = 8.0 and 7.8 Hz, 1 H, H-5″), 7.38 (d, J = 7.9 Hz, 1 H, H-4″ or -6″), 7.58 (d, J = 7.9 Hz, 1 H, H-4″ or -6″), 7.70 (m, 1 H, exchangeable with D2O, NH), 7.76 (s, 1 H, H-2″), 8.02 (s, 1 H, H-8), 8.05 (br s, 1 H, exchangeable with D2O, NH).
N6-(3-Iodobenzyl)-2-(methylthio)-9-[5-(methylcarbamoyl)-β-d-ribofuranosyl]adenine (15)
A solution of 13 (15 mg, 0.029 mmol) and sodium thiomethoxide (4.0 mg, 0.057 mmol) in anhydrous ethylene glycol dimethyl ether (2 mL) was heated at 80 °C, under nitrogen atmosphere, for 3 days. After cooling to room temperature, the reaction mixture was neutralized with glacial acetic acid and evaporated to dryness. The residue was purified on a preparative TLC (CH2Cl2–MeOH, 9.5:0.5) to give 15 (5.6 mg, 36.5%) as a yellow solid: 1H NMR (DMSO-d6) δ 2.43 (s, 3 H, SH3), 2.74 (d, J = 4.3Hz, 3 H, NHCH3), 3.48 (br s, 2 H, 2 × OH), 4.19 (m, 1H, H-3′), 4.31 (s, 1 H, H-4′), 4.62 (br s, 3 H, CH2 & H-2′), 5.87 (d, J = 7.9 Hz, 1 H, H-1′), 7.11 (pseudo t, J = 8.0 and 7.8 Hz, 1 H, H-5″), 7.90 (d, J = 7.9 Hz, 1 H, H-4″ or -6″), 7.58 (d, J = 7.9 Hz, 1 H, H-4″ or -6″), 7.76 (s, 1H, H-2″), 8.24 (br s, 1 H, NH), 8.35 (s, 1 H, H-8), 8.68 (br s, 1 H, NH).
Methyl 5-(tert-Butyldiphenylsilyl)-β-d-ribofuranoside (17)
To a mixture of methyl β-d-ribofuranoside (16, purchased from Sigma Chemical Co., 460 mg, 2.8 mmol) and anhydrous methylene chloride (20 mL) were added triethylamine (0.468 mL, 3.36 mmol), tert-butyldiphenylchlorosilane (0.9 mL, 3.46 mmol), and DMAP (13.7 mg, 0.112 mmol) successively at room temperature. The reaction mixture was stirred for 18 h at room temperature under nitrogen. The reaction mixture was washed with water (20 mL), saturated ammonium chloride (20 mL), and brine (20 mL), dried over anhydrous MgSO4, filtered, and concentrated to dryness. The residue was separated by silica gel column chromatography (CHCl3–MeOH, 50:1) to yield compound 17 [Rf = 0.48 (CHCl3–MeOH, 10:1), 618 mg, 54.8%] as a thick syrup: 1H NMR (DMSO-d6) δ 0.96 (s, 9 H, t-Bu), 3.22 (s, 3 H, OCH3), 3.61 (dd, J = 11.0 and 5.3 Hz, 1 H), 3.74 (d, J = 4.4 Hz, 1 H), 3.81 (dd, J = 11.0 and 2.7 Hz, 1 H), 3.90 (m, 1 H), 4.00 (m, 1 H), 4.68 (s, 1 H, H-1′), 4.84 (br s, 1 H, exchangeable with D2O, OH), 5.05 (br s, 1 H, exchangeable with D2O, OH), 7.45 and 7.67 (m, 10 H, Ph2).
Methyl 5-(tert-Butyldiphenylsilyl)-2,3-dibenzoyl-β-d-ribofuranoside (18)
To a solution of compound 17 (579 mg, 1.44 mmol) in methylene chloride–pyridine (4:1, 12.5 mL) was added dropwise benzoyl chloride (0.367 mL, 3.16 mmol) at 0 °C. The reaction mixture was stirred for 2.5 h at 0 °C and for 14.5 h at room temperature. Ice was added to quench the reaction, and the mixture was stirred for 1 h. Methylene chloride (100 mL) was added, and two phases were separated. Organic layer was washed with water, saturated ammonium chloride, and brine, dried over anhydrous MgSO4, filtered, and concentrated to dryness to give crude compound 18, which was then purified by silica gel column chromatography (Hx–EtOAc, 5:1 → 1:1) to yield compound 18 [Rf = 0.75 (CHCl3–MeOH, 10:1), 869 mg, 99%] as a thick syrup: 1H NMR (CDCl3) δ 1.05 (s, 9 H, t-Bu), 3.42 (s, 3 H, OCH3), 3.86 (dd, J = 11.1 and 4.8 Hz, 1 H, H-5a), 3.92 (dd, J = 11.1 and 4.7 Hz, 1 H, H-5b), 4.48 (dd, J = 10.6 and 4.6 Hz, 1 H), 5.15 (s, 1 H), 5.63 (d, J = 5.0 Hz, 1 H), 5.82 (pseudo t, J = 5.8 and 5.4 Hz, 1 H), 7.29–8.18 (m, 20 H, Ar).
Methyl 2,3-Dibenzoyl-β-d-ribofuranoside (19)
A solution of compound 18 (849 mg, 1.39 mmol) and 1.0 M tetrabutylammonium fluoride in THF (1.53 mL, 1.53 mmol) was stirred for 2 h at room temperature. After evaporation of the solvent, the residue was purified by silica gel column chromatography (Hx–EtOAc, 1:1) to yield compound 19 [Rf = 0.50 (Hx-EtOAc, 1:1), 461 mg, 89%] as a thick syrup: 1H NMR (DMSO-d6) δ 3.38 (s, 3 H, OCH3), 3.62 (m, 2 H, H-5), 4.37 (q, J = 5.2 Hz, 1 H, H-4), 5.02 (pseudo t, J = 6.0 and 5.3 Hz, 1 H, exchangeable with D2O, 5-OH), 5.18 (s, 1 H, H-1), 5.45 (m, 1 H, H-2), 5.53 (t, J = 5.2 Hz, 1 H, H-3), 7.42–7.88 (m, 10 H, Ar).
1-O-Methyl 2,3-Dibenzoyl-β-d-ribofuronic Acid (20)
A mixture of compound 19 (374.8 mg, 1.01 mmol), ruthenium-(IV) oxide (10 mg), and sodium periodate (1161 mg, 5.43 mmol) in CHCl3–CH3CN–H2O (2:2:3, 14 mL) was stirred vigorously for 2.5 h at room temperature. Chloroform (20 mL) was added, and semisolid was removed by filtration. The two layers of filtrate were separated, and aqueous layer was extracted with chloroform (2 × 40 mL). Combined organic layer and extracts were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated to dryness. After drying in vacuo overnight, 1-O-methyl-2,3-dibenzoyl-β-d-ribofuranuronic acid (20, 340 mg, 89.5%) was obtained as a thick syrup: 1H NMR (CDCl3) δ 3.56 (s, 3 H, OCH3), 4.90 (d, J = 6.1 Hz, 1 H, H-4), 5.25 (s, 1 H, H-1), 5.66 (d, J = 5.0 Hz, 1 H, H-2), 6.00 (pseudo t, J = 5.7 and 5.4 Hz, 1 H, H-3), 7.32–7.99 (m, 10 H, Ar).
Methyl 1-O-Methyl-2,3-dibenzoyl-β-d-ribofuranuronate (21)
N-Ethyl-N′-(diaminopropyl)carbodiimide (EDAC, 198 mg, 1.04 mmol) was added to a solution of acid (20, 0.16 g, 0.414 mmol) in MeOH (3 mL), and the reaction mixture was stirred for 3 h at room temperature. After the solvent was removed by rotary evaporation, the residue was dissolved in chloroform (50 mL), washed with water (30 mL) and brine (30 mL), dried over anhydrous MgSO4, filtered, and concentrated to dryness. The residue was purified on a preparative TLC (Hx–EtOAc, 1:1) to yield methyl 1-O-methyl-2,3-dibenzoyl-β-d-ribofuronate [21, Rf = 0.77 (Hx–EtOAc, 1:1), 120 mg, 72.3%] as a colorless solid. 1H NMR (CDCl3) δ 3.52 (s, 3 H, 1-OCH3), 3.82 (2, 3 H, 5-OCH3), 4.84 (d, J = 6.2 Hz, 1 H, H-4), 5.21 (s, 1 H, H-1), 5.62 (d, J = 4.8 Hz, 1 H, H-2), 6.01 (pseudo t, J = 5.7 and 5.5 Hz, 1 H, H-3), 7.32–7.99 (m, 10 H, Ar).
N,1-O-Dimethyl-2,3-dibenzoyl-β-d-ribofuranuronamide (22)
A mixture of methyl ester 21 (35 mg, 0.087 mmol) and 2.0 M methylamine in THF (3 mL) was heated for 15 h at 50 °C in a sealed tube. The volatiles were removed by evaporation, and the residue was reacted with benzoyl chloride (0.15 mL, 1.29 mmol) in methylene chloride–pyridine (2:1, 6 mL) for 3 h at room temperature. After workup as procedure for compound 18, the residue was separated by preparative TLC (Hx–EtOAc, 1:1) to yield compound 22 [Rf = 0.28 (Hx–EtOAc, 1:1) or 0.77 (CHCl3–MeOH, 10:1), 25 mg, 72%] as a syrup: 1H NMR (CDCl3) δ 2.92 (d, J = 5.0 Hz, 3 H, NHCH3), 3.58 (s, 3 H, 1-OCH3), 4.82 (d, J = 5.5 Hz, 1 H, H-4), 5.24 (s, 1 H, H-1), 5.60 (m, 1 H, H-2), 5.87 (t, J = 5.1 Hz, 1 H, H-3), 6.68 (br m, 1 H, NH), 7.33–7.99 (m, 10 H, Ar).
N-Methyl-1-O-acetyl-2,3-dibenzoyl-α-d-ribofuronamide (23a) and N-methyl-1-O-acetyl-2,3-dibenzoyl-β-d-ribofuranuronamide (23b)
To a solution of 22 (1.533 g, 3.84 mmol) and acetic anhydride (3.8 mL, 40.3 mmol) in glacial acetic acid (19 mL) was added dropwise concentrated H2SO4 (1.125 mL, 21.1 mmol), and the reaction mixture was stirred for 15 h at room temperature. After water (30 mL) was added slowly, the mixture was extracted with methylene chloride (150 mL × 3) and the organic layer was washed with saturated NaHCO3 and brine, dried over anhydrous MgSO4, filtered, and connectrated to dryness. The residue was purified by silica gel column chromatography (CHCl3–MeOH, 20:1) to give a mixture of 23a and 23b [Rf = 0.71 and 0.76 (CHCl3–MeOH, 20:1), respectively, 0.55 g, 33.5%] as a foam. Analytical samples were separated by preparative TLC (CHCl3–MeOH, 20:1): 1H NMR (CDCl3) (compound 23a) δ 2.10 (s, 3 H, OAc), 2.91 (d, J = 4.9 Hz, 3 H, -NHCH3), 4.96 (s, 1 H, H-4), 5.45 (pseudo t, J = 5.5 and 5.0 Hz, 1 H, H-2), 6.08 (d, J = 5.9 Hz, 1 H, H-3), 6.71 (d, J = 4.7 Hz, 1 H, H-1), 6.72 (br s, 1 H, NH), 7.28 (pseudo t, J = 7.8 and 7.7 Hz, 2 H, Ar), 7.48 (q, J = 7.8 Hz, 3 H, Ar), 7.62 (pseuso t, J = 7.7 and 6.9 Hz, 1 H, Ar), 7.79 (d, J = 7.4 Hz, 2 H, Ar), 8.13 (d, J = 7.8 Hz, 2 H, Ar); (compound 23b) δ 2.17 (s, 3 H, OAc), 2.90 (d, J = 4.9 Hz, 3 H, NHCH3), 4.89 (d, J = 6.2 Hz, 1 H, H-4), 5.73 (d J = 4.9 Hz, 1 H, H-2), 5.96 (t, J = 5.9 Hz, 1 H, H-3), 6.43 (s, 1 H, H-1), 6.50 (br s, 1 H, NH), 7.37 (pseudo t, J = 7.8 and 7.6 Hz, 4 H, Ar), 7.48–7.58 (m, 2 H, Ar), 7.93 (d, J = 8.1 Hz, 2 H, Ar), 7.98 (d, J = 7.3 Hz, 2 H, Ar).
2-Chloro-N6-(3-iodobenzyl)adenine (24a)
A solution of 2,6-dichloropurine (purchased from Aldrich Chemical Co., 1 g, 5.3 mmol), 3-iodobenzylamine hydrochloride (1.7 g, 5.8 mmol), and triethylamine (2.2 mL, 15.35 mmol) in ethanol (10 mL) was stirred for 5 days at room temperature. The colorless solid formed was collected by suction, washed with small amount of cold ethanol, and dried to give compound 24a (1.16 g, 60%): mass (EI) 385 (M+); 1H NMR (DMSO-d6) δ 4.59 (br s, 2 H, CH2), 7.13 (pseudo t, J = 8.2 and 7.5 Hz, 1 H, Bn), 7.36 (d, J = 7.5 Hz, 1 H, Bn), 7.61 (d, J = 7.5 Hz, 1 H, Bn), 7.74 (s, 1 H, Bn), 8.14 (s, 1 H, H-8), 8.76 (br s, 1 H, exchangeable with D2O, NH), 13.14 (br s, 1 H, exchangeable with D2O, NH); UV (MeOH) λmax, 281.7, 257.5, 232.5 nm.
2-Chloro-N6-(3-iodobenzyl)-9-[5-(methylcarbamoyl)-2,3-di-O-benzoyl-β-d-ribofuranosyl]adenine (25)
A mixture of 2-chloro-N6-(3-iodobenzyl)adenine (24a, 165 mg, 0.43 mmol), ammonium sulfate (catalytic amount), and HMDS (15 mL) was refluxed for 4 h under nitrogen to provide the silylated derivative 24b. The clear solution was concentrated to dryness in vacuo with exclusion of moisture, and the residue was dissolved in dry dichloroethane (6 mL). A solution of 23 (141 mg, 0.33 mmol) in dry dichloroethane (6 mL) and TMSOTf (83 µL, 0.43 mmol) were added, and the reaction mixture was stirred for 0.5 h at room temperature and refluxed for 62 h under nitrogen. Saturated NaHCO3 (10 mL) was added, and the mixture was stirred for 15 min. Two layers were separated, and the aqueous layer was extracted with methylene chloride (50 mL × 3), washed with brine, dried over anhydrous MgSO4, filtered, and concentrated to dryness. The residue was purified on a preparative TLC (CHCl3–MeOH, 20:1) to give 25 [Rf = 0.58 (CHCl3–MeOH, 20:1), 83 mg, 33 %] as a foam: MS (CI NH3) 753 (M++l); 1H NMR (CDCl3) δ 3.10 (d, J = 4.6 Hz, 3 H, NHCH3), 4.79 (br s, 2 H, CH2), 4.97 (s, 1 H, H-4′), 6.08 (m, 1 H, H-3′), 6.15–6.25 (m, 3 H, H-2′, 1′, NH), 7.06–8.06 (m, 15 H, Ar), 8.52 (br s, 1 H, NH).
2-Chloro-N6-(3-iodobenzyl)-9-(2,3,5-tri-O-benzoyl-β-d-ribofuranosyl)adenine (26)
A mixture of 2-chloro-N6-(3-iodobenzyl)adenine (24a, 0.84 g, 2.18 mmol), ammonium sulfate (catalytic amount), and HMDS (20 mL) was refluxed for 5 h under nitrogen to provide the silylated derivative 24b. The clear solution was concentrated to dryness in vacuo with exclusion of moisture, and the residue was dissolved in dry dichloroethane (6 mL). A solution of acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranoside (purchased from Janssen Chimica Chemical Co., 1 g, 1.98 mmol) in dry dichloroethane (12 mL) and TMSOTf (0.42 mL, 2.18 mmol) were added, and the reaction mixture was stirred for 20 min at room temperature and refluxed for 14 h under nitrogen. After similar workup for compound 25, the residue was purified by silica gel column chromatography (Hx–EtOAc, 2:1) to give 26 [Rf = 0.11 (Hx–EtOAc, 3:1), 1.495 g, 91%] as a colorless foam: 1H NMR (CDCl3) δ 4.69–4.92 (m, 5 H, CH2, H-4′, H-5′), 6.15 (m, 3 H, H-2′, H-3′, NH), 6.45 (d, J = 4.3 Hz, 1 H, H-1′), 7.07 (pseudo t, 1 H, Bn), 7.31–8.10 (m, 20 H, Ar).
Methods for Receptor Binding and Adenylate Cyclase Measurement
Procedures for preparation of rat brain membranes and CHO cell membranes were as reported.10,11,14 For binding experiments, membrane homogenates were frozen and stored at −20 °C for ≤2 months. Adenosine deaminase (ADA) was from Boehringer Mannheim (Indianapolis, IN). [3H]R-PIA was from Amersham (Arlington Heights, IL), and [3H]CGS 21680 was from DuPont NEN (Boston, MA). [125I]-AB-MECA was prepared as described by Olah et al.14
Binding of [125I]AB-MECA to CHO cells stably transfected with the A3 receptor clone was performed essentially as described.11,14 Assays were performed in 50 mM Tris/10 mM MgCl2/1 mM EDTA buffer (adjusted to pH 8.26 at 5 °C) in glass tubes and contained 100 µL of the membrane suspension, 50 µL of [125I]AB-MECA (final concentration 0.3 nM), and 50 µL of inhibitor. Inhibitors were routinely dissolved in DMSO and were then diluted with buffer; final DMSO concentrations never exceeded 1%; this concentration did not influence [125I]-AB-MECA binding. Incubations were carried out in duplicate for 1 h at 37 °C, and were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). Tubes were washed three times with 3 mL of buffer. Radioactivity was determined in a Beckman gamma 5500B γ-counter. Nonspecific binding was determined in the presence of 200 µM NECA. Ki values were calculated according to Cheng–Prusoff,21 assuming a Kd for [125I]AB-MECA of 1.48 nM.6
Binding of [3H]PIA to A1 receptors from rat cortical membranes and of [3H]CGS 21680 to A2 receptors from rat striatal membranes was performed as described previously.8,11 Adenosine deaminase (2 units/mL) was present during the preparation of brain membranes. Additional deaminase was not added during incubation with the radioligand.
Competition for binding of [3H]NBTI was carried out by a modification of the procedure of Marangos et al.24 Rat striatal membranes, prepared as above, were subjected to incubation for 30 min at 23 °C with 0.3 nM [3H]NBTI and varying concentrations of the nucleoside derivative in Tris buffer, pH 7.4, in a total of 0.5 mL. For nonspecific binding 5 µM S-(p-nitrobenzyl)-6-thioguanosine (Sigma, St. Louis, MO) was added, and specific binding was 95% of total. A Kd value of 0.15 nM was used in the calculation of Ki values.24 Specific binding was 95% of total.
Adenylate cyclase was assayed in membranes from CHO cells stably expressing the rat A3 receptor, prepared as above, using a previously reported method.10 The method involved addition of [α-32P]ATP to membranes in the presence of forskolin to stimulate adenylate cyclase and papaverine as a phosphodiesterase inhibitor. The reaction was terminated by addition of a stop solution containing 20 000 cpm/mL [3H]cyclic AMP. The total radiolabeled cyclic AMP was isolated on columns of Dowex 50 ion-exchange resin and alumina. Maximal inhibition of adenylate cyclase activity corresponded to ~40% of total activity under conditions of stimulation (typically by 6–8-fold) in the presence of 1 µM forskolin. IC50 values were calculated using InPlot (Graphpad, San Diego, CA).
Table 3.
Inhibition by Various N 6-Benzyladenosine Derivatives of the Specific Binding of [3H]-S-(4-Nitrobenzyl)-6-thioinosiante at Adenosine Uptake Sites in Rat Brain Membranes and the Selectivity Ratio for Affinity at Cloned Rat A3 Receptors (Ki values from Table 1)
| compd | Ki (NBTI)a |
Ki (NBTI)/Ki ([125I]AB-MECA |
|---|---|---|
| N 6-benzyladenosine | 203 ± 93 | 1.69 |
| 1 | 28200 ± 10700 | 22000 |
| 13 | 15200 ± 5200 | 46000 |
| 15 | 49500 ± 633 | 22000 |
Expressed in nanomolar as Ki ± SEM for three or four determinations, each done in triplicate. Rat striatal membranes were incubated for 30 min at 23 °C with 0.3 nM [3H]NBTI and varying concentrations of the nucleoside derivative in Tris buffer, pH 7.4 in a total of 0.5 mL. Nonspecific binding was determined in the presence of 5 µM S-(p-nitrobenzyl)-6-thioguanosine.
Acknowledgment
We thank Dr. Timothy M. Palmer (Duke University) for cyclase measurements.
Abbreviations
- AB-MECA
N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide
- APNEA
N6-[2-(4-aminophenyl)ethyl]adenosine
- CGS 21680
2-[4-[(2-carboxyethyl)-phenyl]ethyl-amino]-5′-N-(ethylcarbamoyl)adenosine
- CHO
Chinese hamster ovary
- DMAP
4-(N,N-dimethylamino)pyridine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EDAC
1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
- HMDS
1,1,1,3,3,3-hexamethyldisilazane
- IB-MECA
N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- NBTI
S-(4-nitrobenzyl)-6-thioinosine
- NECA
5′-N-(ethylcarbamoyl)adenosine;
- PIA
(R)-N6-(phenylisopropyl)adenosine
- TBAF
tetrabutylammonium fluoride
- TBDPSiCl
tert-butyldiphenylsilyl chloride
- THF
tetrahydrofuran
- TMSOTf
trimethylsilyl trifuoromethanesulfonate
- Tris
tris(hydroxymethyl)-aminomethane
References
- 1.Zhou QY, Li CY, Olah ME, Johnson RA, Stiles GL, Civelli O. Molecular cloning and characterization of an adenosine receptor - the A3 adenosine receptor. Proc. Natl. Acad. Sci. USA. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bai TR, Weir T, Walker BAM, Salvatore CA, Johnson RG, Jacobson MA. Comparison and localization of adenosine A3 receptor expression in normal and asthmatic lung. Drug. Dev. Res. 1994;31:244. Abstract 1011. [Google Scholar]
- 3.Meyerhof W, Müller-Brechlin R, Richter D. Molecular cloning of a novel putative G-protein coupled receptor expressed during rat spermiogenesis. FEBS Lett. 1991;284:155–160. doi: 10.1016/0014-5793(91)80674-r. [DOI] [PubMed] [Google Scholar]
- 4.Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3AR is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem. 1993;268 168871–6890. [PubMed] [Google Scholar]
- 5.Ali H, Cunha-Melo JR, Saul WF, Beavan MF. Activation of phospholipase C via adenosine receptors provides synergistic signals for secretion in antigen stimulated RBL-2H3 cells. J. Biol. Chem. 1990;265:745–753. [PubMed] [Google Scholar]
- 6.Fozard JR, Carruthers AM. Adenosine A3 receptors mediate hypotension in the angiotensin II-supported circulation of the pithed rat. Br. J. Pharmacol. 1993;109:3–5. doi: 10.1111/j.1476-5381.1993.tb13522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson KA, Nikodijević O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, Daly JW. A role for central A3-adenosine receptors: Mediation of behavioral depressant responses. FEBS Lett. 1993;336:57–60. doi: 10.1016/0014-5793(93)81608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Lubitz DKJE, Lin RC-S, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur. J. Pharmacol. doi: 10.1016/0014-2999(94)90523-1. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu GS, Richards SC, Olsson RA, Mullane K, Walsh RS, Downey JM. Evidence that the adenosine A3 receptor can mediate preconditioning’s protection in isolated rabbit hearts. Cardiovasc. Res. 1994;28:1057–1061. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- 10.(a) van Galen PJM, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol. Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]; (b) van Bergen A, van Galen PJM, Stiles GL, Jacobson KA. A3 receptors: Structure activity relationships and molecular modeling. ACS 206th National Meeting; Aug. 1993; Chicago, IL. Abstract MEDI217. [Google Scholar]
- 11.Gallo-Rodriguez C, Ji X-D, Melman N, Siegman BD, Sanders LH, Orlina J, Pu Q-L, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure-activity relationships at A3-adenosine receptors. J. Med. Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linden J, Taylor HE, Robeva AS, Tucker AL, Stehle JH, Rivkees SA, Fink JS, Reppert SM. Molecular cloning and functional expression of a sheep A3 adenosine receptor with widespread tissue distribution. Mol. Pharmacol. 1993;44:524–532. [PubMed] [Google Scholar]
- 13.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. USA. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 15.Schwabe U, Trost T. Characterization of adenosine receptors in rat brain by (−) [3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1980;313:179–187. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 16.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J. Pharmacol. Exp. Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 17.Baker R, Fletcher HG., Jr 2,3,5-Tri-O-benzyl-d-ribosyl and -L-arabinosyl bromides. J. Org. Chem. 1961;26:4605–4609. [Google Scholar]
- 18.Chaudhary SK, Hernandez O. 4-Dimethylaminopyridine: an efficient and selective catalyst for the silylation of alcohols. Tetrahedron Lett. 1979;20(2):99–102. [Google Scholar]
- 19.Singh AK, Varma RS. Ruthenium tetroxide: a mild reagent for the oxidation of 2′,3′-O-isopropylidene purine nucleosides. Tetrahedron Lett. 1992;33(17):2307–2310. [Google Scholar]
- 20.von Lubitz D, Jacobson KA. Neurodegenerative disorders and treatment with selective agents acting at A1 and A3 receptors: a problem or a bright future? Drug Devel. Res. 1994;31:332. Abstract 1224. [Google Scholar]
- 21.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 22.Hide I, Padgett WL, Jacobson KA, Daly JW. A2a-Adenosine receptors from rat striatum and rat pheochromocytoma PC12 cells: Characterization with radioligand binding and by activation of adenylate cyclase. Mol. Pharmacol. 1992;41:352–359. [PMC free article] [PubMed] [Google Scholar]
- 23.Stiles GL, Daly DT, Olsson RA. Characterization of the A1 adenosine receptor - adenylate cyclase system of cerebral cortex using an agonist photoaffinity probe. J. Neurochem. 1986;47:1020–1025. doi: 10.1111/j.1471-4159.1986.tb00715.x. [DOI] [PubMed] [Google Scholar]
- 24.Marangos PJ, Patel J, Clark-Rosenberg R, Martino AM. Nitrobenzylthioinosine binding as a probe for the study of adenosine uptake sites in brain. J. Neurochem. 1982;39:184–191. doi: 10.1111/j.1471-4159.1982.tb04717.x. [DOI] [PubMed] [Google Scholar]
- 25.Ji X-D, von Lubitz D, Olah ME, Stiles GL, Jacobson KA. Species differences in ligand affinity at central A3-adenosine receptors. Drug Devel. Res. 1994;33:51–59. doi: 10.1002/ddr.430330109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brackett LE, Daly JW. Functional characterization of the A2b adenosine receptor in NIH 3T3 fibroblasts. Biochem. Pharmacol. 1994;47:801–814. doi: 10.1016/0006-2952(94)90480-4. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson KA, Barrington WW, Pannell LK, Jarvis MF, Ji X-D, Williams M, Hutchison AJ, Stiles GL. Agonist-derived molecular probes for A2-adenosine receptors. J. Mol. Recognition. 1989;2:170–178. doi: 10.1002/jmr.300020406. [DOI] [PMC free article] [PubMed] [Google Scholar]