

Abstract
Innate lymphoid cells (ILC)-22 protect the intestinal mucosa from infections by secreting interleukin-22 (IL-22). They include NKp46+ and Lymphoid Tissues inducer (LTi)-like subsets. Both express the aryl-hydrocarbon receptor (AHR), a sensor for environmental, dietary and endogenous aromatic compounds. We show that AHR-/- mice have a marked ILC22 deficit, resulting in diminished IL-22 secretion and inadequate protection against intestinal bacterial infections. AHR-/- mice also lack post-natally-imprinted cryptopatches (CP) and isolated lymphoid follicles (ILF), but not embryonically-imprinted Peyer's Patches (PP). AHR induces Notch, which is required for NKp46+ILC, while LTi-like ILC, CP and ILF are partially dependent on Notch signaling. These results establish that AHR is essential for ILC22 and post-natal intestinal lymphoid tissues and reveal heterogeneity of ILC22 subsets in their developmental requirements and their impact on the generation of intestinal lymphoid tissues.
The mucosa of the gastrointestinal tract is a critical interface between the host, its food and gut-associated microbial ecosystem. The immune system maintains the integrity of the gut mucosa through multiple mechanisms that concurrently provide protection from potentially harmful microbes and induce tolerance to nutrients, commensals and self-antigens1-4. A key protective factor is interleukin 22 (IL-22), which acts on mucosal epithelial cells, inducing their survival, proliferation and secretion of antimicrobial peptides5, 6. The protective role of IL-22 is evident in genetically targeted IL-22-deficient mice, which are highly susceptible to gastrointestinal infection by the pathogen C. rodentium7.
Initial studies demonstrated that IL-22 is a product of CD4 T helper 17 (TH17) cells, well known for their capacity to secrete IL-17, which is critical for defense against extracellular pathogens and fungi8. However, recent studies showed that a population of non-T and non-B lymphocytes specializes in the secretion of large amounts of IL-229. These innate lymphoid cells, often called ILC2210, have been found in all lymphoid tissues located in the lamina propria (LP) of the intestinal mucosa, including cryptopatches (CP), isolated lymphoid follicles (ILF) and lymphoid follicles organized into Peyer's patches (PP). IL-22 is secreted in response to the inflammatory cytokine IL-23, which is produced by intestinal dendritic cells (DC) and macrophages when they encounter microbial products that cross the epithelial barrier.
ILC22 include several cell subsets with distinct phenotypic markers, reflecting functional and/or developmental heterogeneity10. In the mouse, one subset expresses the NK cell marker NKp46, but very little or none of the prototypic NK cell marker NK1.1. We will refer to this subset as NKp46+ILC. Another subset resembles lymphoid tissue inducer (LTi) cells, which promote prenatal organogenesis of lymphoid organs11. We will refer to these cells as LTi-like ILC, which include CD4+ and CD4- subsets9, 12, 13. The LP of the large intestine contains yet another population of ILC, distinguished by expression of stem cell antigen-1 (SCA-1), high levels of Thy1 and production of IL-1714. The development of ILC subsets requires the transcription factor RORγt10, 14, 15 as well as cytokine signals and transcription factors that have been implicated in the development of other lymphocyte populations, such as IL-7, the common gamma chain of the cytokine receptors (γc) and Id210, 16, 17.
Recent evidence indicates that environmental factors play a role in the development of IL-22 producing ILC. NKp46+ILC are not found in some germ-free mice, which implies that intestinal microbiota contribute to their generation16, 18. Furthermore, human NKp46+ and mouse LTi-like ILC express high levels of the transcription factor aryl hydrocarbon receptor (AHR)13, 19. AHR is a cytosolic sensor of small polycyclic aromatic compounds, known as xenobiotics, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)20-24. Situated in the cytoplasm in an inactive complex, AHR translocates into the nucleus upon ligand binding and recognizes promoters containing specific enhancer sequences termed xenobiotic responsive elements (XRE), inducing de novo transcription of multiple target genes. These genes comprise xenobiotic metabolizing enzymes including the cytochrome P450 superfamily members Cyp1a1, Cyp1a2, and Cyp1b1.
In addition to xenobiotics, AHR binds endogenous ligands, including metabolites of tryptophan, metabolites of arachidonic acid, as well as dietary compounds, such as natural flavonoids and indole-3-carbinol derivatives20-26. The marked developmental defects in AHR-/- mice imply that AHR is an important link between endogenous and/or exogenous aromatic compounds and normal development. Specifically, within the immune system, recent studies have shown that AHR participates in the differentiation of regulatory T cells, including FoxP3+ Treg27-30 and Tr131. In addition, AHR enhances IL-17A and IL-17F production by TH1727, 32, 33, contributes to the development of TH1734 and intraepithelial intestinal γδT cells25 and is required for the TH17 ability to produce IL-2227, 32.
The observation that human NKp46+ and mouse LTi-like ILC express AHR13, 19, prompted us to investigate the impact of AHR deficiency on the function and differentiation of ILC22. Here we show that AHR-/- mice have a marked defect of ILC22, resulting in diminished secretion of IL-22 and inadequate protection against bacterial intestinal infections. Remarkably, AHR-/- mice also lack CP and ILF, which develop post-natally, but not PP, which are embryonically-imprinted, demonstrating a role of ILC22 in the generation of diffuse intestinal lymphoid tissues. Mechanistically, we show that AHR acts in part by inducing Notch, which is required for NKp46+ ILC. LTi-like ILC, CP and ILF, which are severely affected by AHR loss, are less impaired in the absence of Notch signaling, possibly suggesting compensation by alternative signaling pathways. These results establish AHR as a critical transcription factor for the development of ILC and post-natal intestinal lymphoid tissues. Moreover, this data reveals heterogeneity of ILC subsets in their developmental requirements and their impact on the generation of intestinal lymphoid tissues.
Results
Innate IL-22-production in the intestinal mucosa requires AHR
We initially investigated the impact of AHR on innate, cytokine-driven IL-22 production. We isolated cells from the small intestine LP (siLP), the PP and the mesenteric lymph nodes (MLN) of AHR-/- and wild type (WT) mice, stimulated them in vitro with IL-23 and measured IL-22 released in the culture supernatant. Cells from siLP and PP of WT mice produced large amounts of IL-22; in contrast, cells from the siLP of AHR-/- mice released virtually no IL-22; moreover, cells from the PP of AHR-/- mice released approximately half as much IL-22 than did cells from WT mice (Fig. 1a). Cells isolated from MLN produced minimal amounts of IL-22, whether they were isolated from AHR-/- or WT mice. Thus, AHR is essential for IL-22-production in response to IL-23 in the intestinal LP and, in part, in PP.
Figure 1.
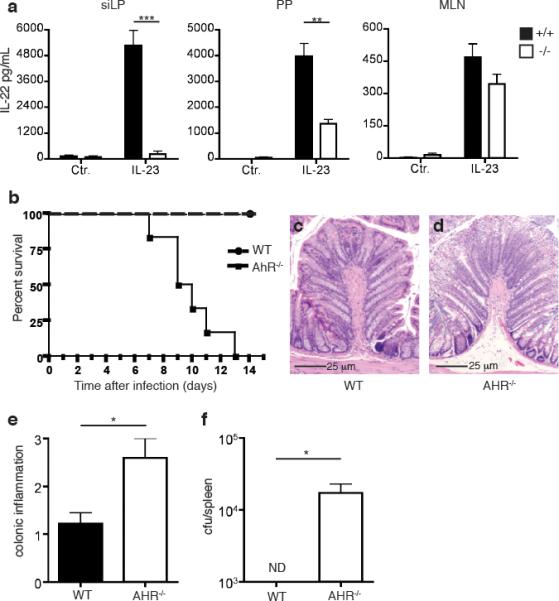
AHR is essential for IL-22-production in the intestinal LP and resistance to C. rodentium infection. (a) Cells were isolated from siLP, PP, and MLN of AHR-/- and WT mice. Identical numbers of cells were stimulated with IL-23. IL-22 released in culture supernatant was measured by ELISA. One experiment representative of two is shown. (b) Survival rates after C. rodentium infection. AHR-/- mice and WT mice were orally inoculated with C. rodentium. Data are from 2 independent experiments (n=6). (c,d) Histological analysis from H&E staining of representative colons from WT (c) and AHR-/- (d) mice 6 days post inoculation reveals increased mucosal hyperplasia and epithelial erosion in AHR-/- colons. Scale bars 25μm. (e) Histological scores for colons from WT and AHR-/- mice from (c and d). (f) C. rodentium titers in the spleen 6 days after inoculation.
Consistent with the known role for IL-22 in the early host response to gastro-intestinal bacterial infections, AHR-/- mice infected with C. rodentium rapidly succumbed to infection, with 100% mortality within 2 weeks (Fig. 1b). Histological analysis of the colon of infected AHR-/- mice on day 6 post-infection showed increased inflammatory cellular infiltration as well as mucosal hyperplasia in comparison to colon from WT littermates (Fig. 1c-e). Moreover, AHR-/- mice failed to contain C. rodentium infection in the gut, verified by bacterial translocation and replication in the spleens of AHR-/- mice that was not evident in WT mice (Fig. 1f). The data demonstrates that AHR deficiency causes a defect in IL-23-driven IL-22 production in the gut that facilitates infection and translocation of pathogenic bacteria. It is noteworthy that the relatively rapid death of AHR-/- mice following bacterial infection is similar to that observed in IL-22-/- mice and in mice treated with neutralizing anti-IL-22 antibodies within the first week of infection7. In contrast, RAG-deficient mice that lack adaptive sources of IL-22 survive longer following infection. Thus, the susceptibility of AHR-/- mice to C. rodentium infection observed here most likely reflects a prominent deficit of ILC22 response, whereas a defective TH17 response may play only a secondary role.
AHR-deficient mice lack intestinal NKp46+ILC
Since IL-22 production in response to IL-23 is a feature of different ILC subsets, we investigated the impact of AHR deficiency on individual ILC subsets. Although human NKp46+ILC express high levels of AHR19, it was not known whether this is also the case for their murine counterpart. We first confirmed that NKp46+ILC can be identified as CD3-NKp46+NK1.1-/loRORγt+ cells, whereas conventional NK cells correspond to CD3-NKp46+NK1.1+RORγt- cells (Supplementary Fig. 1)16, 18, 19, 35. Interestingly, we found that NKp46+ILC are particularly abundant in locations close to the intestinal lumen, such as mucosal LP and PP, but undetectable in the MLN. Then we measured AHR expression by RT-PCR analysis of NKp46+ILC and conventional NK cells sorted from intestinal LP and demonstrated higher expression of AHR in NKp46+ILC compared with conventional NK cells (Supplementary Fig. 2).
We next tested the impact of AHR deficiency on NKp46+ILC. We isolated cells from siLP, PP and MLN of WT and AHR-/- mice, stimulated them with IL-23 and measured the intracellular content of IL-22 within the CD3-NKp46+ population. IL-22 was evident in CD3-NKp46+ cells from the siLP and PP of WT mice; in the siLP and PP of AHR-/- mice, however, the CD3-NKp46+ population contained markedly fewer or no IL-22-producing cells (Fig. 2a, b). Since we did not detect any IL-22 in CD3+ T cells in response to IL-23 (data not shown), the IL-23 responsive AHR-dependent production of IL-22 is mostly restricted to intestinal ILC22.
Figure 2.

IL-22 producing-NK like cells are markedly reduced in AHR-/- mice. (a, b) siLP (a) and PP (b) cells were isolated from AHR-/- or WT mice and stimulated with IL-23. Intracellular content of IL-22 was determined in CD3-CD19-NKp46+ cells. Numbers above bracketed lines indicate percent of IL-22+ cells (mean ± s.d.) Data are representative of 2 independent experiments (n=3). (c) Cells were isolated from siLP and PP of WT and AHR-/- mice, stained for CD3, CD19, NKp46 and NK1.1 and analyzed by flow cytometry. Numbers above outlined areas indicate percent cells gated on CD3-CD19- cells. (d) Quantification of NKp46+NK1.1-, NKp46+NK1.1lo, and NKp46+NK1.1+ subsets in siLP and PP of AHR-/- and WT mice (y-axis = percent cells). Data points represent individual mice and indicate frequency of cells gated on CD3-CD19- cells. (e,f) Representative flow cytometry analysis of cells isolated from the siLP (e) and PP (f) of AHR-/- and WT animals. Cells are gated on CD3-CD19-CD4- and numbers in the quadrants represent percent cells in each.
To determine whether AHR deficiency affects the function or the differentiation of NKp46+ILC, we assessed the frequency of NKp46+ILC versus conventional NK cells within the CD3-NKp46+ population from AHR-/- and WT mice. Surprisingly, we found an almost complete absence of NKp46+ILC in the siLP and a marked reduction in the PP of AHR-/- mice (Fig. 2c, d). In contrast, conventional NK cells were equally represented in AHR-/- and WT littermates. NKp46+ILC have also been distinguished from conventional NK cells based on preferential expression of the α chain of IL-7 receptor (CD127)10. Using this phenotypic criterion, we confirmed a dramatic reduction in CD3-NKp46+CD127+ cells in AHR-/- mice as compared to WT controls (Fig. 2e, f). Thus, lack of AHR results in absence of NKp46+ILC in the siLP and marked reduction in the PP.
AHR deficiency impairs LTi-like ILC, CP and ILF in the intestinal mucosa
Since AHR has been shown to be highly expressed in mouse LTi-like ILC13, we next asked whether the defect of IL-22-producing cells in AHR-/- mice also involves LTi-like ILC. Flow cytometric analysis of cells isolated from the small intestine LP revealed markedly fewer CD4+LTi-like ILC (defined as CD3-NKp46-CD127+CD4+) in AHR-/- mice than in WT controls (Fig. 3a). Intracellular staining corroborated that the number of CD4+LTi-like ILC producing IL-22 was significantly decreased in the siLP of AHR-/- mice (Fig. 3b). In contrast, there were no differences between the CD4+LTi-like populations from AHR-/- and WT control mice in the PP. AHR deficiency also impacted the presence of CD127+CD4-RORγt+LTi-like ILCs in the siLP as their frequencies were dramatically reduced in AHR-/- mice (Supplementary Fig. 3) We conclude that AHR is required for the presence of both NKp46+ and LTi-like ILC in the small intestine LP.
Figure 3.
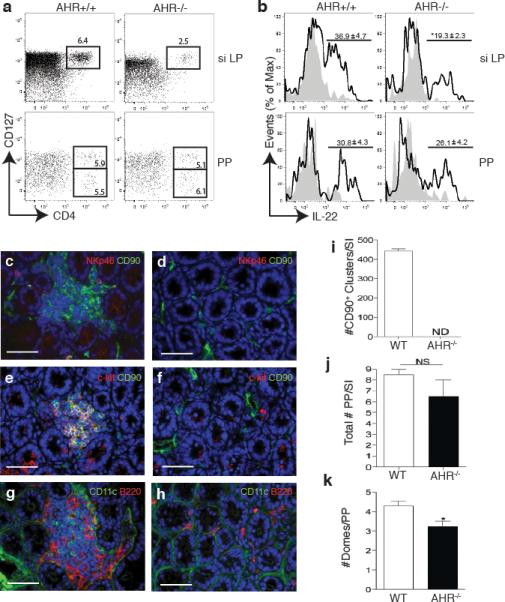
AHR-/- mice lack CD4+LTi-like ILC as well as CP and ILF in the small intestine. (a) Flow cytometry analysis of cells isolated from the PP and siLP of AhR-/- or WT mice. Plots show cells gated on CD3-CD19-NKp46- and numbers above outlined areas indicate percent cells in each. Data are representative of 2 independent experiments (n=4). (b) Intracellular IL-22 expression in IL-23 stimulated LTi-like ILC from siLP or PP (gated on CD3-CD19-NKp46-CD4+). Numbers above bracketed lines represent percent IL-22+ cells (mean ± s.e.m.). Data are representative of 2 independent experiments (n=4). (c,d,e,f,g,h) Immunohistochemistry to identify cryptopatch CD90+c-kit+ clusters as well as CD11c+ and B220+ cells associated with CP and ILF in the small intestine. (c,d) CD90 in green, NKp46 in red in WT (c) and AHR-/- mice (d). (e,f) CD90 in green, c-kit in red in WT (e) and AHR-/- mice (f). (g,h) CD11c in green and B220 in red in WT (g) and AHR-/- mice (h). Scale bar for (c to h) 50μm. (i) Quantification of total CD90+ clusters along the small intestine (SI). (j) Frequency of PP along the small intestine (SI). ND – not detected. NS – not significant. (k) Quantification of the number of domes per PP along the whole intestine.
RORγt+ LTi cells are essential for supporting lymphoid organogenesis11, 15. Because AHR-/- mice had a marked reduction of LTi-like ILC in the intestinal LP, we hypothesized that AHR deficiency may selectively affect lymphoid tissues at this site. Immunohistochemical analysis of the small intestine from AHR-/- and WT mice exposed a remarkable lack of both CP and mature ILF in the LP of AHR-/- mice (Fig. 3c-h). CP, which were visualized as clusters of CD90+ and c-Kit+ cells in the intestinal crypts of WT mice, were completely absent in the small intestine of AHR-/- mice (Fig. 3i), as were the CD11c+ and B220+ cell clusters associated with ILF. In contrast, PP were substantially conserved and similar in AHR-/- and WT mice, although we did observe a reduction in the number of domes within each PP in the AHR-/- mice (Fig. 3j and k). These data demonstrate that AHR deficiency causes a selective defect in the formation of CP and ILF in the small intestine LP but does not markedly affect the PP. Interestingly, immunohistochemical analysis of the large intestine of AHR-/- mice revealed the absence of colonic cryptopatches (Supplementary Fig. 4), whereas caecum patches were preserved. Thus, AHR deficiency seems to affect the diffuse post-natally imprinted lymphoid tissues throughout the intestine whereas the aggregated embryonically imprinted lymphoid follicles are relatively unaffected.
The developmental defect of ILC in AHR-/- mice is cell autonomous
Since AHR is broadly expressed in cells of both non-hematotopoietic and hematopoietic lineage, we wanted to know whether the impairment of IL-22-producing innate lymphoid cells observed in AHR-/- mice is cell autonomous, as suggested by the high AHR expression in NKp46+ILC and LTi-like ILC, or cell-extrinsic. First, analysis of LP and PP revealed no significant differences in CD8+ and CD4+ T cell subsets in either AHR-/- or AHR+/+ mice (Fig. 4a). CD11c+ MHC class II+ DC populations were intact in AHR-/- mice as well (Fig. 4b). B cells showed a slight non-significant reduction; Foxp3+CD4+ Tregs were also conserved (Supplementary Fig. 5). Only intraepithelial (but not lamina propria) γδ T cells were reduced, as recently reported25; however, these cells do not produce IL-22. Thus, AHR deficiency seems to predominantly affect the development of NKp46+ILC and LTi-like ILC as well as intestinal γδ T cells within the hematopoietic compartment.
Figure 4.

The developmental defect of AHR-/- mice is cell-intrinsic. (a,b) siLP from AHR-/- and AHR+/+ mice were harvested and analyzed by FACS for CD4+ and CD8+ T cells (a) and dendritic cell populations (b). Cells are gated on CD3+ lymphocytes (a) and on CD19-CD11c+ populations (b). (c) siLP cells from AHR-/- into wildtype and wildtype into wildtype bone marrow (BM) chimeras were harvested and analyzed by FACS. Cells are gated on CD45.2+CD3-CD19-CD4-NKp46+. The bottom bar represents percent of NKp46+NK1.1- cells from AHR-/- reconstituted cells, and top bar represent percent of NKp46+NK1.1- cells from AHR+/+ reconstituted cells. Right panel represents fold increase in IL-22+ cells from IL-23 stimulated cells gated on CD3-CD19-NKp46+ cells from AHR-/- and AHR+/+ chimeras (n=4). AHR-/- BM reconstituted mice show reduced frequencies of NKp46+ILC compared to WT BM reconstituted mice. The reconstitution of WT NKp46+ILC in irradiated WT mice was incomplete. (d) siLP cells from mixed AHR-/- and WT bone marrow chimeras were analyzed by gating on CD45.2+CD3-CD19-CD4-NKp46+ (AHR-/-) cells and CD45.2-CD3-CD19-CD4-NKp46+ (AHR+/+) cells and reconstitution of NKp46+ILC were compared. Bottom bar represent percent of NKp46+NK1.1- cells in AHR-/- cells, and top bar represent percent of NKp46+NK1.1- cells in AHR+/+ cells. Right panel represents fold increase in IL-22+ from IL-23 stimulated cells gated on CD3-CD19-NKp46+ from AHR-/- and AHR+/+ mixed chimeras. (n=3) (e,f) Cells isolated from the siLP of control (AHR-RORγt-cre-/-) and AHR conditional deletion (AHR–RORγt-cre-/Tg) mice were harvested and analyzed by FACS for NKp46+ ILC (e) and CD4+ LTi-like ILC (f) populations. Graphs show absolute numbers of cells. (n=4) Cells were also compared to absolute numbers of NKp46+ ILC (e) and CD4+ LTi-like ILC (f) populations from AHR-/- siLP from (supplementary fig. 4)
To corroborate that AHR in hematopoietic cells is essential for the development of NKp46+ and/or LTi-like ILC, we performed bone marrow chimera experiments. Lethally irradiated CD45.1 WT mice were reconstituted with CD45.2 AHR-/- bone marrow cells (AHR-/-⇒WT BM chimeras) or wild type bone marrow cells (WT⇒WT BM chimeras). Recipients of AHR-/- bone marrow showed a reduction of NKp46+ILC in the LP, suggesting a major impact of hematopoietic AHR on ILC development (Fig. 4c). To examine the effect of AHR deficiency in a competitive environment, we also performed mixed bone marrow chimeras where lethally irradiated mice were reconstituted with a 50/50 mix of CD45.2+ AHR-/- and CD45.1+ AHR+/+ bone marrow. Again, AHR-/- bone marrow cells failed to reconstitute NKp46+ILC populations while WT bone marrow were able to reconstitute them (Fig. 4d). Moreover, reconstituted NKp46+ILC from WT bone marrow produced IL-22 in response to IL-23. All bone marrow chimera experiments showed normal and even distribution of reconstitution in the spleen (Supplementary Fig. 6a). CD3+CD19+ lymphocytes in the siLP also showed even distribution and reconstitution in the bone marrow chimeras (Supplementary Fig. 6b). Furthermore we observed that while AHR-/- cells colonized the CP and ILF after mixed bone marrow transfers, the LTi like cells within the CP or ILF were largely of wildtype origin (Supplementary Fig. 6c). Thus, the data suggested that the generation of NKp46+ILC in the intestinal LP requires AHR expression in the hematopoietic compartment.
To further substantiate that the developmental defect was specifically due to lack of AHR expression in NKp46+ and LTi-like ILC, we utilized mice with a conditional deletion of AHR in RORγt-expressing cells. AHRfl/fl mice were crossed to RORγt-Cre transgenic mice to generate AHR-RORγt-cre-/- and AHR–RORγt-cre-/Tg. Similar to the phenotype observed in AHR-/- mice, there was a severe defect of NKp46+ILC and CD4+LTi-like ILC in mice with a specific deletion of AHR in RORγt expressing cells (Fig. 4e and f). This result, together with the high expression of AHR in NKp46+ILC, the lack of major developmental defects of lamina propria γδ T cells, DC and B cells in AHR-/- mice, and bone marrow reconstitution experiments demonstrates that the defect of ILC22 in AHR-/- mice is cell autonomous.
Germ-free mice exhibit normal content of intestinal ILC
Since previous studies indicated that the development of ILC22 requires the presence of intestinal microbiota16, 18, we hypothesized that the flora may be required to generate food catabolites, which act as AHR ligands inducing the development of IL-22-producing ILC. However, we found that the NKp46+ILC are intact in germ-free mice and noted no obvious differences in the frequency of these cells in the small intestine LP from mice housed in a germ-free versus a conventional barrier facility (Supplementary Fig. 7). This result is corroborated by a recently published study that also reported NKp46+ILC in germ-free mice36. Therefore, we surmise that the AHR ligands that drive the differentiation of ILC do not require the enzymatic activities of the intestinal microbiota, but are entirely derived from the endogenous ligands, such as the tryptophan catabolite kynurenine26, and/or natural ligands in the diet that have been recently shown to activate AHR in other cell compartments, such as intraepithelial γδ T cells25.
Notch signaling is required for intestinal NKp46+ILC
AHR has been shown to induce the transcription of multiple target genes, some of which could be involved in lymphoid cell development21, 22, 37. To narrow down the AHR target genes of potential interest, we compared known lists of AHR target genes with genes that are selectively transcribed in AHRhigh NKp46+ILC but not in AHRlow conventional NK cells. Among a few AHR target genes selectively expressed in AHRhigh NKp46+ILC, we found the receptor for IL-1 (IL-1R)19, CYP24A1, NOTCH1 and HES1, a basic helix-loop-helix repressor induced by Notch signaling (Supplementary Fig. 8). IL-1R was an attractive candidate, since IL-1β has been shown to promote the expansion of NKp46+ILC, at least in vitro38. However, we found no obvious differences between the CD3-CD19-NKp46+NK1.1- population in WT vs. IL-1R1-/- mice in either the siLP or the PP (Fig. 5a and b). Moreover, mice lacking SIGIRR, a cell surface receptor that behaves as an intracellular decoy for IL-1β signaling, had no apparent increase in NKp46+ILC, despite having exaggerated IL-1 signaling (Supplementary Fig. 9).
Figure 5.

AHR activation induces the upregulation of Notch. (a) Cells isolated from the PP and the siLP of IL-1R1-/- and WT mice were stained for CD3, CD19, NKp46, NK1.1 and analysed by flow cytometry. Cells are gated on CD3-CD19-cells and numbers above outlined areas indicate percent cells in each. (n=4). (b) Cells from PP and siLP were isolated from IL-1R1-/- or WT mice and stimulated with IL-23. Intracellular IL-22 was analyzed by flow cytometry. The values above bracketed lines indicate percentages of IL-22+ cells in CD3-CD19-NKp46+ cells (mean ± s.e.m.). Data are representative of 2 independent experiments (n=3 siLP or n=4 PP). (c) Mice were treated with 30ug/kg TCDD or DMSO by oral gavage and 3hrs later the liver was harvested and RNA was extracted for qPCR analysis of Notch 1, Notch 2 and Cyp1a1 expression. (d) Mice were treated with 30ug/kg TCDD or DMSO by oral gavage as in (c) and cells from the small intestine lamina propria were harvested and RNA was extracted for qPCR analysis of Notch 1, Notch 2 and Cyp1a1 expression. (e) NKL cell line transduced with a constitutively active AHR expression plasmid was analyzed by flow cytometry for expression of Notch 1 and Notch 2. Shaded histograms represent cells gated on GFP- untransduced cells and clear histograms represent cells gated on GFP+ transduced cells.
On the other hand, the Notch1 and Notch2 genes contain several XRE in their promoters and are induced by TCDD treatment22, 37, 39. Moreover, the Notch2 gene is expressed in c-kit+ LTi-like ILC of CP40. Therefore we investigated whether AHR may support the presence of NKp46+ and/or LTi-like ILC in the intestinal LP through induction of Notch receptors. We observed that administration of the AHR ligand TCDD by gavage induces the expression of Notch1 and Notch2 transcripts in the liver and in the gut in an AHR-dependent fashion (Fig. 5c, d). Moreover, overexpression of a constitutively active form of AHR in a NK cell line induced upregulation of Notch2, corroborating the regulation of Notch by AHR (Fig. 5e). We next analyzed intestinal ILC in mice that selectively lack the expression of RBP-Jk in the hematopoietic compartment (RBP-Jk × Vav1-Cre). RBP-J is a DNA-binding protein that associates with the intracellular regions of all Notch isoforms, mediating the transcriptional output of Notch signaling. Strikingly, analysis of intestinal ILC in RBP-Jk × Vav1-Cre mice revealed a marked reduction in the frequency of NKp46+ILC in the LP, but not in the PP, similar to what was seen in AHR-/- mice (Fig. 6a, c and Supplementary Fig. 10), whereas conventional NK cells were intact (data not shown). While the developmental defect in NKp46+ILC was dramatic, the reduction in the frequency of CD4+LTi-like ILC was less evident, although statistically significant (Fig. 6b, c). The partial preservation of CD4+LTi-like ILC was paralleled by conservation of CP and ILF (Fig. 6d, e). These results suggest that AHR sustain NKp46+ and, in part, LTi-like ILC through activation of the Notch pathway.
Figure 6.

IL-22 producing NK-like cells are reduced in RBP-Jκf/f × Cre-Vav1 mice. (a) siLp from RBP-Jκf/f and RBP-Jκ-/- mice were harvested and analyzed by FACS for NKp46+ILC. The cells in the top panel are gated on CD3-CD19-. Intracellular staining for IL-22 on the bottom panels show cells gated on CD3-CD19-NKp46+ cells. (b) siLP from RBP-Jκf/f and RBP-Jκ-/- mice were harvested and analyzed by FACS for CD4+ LTi-like ILC. Cells are gated on CD3-CD19-. (c) Quantification of the frequency of NKp46+ and LTi-like ILC from (a and b). (d) Immunohistochemistry to identify CD11c+ and B220+ cells associated with CP and ILF in the small intestine. CD11c in green and B220 in red in RBP-Jκf/f and RBP-Jκ-/- mice. Scale bar 50μm. (e) Quantification of total CD90+ clusters along the small intestine (SI).
Discussion
Our findings establish that AHR is required for the presence of ILC22 in the intestinal LP, including the NKp46+ and CD4+LTi-like ILC. The rapid death of AHR-/- mice following C. rodentium infection corroborates the importance of these early innate sources of IL-22 in defending the intestinal mucosal barrier against pathogenic bacteria. Ramarkably, AHR signaling in vivo is critical for the development of ILC22 and not simply for their ability to secrete IL-22, as ILC22 are completely deficient in AHR-/- mice. The requirement for AHR expression was cell autonomous as ILC22 deficiencies were reproduced in conditional deletion of AHR in RORγt-expressing cells. Notably, AHR and ILC22 deficiencies also resulted in the absence of CP and ILF in the intestinal LP, whereas it had a limited impact on PP. This unique phenotype clearly demonstrates the important and specific requirement of AHR signaling in RORγt+ ILCs for the organogenesis of post-natal lymphoid tissues. While CP and ILF formation is complete following the first few weeks after birth, PP develop prenatally, not only suggesting a temporal or environmental separation of these two processes, but perhaps heterogeneity in the cell types responsible for their organogenesis. Thus, our study corroborates the difference of CP and ILF with PP in terms of organogenesis, which was originally suggested by the analysis of mice lacking key molecules for lymphoid development41.
Since AHR deficiency selectively affects intestinal lymphoid tissues that develop in the first few weeks after birth, this strongly implies that AHR ligands must become available in the post-natal environment. While AHR ligands in the post-natal environment may be derived from the products of intestinal microbiota as the intestine becomes colonized following birth, we have found that ILC22 are intact in germ-free mice. This result is corroborated by a recently published study that also reported the presence of NKp46+ILC in germ-free mice36. AHR ligands that promote ILC22 generation may also include dietary compounds such as those contained in cruciferous vegetables that have been recently reported to drive the expansion of other AHR+ T cell subsets20, 25. However, our preliminary studies with synthetic diets that seemingly lack vegetable products do not show significant impairment of intestinal ILC22 (Supplementary Fig. 11). While attempting to generate a diet or environmental conditions completely devoid of potential AHR ligands may be difficult, perhaps another explanation is that the AHR ligand that drives the differentiation of ILC22 may be endogenous, such as the tryptophan catabolite kynurenine26. It will be important to establish the relative impact of endogenous ligands versus nutrients, supplements and additives/preservatives in the post-natal activation of AHR.
AHR induces expression of IL-1R and Notch21, 22, 37. We showed that defective or exaggerated IL-1 signaling has no impact on intestinal ILC. In contrast, blockade of Notch signaling in RBP-Jk × Vav1-Cre mice leads to a noteworthy defect in ILC. This result establishes Notch signaling as a crucial pathway downstream of AHR in the generation of ILC. Interestingly, while AHR deficiency impaired two subsets of ILC, the CD4+LTi-like and the NKp46+ILC, a defect of Notch signaling preferentially impaired the differentiation of NKp46+ILC and thus did not fully phenocopy AHR-/- mice. It is possible that additional signals downstream of AHR partially compensate lack of Notch signaling in LTi-like ILC. While the CP and ILF were absent in AHR-/- mice, these structures were preserved in RBP-Jk × Vav1-Cre mice, as were CD4+LTi-like ILC. Thus, LTi-like ILC may be sufficient to maintain CP and ILF in the small intestine perhaps through AHR-driven IL-22, which was recently shown to maintain colonic CP and ILF following bacterial infection through a yet unknown mechanism42. These results establish Notch signaling as a crucial pathway downstream of AHR in the generation of NKp46+ILC.
Since we evaluated mice lacking Notch signaling in all hematopoietic cells, it remains unclear whether Notch is required in mature NKp46+ILC or in their progenitors. Notch signaling may be essential for the survival of mature NKp46+ILC, consistent with the recent demonstration that Notch promotes survival of IL-17-producing γδ T cells43. It may also facilitate the generation of NKp46+ILC from progenitors and the expression of chemokine receptors that promote their migration to the lamina propria. In support of this possibility, it was recently shown that Notch promotes the differentiation of TH1744, 45 as well as the development of adult, but not fetal, RORγt+ ILC46. Finally, one study suggests that Notch signaling may also induce yet unknown endogenous AHR ligands, suggesting a possible AHR-Notch-AHR feed-forward loop47.
Whether NKp46+ILC are the progeny of LTi-like ILC or a closely related cell lineage has been matter of debate10, 12, 48. While our data cannot resolve this debate, it is clear from our study that both cell types contribute to IL-22 secretion but have different function in lymphoid organogenesis in the gut. Lack of both LTi-like and NKp46+ILC in AHR-/- mice leads to a severe impairment of CP and ILF, whereas preferential loss of NKp46+ILC only has a limited impact on intestinal lymphoid tissues. Thus, LTi-like ILC seem to have a predominant role in inducing CP and ILF. Furthermore, the requirement of LTi-like ILC for CP and ILF but not PP emphasizes the heterogeneity of inducer cells and signaling pathways involved in the organogenesis of distinct gut-associated lymphoid tissues41, 49.
Methods
Mice
All mice were bred and maintained at an SPF facility at Washington University School of Medicine and animal protocols were reviewed and approved by the Washington University animal studies committee. AHR-/- mice were previously reported24. AHR-/- mice were generated from heterozygous breeders and WT or AHR+/+ refers to littermates. RPB-Jkf/f were crossed to Vav1-cre mice to generate RPB-Jkf/f × Vav1-cre-/tg (RPB-Jk-/-) and RPB-Jkf/f × Vav1-cre-/- (RPB-Jkf/f) mice. AHRfl/fl were crossed to RORγt-cre transgenic mice to generate AHRfl/fl × RORγt-cre-/- (AHR-RORγt-cre-/-) and AHRfl/fl × RORγt-cre-/tg (AHR–RORγt-cre-/Tg) Conditional deletions were heterozygous for cre expression. Mice used for experiments were between 6-8 weeks of age and on a C57BL/6 background (each mutation was backcrossed to C57BL/6 at least 6 times). Control mice were littermates. The origins of the various strains of mice are described in the supplemental methods.
Cell preparation, isolation and flow cytometry
The preparation of PP, siLP, large intestine lamina propria (liLP) is described in the supplemental methods. For surface staining, fluorophore-labeled mAbs specific for CD3, CD19, NK1.1, CD4 were obtained from BD PharMingen and CD127 from Ebioscience. Anti-mouse NKp46 was obtained from R&D. For intracellular cytokine staining, immediately after isolation, the cells were incubated for 1hr at 37°C with 40ng/mL IL-23 (Peprotech); then monensin was added (1:1000 dilution, Ebioscience) and incubation was continued for 6 hrs. Intracellular cytokine staining was performed with IC Fixation Buffer and Permeabilization Buffer (Ebioscience) per the manufacturer's instructions. Mouse IL-22 was obtained from Ebioscience. Cells were run on either a FACSCalibur or FACS Canto II (BD Biosciences) and analyzed with FlowJo (Tree Star, Inc.).
ELISAs
106 cells/mL were stimulated with 40ng/mL IL-23 for 24hrs at 37°C. IL-22 concentrations in cell culture supernatants were measured using the murine IL-22 ELISA kit obtained from Antigenix America per manufacturer's instructions.
Quantification of mRNA levels by Real-time PCR
cDNA was synthesized using Superscript III reverse transcriptase (Invitrogen). Real-time quantitative PCR reactions were performed on an ABI Prism 7000 (Applied Biosystems) using the iQ™ SYBR Green supermix (Bio-rad) and specific primers. Target mRNA expression was calculated and normalized to the expression of HPRT. AHR specific primers: forward 5 ’-CGCGGGCACCATGAGCAG- 3’; reverse 5’ –CTGTAACAAGAACTCTCC- 3’, Notch1 forward 5’ –ATGTCAATGTTCGAGGACCAG- 3’; reverse 5’–TCACTGTTGCCTGTCTCAAG-3 ’ ; Notch2 forward 5’– GTGGCAGAGTTGATCAATTG- 3’; reverse 5’–ATATCTCTGTTGGCCCCATTC-3’; Cyp1a1 forward 5’–GGTTAACCATGACCGGGAACT-3 ’; reverse 5’– TGCCCAAACCAAAGAGAGTGA- 3’.
C. rodentium infection
AHR-/- or WT mice were fasted for 8hrs before intraoral inoculation of 2×109 C. rodentium strain DBS100 (ATCC) as described7. Survival was monitored for 21 days. The distal colon was harvested and fixed in 10% formalin overnight at room temperature, then embedded in paraffin, sectioned, and stained with H&E. Slides were prepared by the digestive Diseases Research Core Center (DDRCC). The spleen was weighed, homogenized and serial dilutions were plated in triplicates onto MacConkey agar plates at 37 degrees for 24hrs for measurement of colony forming units. The histologic severity of colitis was scored from 0-5 as described7. Sections were graded on the criteria of inflammatory cellular infiltration, mucosal hyperplasia, mucosal erosion and/or ulceration, crypt abscessation, and loss of crypts and goblet cells.
Immunohistochemistry
Small intestines and colons were removed intact and prepared for immunohistochemistry as previously described50. Intestines were embedded in optimal cutting temperature compound (Sakura Finetek, Torrence, CA) frozen, and serial 8-um sections were cut at an axis perpendicular to the villi for evaluation. Frozen sections were stained with indicated antibodies. Pseudo-colored black and white images from fluorescent microscopy were obtained with an Axioskop 2 microscope using Axiovision software (Carl Zeiss MicroImaging, Thornwood, NY) Quantification of CD90+ clusters was performed by the whole mount technique as described50.
Bone Marrow (BM) chimera generation
Recipient C57BL/6 SJL mice were γ-irradiated with 10Gy. After an overnight rest, mice were reconstituted with 5 × 106 BM cells per mouse that had been harvested from the femurs and tibias of gender and age-matched AHR-/- or WT donors. After 8 weeks, the spleens and siLPs were harvested for analysis. The mixed bone marrow chimera experiments were performed as previously described50. C57BL/6 SJL mice were γ-irradiated with two doses of 5Gy. After an overnight rest, mice were reconstituted with 5 × 106 AHR-/- BM cells mixed with 5 × 106 AHR+/+ BM cells. After 12 weeks, the spleens and siLPs were harvested for analysis. The number of cells recovered from the siLP from each experiment were similar and between 5-10×106 cells/intestine.
TCDD treatment
C57BL/6 mice were treated with 30μg/kg of TCDD or DMSO control by oral gavage. After 3 hrs the mice were sacrificed and the liver was harvested for mRNA analysis. The liver sample was harvested into RNAlater (Qiagen) and mRNA was extracted using RNeasy Mini Kit (Qiagen). The small intestine was also harvested, siLP cells were isolated and mRNA was extracted using the RNeasy Mini Kit.
Generation of cells expressing constitutively active (CA)-AHR
A DNA fragment encoding CA-AHR was subcloned into pMX-ires-GFP and transfected into PLATE-E cells to generate a recombinant retrovirus, which was used to infect human NKL cells. Cells stably expressing CA-AHR were FACS-sorted based on the expression of GFP. The details and modifications CA-AHR plasmid are specified in supplemental methods.
Statistical analysis
Statistical significance was determined with the unpaired Student's t-test. Prism GraphPad software v4.0 and v5.0 was used for statistical analyses. *P < 0.05, **P < 0.01 and ***P < 0.001.
Supplementary Material
Acknowledgments
We would like to thank Susan Gilfillan and Philip Ahern for critical comments. The germ-free mice were a kind gift from Prof. Jeffrey Gordon, funded by a Digestive Disease Research Core Center grant, P30DK052574, from the National Institute of Diabetes and Digestive and Kidney Disease. We would also like to acknowledge David O'Donnell and Maria Karlsson for germ-free husbandry. J. L. is supported by Kirschstein-NRSA training grant. MC is supported by NIH grant R01 DE021255-01 and NIAID Center for HIV/AIDS Vaccine Immunology grant A1067854. RDN is supported by NIH grant R01 DK064798.
References
- 1.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–69. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 2.Asquith M, Powrie F. An innately dangerous balancing act: intestinal homeostasis, inflammation, and colitis-associated cancer. J Exp Med. 2010;207:1573–7. doi: 10.1084/jem.20101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hand T, Belkaid Y. Microbial control of regulatory and effector T cell responses in the gut. Curr Opin Immunol. 2010;22:63–72. doi: 10.1016/j.coi.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eberl G, Lochner M. The development of intestinal lymphoid tissues at the interface of self and microbiota. Mucosal Immunol. 2009;2:478–85. doi: 10.1038/mi.2009.114. [DOI] [PubMed] [Google Scholar]
- 5.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zenewicz LA, Flavell RA. IL-22 and inflammation: leukin’ through a glass onion. Eur J Immunol. 2008;38:3265–8. doi: 10.1002/eji.200838655. [DOI] [PubMed] [Google Scholar]
- 7.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–9. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 8.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 9.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–90. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 10.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–7. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 11.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–74. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 12.Sawa S, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665–9. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- 13.Takatori H, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buonocore S, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–5. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eberl G, Littman DR. The role of the nuclear hormone receptor RORgammat in the development of lymph nodes and Peyer's patches. Immunol Rev. 2003;195:81–90. doi: 10.1034/j.1600-065x.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 16.Satoh-Takayama N, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–70. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Satoh-Takayama N, et al. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J Exp Med. 2010;207:273–80. doi: 10.1084/jem.20092029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanos SL, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cella M, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–5. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stockinger B, Hirota K, Duarte J, Veldhoen M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin Immunol. 2011;23:99–105. doi: 10.1016/j.smim.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Kerkvliet NI. AHR-mediated immunomodulation: the role of altered gene transcription. Biochem Pharmacol. 2009;77:746–60. doi: 10.1016/j.bcp.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology. 2009;127:299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009;30:447–54. doi: 10.1016/j.it.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–16. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, et al. Exogenous Stimuli Maintain Intraepithelial Lymphocytes via Aryl Hydrocarbon Receptor Activation. Cell. 2011;147:629–40. doi: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 26.Opitz CA, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 27.Quintana FJ, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen NT, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107:19961–6. doi: 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quintana FJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:20768–73. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Apetoh L, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11:854–61. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veldhoen M, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 33.Veldhoen M, Hirota K, Christensen J, O'Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–9. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105:9721–6. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luci C, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol. 2009;10:75–82. doi: 10.1038/ni.1681. [DOI] [PubMed] [Google Scholar]
- 36.Sawa S, et al. RORgammat(+) innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–6. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 37.Boverhof DR, et al. Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicological sciences : an official journal of the Society of Toxicology. 2006;94:398–416. doi: 10.1093/toxsci/kfl100. [DOI] [PubMed] [Google Scholar]
- 38.Hughes T, et al. Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity. 2010;32:803–14. doi: 10.1016/j.immuni.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dere E, Lo R, Celius T, Matthews J, Zacharewski TR. Integration of Genome-Wide Computation DRE Search, AhR ChIP-chip and Gene Expression Analyses of TCDD-Elicited Responses in the Mouse Liver. BMC genomics. 2011;12:365. doi: 10.1186/1471-2164-12-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lugering A, et al. CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol. 2010;160:440–9. doi: 10.1111/j.1365-2249.2010.04103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamada H, et al. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J Immunol. 2002;168:57–64. doi: 10.4049/jimmunol.168.1.57. [DOI] [PubMed] [Google Scholar]
- 42.Ota N, et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat Immunol. 2011;12:941–8. doi: 10.1038/ni.2089. [DOI] [PubMed] [Google Scholar]
- 43.Shibata K, et al. Notch-Hes1 pathway is required for the development of IL-17-producing {gamma}{delta} T cells. Blood. 2011;118:586–93. doi: 10.1182/blood-2011-02-334995. [DOI] [PubMed] [Google Scholar]
- 44.Mukherjee S, Schaller MA, Neupane R, Kunkel SL, Lukacs NW. Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J Immunol. 2009;182:7381–8. doi: 10.4049/jimmunol.0804322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keerthivasan S, et al. Notch signaling regulates mouse and human Th17 differentiation. Journal of immunology. 2011;187:692–701. doi: 10.4049/jimmunol.1003658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Possot C, et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nature immunology. 2011;12:949–58. doi: 10.1038/ni.2105. [DOI] [PubMed] [Google Scholar]
- 47.Alam MS, et al. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2010;107:5943–8. doi: 10.1073/pnas.0911755107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vonarbourg C, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736–51. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finke D. Induction of intestinal lymphoid tissue formation by intrinsic and extrinsic signals. Semin Immunopathol. 2009;31:151–69. doi: 10.1007/s00281-009-0163-6. [DOI] [PubMed] [Google Scholar]
- 50.Wang C, McDonough JS, McDonald KG, Huang C, Newberry RD. Alpha4beta7/MAdCAM-1 interactions play an essential role in transitioning cryptopatches into isolated lymphoid follicles and a nonessential role in cryptopatch formation. J Immunol. 2008;181:4052–61. doi: 10.4049/jimmunol.181.6.4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.