

Abstract
Protein-S-glutathionylation (PSSG) is an oxidative modification of reactive cysteines that has emerged as an important player in pathophysiological processes. Under physiological conditions, the thiol transferase, glutaredoxin-1 (Glrx1) catalyses deglutathionylation. Although we previously demonstrated that Glrx1 expression is increased in mice with allergic inflammation, the impact of Glrx1/PSSG in the development of allergic airways disease remains unknown. In the present study we examined the impact of genetic ablation of Glrx1 in the pathogenesis of allergic inflammation and airway hyperresponsiveness (AHR) in mice. Glrx1−/− or WT mice were subjected to the antigen, ovalbumin (OVA), and parameters of allergic airways disease were evaluated 48 h after three challenges, and 48 h or 7 days after six challenges with aerosolized antigen. Although no clear increases in PSSG were observed in WT mice in response to OVA, marked increases were detected in lung tissue of mice lacking Glrx1 48 h following six antigen challenges. Inflammation and expression of proinflammatory mediators were decreased in Glrx1−/− mice, dependent on the time of analysis. WT and Glrx1−/− mice demonstrated comparable increases in AHR 48 h after three or six challenges with OVA. However, 7 days postcessation of six challenges, parameters of AHR in Glrx1−/− mice were resolved to control levels, accompanied by marked decreases in mucus metaplasia and expression of Muc5AC and GOB5. These results demonstrate that the Glrx1/S-glutathionylation redox status in mice is a critical regulator of AHR, suggesting that avenues to increase S-glutathionylation of specific target proteins may be beneficial to attenuate AHR.
Keywords: asthma, lung inflammation, protein S-glutathionylation
chronic inflammatory disorders of the lung, including asthma, are accompanied by changes in the oxidative environment (7). For instance, oxidative inactivation of the antioxidant enzymes copper zinc containing SOD and manganese SOD, have been demonstrated in patients with asthma (12, 13), and changes in the redox status of the tripeptide antioxidant, GSH have also been reported in patients with asthma compared with healthy controls (17, 37). The exact mechanisms whereby oxidants contribute to disease pathogenesis remain unresolved. GSH, which is highly abundant and acts as an antioxidant and reducing agent in cells in lung lining fluid, plays a major role in maintenance of overall redox homeostasis. Consequently, agents that cause oxidative stress are known to decrease the ratio of reduced GSH to GSSG (glutathione disulfide) (35). In addition to changes in reduced to oxidized glutathione ratio, under conditions of oxidative stress, glutathione can become conjugated to reactive cysteines in proteins, a posttranslational modification known as protein S-glutathionylation (PSSG; also known as S-glutathiolation, or protein-mixed disulfides). PSSG is a redox-based posttranslational modification of proteins that changes structure and function of proteins in a reversible and tightly regulated manner (21, 26). Mammalian glutaredoxin (Glrx) enzymes are members of the thioredoxin family of thiol transferases that, under physiological conditions, specifically catalyze deglutationylation reactions, leading to the restoration of the reduced sulfhydryl group of protein cysteines (24).
The functional significance of altered regulation of protein function through PSSG in allergic airways disease remains unknown. We recently demonstrated increases in Glrx1 expression and increases in Glrx activity in lung tissue from mice with antigen-induced allergic inflammation. Increases in Glrx1 immunoreactivity occurred predominantly in the bronchiolar epithelium of mice with allergic airways inflammation (39). Furthermore, in mice lacking the Glrx1 gene (Glrx1−/−), LPS-induced inflammation and macrophage activation were attenuated, in association with enhanced S-glutathionylation (4). Primary lung epithelial cells derived from Glrx1−/− mice also showed markedly attenuated production of inflammatory mediators in response to LPS compared with epithelial cells isolated from wild-type (WT) mice (38). The goal of the present study was to determine the impact of Glrx1 ablation on the development of allergic airways disease, using the well-characterized model of ovalbumin (OVA)-induced Th2 inflammation and airway hyperresponsiveness (AHR). Our results demonstrate that Glrx1−/− mice showed enhanced resolution of AHR and mucus metaplasia, in association with increases in PSSG, suggesting a role for PSSG in ameliorating AHR.
MATERIALS AND METHODS
Glrx1−/− mice were backcrossed for > 10 generations into a BALB/c background (19) (Jackson Laboratories, Bar Harbor, ME) and were housed in the University of Vermont Animal Facility. For all experiments, 8- to 12-wk-old Glrx1−/− and littermate WT BALB/c control mice were used, as approved by the Institutional Animal Care and Use Committee.
OVA model of allergic airway disease.
WT BALB/c and Glrx1 −/− mice were administered 20 μg of the antigen OVA (Grade V; Sigma-Aldrich, St. Louis, MO) with aluminum hydroxide (alum) (Pierce, Rockford, IL) via an intraperitoneal injection on days 0 and 14 as previously described (25, 28). The control group was mock sensitized, receiving intraperitoneal PBS and alum. All groups of mice were challenged using three or six doses of aerosolized 1% OVA in sterile PBS for 30 min on days 21, 22, and 23 (33), or on days 21–26. The animals were harvested 48 h or 7 days postchallenge.
Assessment of AHR.
Mice were anesthetized with intraperitoneal pentobarbital sodium (90 mg/kg), tracheotomized, and mechanically ventilated at 200 breaths/min using a FlexiVent computer-controlled small animal ventilator (SCIREQ, QC, Canada), as previously described (28, 40). Airway responsiveness was quantified by averaging the three highest measurements obtained at each incremental methacholine dose.
Bronchoalveolar lavage and lung processing.
Bronchoalveolar lavage (BAL) (800–1,000 μl) was collected from mice immediately after the completion of measurements of respiratory mechanics. Total and differential cell counts were performed as previously described (28). Briefly, cells were isolated by centrifugation and total cell counts were performed using the Advia 120 automated hematology analyzer system. Cytospins were performed and stained using the Hema3 kit (Fisher Scientific, Kalamazoo, MI). Differential cell counts were performed on a minimum of 300 cells. Right lung lobes were flash frozen for RNA and protein isolation. Left lung lobes were fixed with 4% paraformaldehyde, mounted in paraffin, and 5-μm sections were prepared for histopathology as previously described (33).
Cytokineprofiling.
BAL samples were thawed and assayed for OVA-specific immunoglobulins and inflammatory cytokine levels as previously described (4). ELISA were utilized to assess cytokine content in undiluted BAL according to manufacturer's instructions (R&D Systems, Minneapolis, MN). For simultaneous quantitation of multiple analytes, undiluted BAL fluid or serum was analyzed using a mouse cytokine 23-plex kit on the Bio-Plex suspension array system (Bio-Rad, Hercules, CA) (8).
mRNA analysis.
Total RNA was isolated from pulverized lung tissue using the RNeasy kit (Qiagen, Valencia, CA). One microgram of RNA was used for cDNA synthesis and reverse transcribed for Taqman gene analysis using SYBR green (Bio-Rad). Primers for quantitative RT-PCR include: KC (forward: 5′-GCTGGATTCACCTCAAGAA-3′, reverse: 5′-TGGGGACACCTTTTAGCATC-3′); regulated on activation normal T-expressed and presumably secreted (RANTES) (forward: 5′-ATATGGCTCGGACACCACTC-3′, reverse: 5′-TCCTTCGAGTGACAAACACG-3′); IL-6 (forward: 5′-CTGATGCTGGTGACAACCAC-3′, reverse: 5′-CAGAATTGCCATTGCACAAC-3′); CCL-20 (forward: 5′-AAGACAGATGGCCGATGAAG-3′, reverse: 5′-AGCCCTTTTCACCCAGTTCT-3′); Muc5ac (forward: 5′-CAGTGAATTCTGGAGGCCAACAAGGTAGAG-3′, reverse: 5′-AGCTAAGCTTAGATCTGGTTGGGACAGCAGC-3′); GOB5 (forward: 5′-ACTAAGGTGGCCTACCTCCAA-3′: reverse: 5′-GGAGGTGACAGTCAAGGTGAGA-3′); and IL-5 (forward: 5′-ATGGAGATTCCCATGAGCAC-3′, reverse: 5′-CCCACGGACAGTTTGATTCT-3′). The fold induction was calculated using the housekeeping gene, cyclophilin.
Histopathology.
Periodic acid Schiff staining was performed, and mucus metaplasia was evaluated by obtaining three images of small bronchioles (×20 objective) of similar dimensions from each mouse. Images were blinded and ranked by two independent investigators, at a scale from 0 to 3: 0, no reactivity; 1, minimal staining; 2, moderate staining; 3, prominent staining. The cumulative score from each mouse was then averaged according to treatment group.
Biochemical analysis of protein S-glutathionylation in lung tissue.
Protein S-glutathionylation in lung tissue was determined using the glutathione/glutathione reductase/NADPH/5,5′-dithiobis (2-nitrobenzoic acid) recycling assay, according to procedures described previously (36). In brief, pulverized lung tissue stored at −80°C was homogenized in buffer containing 137 mM Tris·HCl, pH 8.0, 130 mM NaCl, and 1% NP-40. Protein content was determined, and samples were equalized for protein content. Then 200 μg of protein was precipitated with acetone. The pellet was resuspended in 0.1% Triton X-100 and 0.6% sulfasalicylic acid containing buffer and freeze thawed twice. Protein-associated glutathione was released with sodium borohydride, and GSH was determined. Samples that were not treated with sodium borohydride were used as a control. The sodium borohydride sensitive fraction of GSH was calculated and expressed as nanomoles GSH per milligram of protein.
Statistical analyses.
Analyses of all data were performed using the Graph Pad Prism software (Graphpad) by ANOVA or Student's t-test where appropriate. All experiments were repeated twice, and data from combined experiments are presented means ± SE. Histological scoring was analyzed using the Kruskal-Wallis test and Dunn's multiple comparison post hoc tests. Analyses with resultant P values of < 0.05 were accepted as significant.
RESULTS
Variable impact of Glrx1 ablation on OVA-induced airways inflammation.
To model allergic airways disease, we utilized the well-described model of OVA-induced pulmonary inflammation to produce Th2-dominated allergic airways disease. WT and Glrx1−/− mice were immunized with OVA plus alum on days 0 and 14, and challenged with aerosolized OVA on days 21, 22, and 23 and harvested 48 h later (3 Chall.+ 48 h). In addition, separate groups of mice were immunized, and then subjected to six consecutive OVA challenges and harvested 48 h (6 Chall.+ 48 h) and 7 days (6 Chall.+ 7 days) postfinal challenge. We first assessed OVA-specific immunoglobulins in serum from WT and Glrx1−/− mice following sensitization and challenge. Results in Fig. 1 demonstrate comparable increases in levels of IgE (Fig. 1A) and IgG1 (Fig. 1B) in WT and Glrx1−/− mice subjected to sensitization and challenge with OVA compared with alum/OVA controls, demonstrating that Glrx1−/− mice mounted an equally robust immune response to OVA as WT mice. We next assessed the magnitude of airways inflammation in WT and Glrx1−/− mice in response to OVA by enumerating cells obtained via BAL. All mice immunized and challenged with OVA showed a marked influx of cells into the airways (Fig. 2), characterized by increases in eosinophils, and to lesser but significant extent, neutrophils and lymphocytes (Table 1).Inflammation was most robust 48 h after six challenges, and remained strongly elevated, despite 7 days of recovery postfinal challenge. Genetic ablation of Glrx1−/− had a variable impact on OVA-induced inflammation. While inflammation was significantly decreased in the Glrx1−/− mice 48 h following three challenges with OVA, predominantly due to a lower influx of eosinophils at the later time points overall airway inflammation was comparable between WT and Glrx1−/− mice (Fig. 2 and Table 1).
Fig. 1.
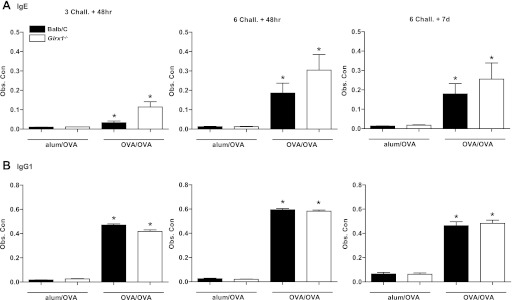
Assessment of ovalbumin (OVA)-specific immunoglobulins in wild-type (WT) or glutaredoxin-1−/− (Glrx1−/−) mice following sensitization and challenge (Chall.) with OVA. Serum immunoglobulin levels were measured by ELISA. Standard curves for OVA-specific IgE (A) and IgG1 (B) were generated from alum/OVA-sensitized BALB/c mouse serum (Obs. Con). For comparisons, values for the most concentrated were set to 10,000 U/ml. Data are expressed as means ± SE from 8 to 10 mice per group. *P < 0.05 (ANOVA) compared with respective alum/OVA control groups. Black bars: BALB/c mice, white bars: Glrx1−/− mice.
Fig. 2.
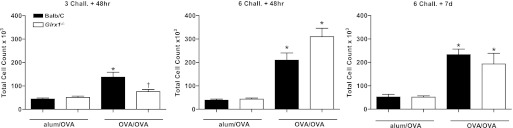
Total cell counts in bronchoalveolar lavage (BAL) from WT or Glrx1−/− mice following sensitization and challenge with OVA. Control mice received an intraperitoneal injection of alum and PBS (alum/OVA) at days 0 and 14. Sensitized mice were administered an intraperitoneal injection of alum and OVA (OVA/OVA) at days 0 and 14. Both groups were nebulized with aerosolized OVA at days 21, 22, and 23, or days 21 through 26. BAL was performed 48 h or 7 days after the last aerosolized OVA challenge. Total cell numbers were determined by Advia, and data are expressed as means ± SE from 8 to 10 mice per group. *P < 0.05 (ANOVA) compared with respective alum/OVA control groups. Black bars: BALB/c mice, white bars: Glrx1−/− mice.
Table 1.
Inflammatory cell profiles in bronchoalveolar lavage (BAL) following ovalbumin (OVA) sensitization and challenge of wild-type (WT) or glutaredoxin-1−/− (Glrx1−/−) mice
| Balb/C |
Glrx1−/− |
|||
|---|---|---|---|---|
| Relative Expression | Alum/OVA | OVA/OVA | Alum/OVA | OVA/OVA |
| 3 Chall. +48 h, 103 | ||||
| MACS | 42.2 ± 5.4 | 29.5 ± 5.4 | 52.4 ± 5.4 | 38.7 ± 4.3 |
| EOS | 0.0 ± 0.0 | 93.2 ± 14.8* | 0.9 ± 0.5 | 27.7 ± 5.4*† |
| PMN | 0.8 ± 0.3 | 11.7 ± 3.9* | 1.2 ± 0.3 | 7.7 ± 2.1* |
| LYMPH | 0.0 ± 0.0 | 3.1 ± 0.5* | 0.5 ± 0.2 | 1.7 ± 0.4 |
| 6 Chall. +48 h, 103 | ||||
| MACS | 36.1 ± 4.9 | 32.2 ± 4.8 | 36.4 ± 4.1 | 49.1 ± 13.2 |
| EOS | 0.2 ± 0.1 | 150.2 ± 30.7* | 0.2 ± 0.1 | 213.8 ± 27.9* |
| PMN | 1.6 ± 0.6 | 21.0 ± 9.3* | 0.5 ± 0.2 | 21.4 ± 8.0* |
| LYMPH | 0.9 ± 0.2 | 6.0 ± 1.4* | 0.4 ± 0.2 | 13.5 ± 5.2* |
| 6 Chall. +7 days, 103 | ||||
| MACS | 49.1 ± 11.1 | 43.7 ± 7.2 | 50.3 ± 5.6 | 44.3 ± 7.2 |
| EOS | 0.6 ± 0.3 | 169.1 ± 21.5* | 0.1 ± 0.0 | 133.0 ± 38.0* |
| PMN | 1.2 ± 0.5 | 1.3 ± 0.7 | 3.5 ± 1.5 | 1.1 ± 0.4 |
| LYMPH | 1.3 ± 0.7 | 6.7 ± 2.5* | 10.7 ± 5.4 | 4.9 ± 3.9* |
A minimum of 300 cells were enumerated by 2 independent investigators. Results were averaged and are expressed as means ± SE from 8 to 10 mice per group. Chall., challenged, MACS, macrophages; EOS, eosinophils; PMN, polymorphonuclear neutrophils (leukocytes); LYMPH, lymphocytes.
P < 0.05 (ANOVA) compared with respective alum/OVA control groups.
P < 0.05 (ANOVA) compared with WT OVA/OVA groups.
OVA-induced expression of proinflammatory mediators is reduced 48 h following 3 challenges in Glrx1−/− mice.
We evaluated the expression of proinflammatory mediators by assessment of mRNA in whole lung homogenates, as well as overall content in BAL fluid. Results in Table 2 demonstrate significant increases in mRNA expression of KC, IL-6, RANTES, CCL-20, and IL-5 in WT mice 48 h after three or six OVA challenges, which tended to decrease 7 days following cessation of the six challenge regimen. The content of CCL-20 and KC was increased in BAL in mice immunized and challenged with OVA at all examined time points (Table 3), while BAL levels of RANTES and IL-6 remained widely unchanged. Forty-eight hours after 3 OVA challenges, Glrx1−/− mice exhibited significant decreases in mRNA expression of KC and CCL-20 in lung tissue (Table 2, top), with accompanying decreases of these chemokines in the BAL (Table 3, top), compared with WT mice. Although small increases in mRNA expression of IL-4 and IL-13 were detected in lung tissue in response to sensitization and challenge with OVA, no clear differences were detected between WT and Glrx1−/− mice (data not shown). Collectively, these findings suggest that ablation of the Glrx1 gene tended to decrease expression of proinflammatory mediators, but did not have a strong impact on overall allergic inflammation, in particular at the later time points that were investigated herein.
Table 2.
Analysis of mRNA expression of NF-κB-dependent inflammatory cytokines in lung homogenates from WT and Glrx1−/− mice subjected to sensitization and challenge with OVA
| Balb/C |
Glrx1−/− |
|||
|---|---|---|---|---|
| Relative Expression | Alum/OVA | OVA/OVA | Alum/OVA | OVA/OVA |
| 3 Chall. +48 h | ||||
| CCL-20 | 1.0 ± 0.3 | 3.8 ± 0.5* | 1.4 ± 0.2 | 1.9 ± 0.3† |
| IL-6 | 1.0 ± 0.2 | 1.5 ± 0.4 | 0.4 ± 0.1 | 1.4 ± 0.3 |
| RANTES | 1.0 ± 0.1 | 0.8 ± 0.1 | 1.0 ± 0.2 | 1.0 ± 0.1 |
| KC | 1.0 ± 0.1 | 7.6 ± 1.2* | 0.9 ± 0.2 | 3.6 ± 0.9† |
| IL-5 | 1.0 ± 0.3 | 9.4 ± 2.8* | 0.9 ± 0.4 | 1.1 ± 0.5† |
| 6 Chall. +48 h | ||||
| CCL-20 | 1.0 ± 0.3 | 2.4 ± 0.7* | 0.6 ± 0.1 | 2.4 ± 0.6 |
| IL-6 | 1.0 ± 0.1 | 4.8 ± 1.1* | 0.6 ± 0.2 | 2.1 ± 0.7 |
| RANTES | 1.0 ± 0.2 | 9.3 ± 2.2* | 1.7 ± 0.5 | 4.1 ± 0.7 |
| KC | 1.0 ± 0.2 | 2.4 ± 1.2 | 2.1 ± 0.8 | 1.3 ± 0.5 |
| IL-5 | 1.0 ± 0.1 | 6.2 ± 1.5* | 2.1 ± 1.6 | 9.8 ± 2.0* |
| 6 Chall. +7d | ||||
| CCL-20 | 1.0 ± 0.4 | 1.8 ± 0.4 | 0.4 ± 0.1 | 1.7 ± 0.6* |
| IL-6 | 1.0 ± 0.3 | 0.7 ± 0.3 | 0.5 ± 0.2 | 1.4 ± 1.0 |
| RANTES | 1.0 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 |
| KC | 1.0 ± 0.1 | 2.5 ± 1.1* | 0.4 ± 0.1 | 1.1 ± 0.2 |
| IL-5 | 1.0 ± 0.2 | 4.4 ± 1.0* | 0.8 ± 0.1 | 3.8 ± 0.6* |
CCL-20, IL-6, RANTES, KC, and IL-5 mRNA from whole lung homogenates of OVA/OVA or alum/OVA mice were analyzed by real-time PCR analyses. Results were normalized to the housekeeping gene cyclophilin. CCL-20, chemokine (C-C motif) ligand-20; RANTES, regulated on activation normal T-expressed and presumably secreted; KC, CXCL1 (neutrophil-activating protein). Data are expressed as fold increases in expression compared with WT alum/OVA controls.
P < 0.05 (ANOVA) compared with respective alum/OVA control groups.
P < 0.05 (ANOVA) compared with WT OVA/OVA groups.
Table 3.
Analysis of content of NF-κB-dependent inflammatory cytokines in BAL from WT and Glrx1−/− mice subjected to sensitization and challenge with OVA
| Balb/C |
Glrx1−/− |
|||
|---|---|---|---|---|
| Relative Expression | Alum/OVA | OVA/OVA | Alum/OVA | OVA/OVA |
| 3 Chall. +48 h, pg/ml | ||||
| CCL-20 | 94.6 ± 26.6 | 183.6 ± 22.9* | 55.7 ± 9.8 | 83.3 ± 14.3† |
| IL-6 | 2.0 ± 0.3 | 2.7 ± 0.4 | 2.0 ± 0.4 | 6.1 ± 1.1* |
| RANTES | 16.5 ± 1.5 | 21.2 ± 4.0 | 17.2 ± 2.5 | 20.4 ± 3.7 |
| KC | 44.0 ± 5.6 | 249.7 ± 15.9* | 29.1 ± 4.6 | 161.7 ± 27.9*† |
| 6 Chall. +48 h, pg/ml | ||||
| CCL-20 | 40.3 ± 16.4 | 95.7 ± 31.8* | 20.2 ± 5.9 | 50.1 ± 16.8* |
| IL-6 | 16.8 ± 3.9 | 16.2 ± 2.5 | 16.1 ± 3.9 | 14.2 ± 2.0 |
| RANTES | 79.7 ± 19.9 | 67.8 ± 8.5 | 76.3 ± 22.5 | 63.8 ± 8.0 |
| KC | 142.0 ± 21.3 | 677.9 ± 147.2* | 128.6 ± 30.5 | 478.7 ± 104.3* |
| 6 Chall. +7 days, pg/ml | ||||
| CCL-20 | 60.2 ± 19.3 | 131.2 ± 47.8* | 86.2 ± 13.3 | 40.7 ± 32.2 |
| IL-6 | 19.4 ± 3.7 | 18.2 ± 4.4 | 17.9 ± 4.5 | 10.5 ± 1.7 |
| RANTES | 111.1 ± 16.6 | 69.9 ± 12.5 | 82.2 ± 14.2 | 43.0 ± 9.5 |
| KC | 227.9 ± 80.5 | 667.3 ± 164.0* | 110.4 ± 19.8 | 502.0 ± 116.9* |
BAL fluid was analyzed by ELISA for cytokines: CCL-20, IL-6, RANTES, and KC. Data are expressed as means ± SE from 8 to 10 mice per group.
P < 0.05 (ANOVA) compared with respective alum/OVA control groups.
P < 0.05 (ANOVA) compared with WT OVA/OVA groups.
Ablation of Glrx1 causes enhanced resolution of OVA-induced AHR and mucus metaplasia.
To address the functional consequences of the lack of Glrx1, a forced oscillation technique was used to evaluate alterations in respiratory mechanics in response to OVA sensitization and challenge. As expected, in response to OVA sensitization and challenge, airway resistance (Fig. 3A), tissue dampening (Fig. 3B), and elastance/stiffness (Fig. 3C) were significantly increased in WT animals, with the most prominent increases occurring 48 h after three challenges with OVA. It is worth noting that 1 wk postcessation of OVA challenges, all parameters of AHR remained significantly increased in WT animals. At 48 h after 3 challenges, Glrx1−/− mice demonstrated comparable increases over respective controls in airway resistance and tissue dampening at 12.5 and 25 mg/ml of Mch. Although tissue elastance tended to increase in Glrx1−/− mice in response to sensitization and challenge with OVA, compared with alum/OVA controls, these increases did not reach statistical significance. Glrx1−/− mice showed significant attenuation in tissue dampening 48 h after six challenges with OVA, compared with WT controls. Importantly, parameters of AHR were significantly decreased in Glrx1−/− mice compared with WT groups 7 days postcessation of the 6-day challenge regimen (Fig. 3, A–C, bottom).
Fig. 3.



Assessment of airway hyperresponsiveness (AHR) in WT or Glrx1−/− mice following sensitization and challenge with OVA. Changes in respiratory mechanics were analyzed in mock-sensitized (alum/OVA) or OVA-sensitized (OVA/OVA) WT (black bars) or Glrx1−/− mice (white bars) 48 h following 3 or 6 challenges with aerosolized OVA, or 7 days following the 6th daily aerosolized OVA challenge. Ascending doses of methacholine were administered to determine airway resistance (RN; A), tissue dampening (G; B), and elastance/stiffness (H; C) parameters. Data are expressed as % change from baseline measurements ± SE for each of the parameters measured from 8 to 10 mice per group. *P < 0.05 (ANOVA) compared with respective alum/OVA control groups. †P < 0.05 (ANOVA) compared with WT OVA/OVA groups.
Defining features of asthma include excess mucus secretion, goblet cell hyperplasia and submucosal gland hypertrophy (27, 40), which have been shown to contribute to airway closure and hyperresponsiveness (14, 41). mRNA analysis of the mucus genes MUC5ac and GOB5, revealed similar increases in expression in response to OVA in WT and Glrx1−/− mice 48 h after six challenges with OVA (Fig. 4, A and B). Decreases in MUC5ac mRNA were apparent in Glrx1−/− compared with WT mice 7 days after the 6-day OVA challenge regimen (Fig. 4A). IL-13 levels in BAL fluid recovered from WT mice sensitized and challenged with OVA, were significantly elevated over alum/OVA controls in WT mice 48 h after 3 challenges (Fig. 5A). Although no clear increases in IL-13 content in the BAL occurred in OVA-sensitized and -challenged mice compared with the alum/OVA group, at the later time points, IL-13 content in BAL from Glrx1−/− mice was slightly decreased 48 h after six challenges, and significantly decreased after 7 days compared with WT groups (Fig. 5A, middle and right). Consistent with these observations, histopathological analysis of lungs from WT and Glrx1−/− mice revealed comparable mucus metaplasia 48 h after three and six aerosolized OVA challenges (Fig. 5B). Although significant increases in mucus metaplasia remained 7 days following cessation of six challenges in WT mice, PAS positivity was significantly reduced in Glrx1−/− mice (Fig. 5B, right, Fig. 5C). In aggregate, these findings demonstrate that Glrx1−/− mice display enhanced resolution of AHR and mucus metaplasia, compared with WT counterparts.
Fig. 4.
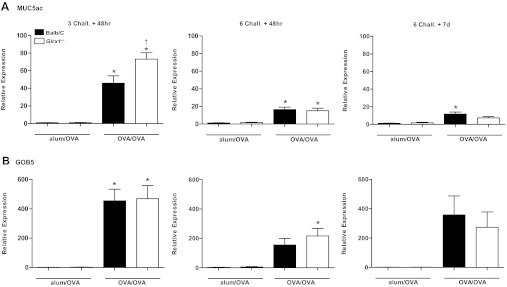
Evaluation of GOB5, MUC5ac, and IL-13 gene expression in WT and Glrx1−/− mice subjected to sensitization and challenge with OVA. RNA from lung homogenates of OVA-sensitized and challenged mice (OVA/OVA) or mock-sensitized mice (alum/OVA) was analyzed by real-time PCR, and results were normalized to the housekeeping gene cyclophilin. Data are expressed as fold increases in expression compared with WT alum/OVA controls. *P < 0.05 (ANOVA) compared with respective alum/OVA control groups. †P < 0.05 (ANOVA) compared with WT OVA/OVA groups. Black bars: BALB/c mice, white bars: Glrx1−/− mice.
Fig. 5.

Evaluation of IL-13 content in BAL and mucus metaplasia in WT and Glrx1−/− mice subjected to sensitization and challenge with OVA. A: IL-13 content in the BAL was determined by ELISA. Data are expressed as means ± SE from 8 to 10 mice per group. *P < 0.05 (ANOVA) compared with respective alum/OVA control groups. †P < 0.05 (ANOVA) compared with WT OVA/OVA groups. B: mucus metaplasia was assessed in periodic acid Schiff-stained (PAS) bronchiolar epithelium from WT and Glrx1−/− mice sensitized and challenged with OVA (OVA/OVA). Mucus metaplasia was evaluated by obtaining 3 images of small bronchioles (×20 objective) of similar dimensions from each mouse. Average scores of 2 independent evaluations were determined. Data are expressed as means ± SE from 8 to 10 mice per group. *P < 0.05 (Kruskal-Wallis) compared with respective alum/OVA control groups. †P < 0.05 (Kruskal-Wallis) compared with WT OVA/OVA groups. Black bars: BALB/c mice, white bars: Glrx1−/− mice.
Absence of Glrx1 leads to increases in protein S-glutathionylation in response to sensitization and challenge with OVA.
Under physiological conditions, the main function of Glrx1 is to deglutathionylate proteins restoring protein sulfhydryl groups. In response to sensitization and challenge with OVA, increases in endogenous Glrx1 content occurred in WT mice over time (Fig. 6A). In WT mice, no clear increases in overall PSSG content occurred in response to OVA, compared with alum/OVA controls at any of the time points investigated. We next assessed whether alterations in PSSG content occurred in lung tissue from mice lacking Glrx1 compared with WT animals. Ablation of Glrx1 resulted in slight increases in PSSG content, in the alum/OVA group compared with WT mice. Slight, but significant increases in PSSG content were apparent 48 h after 3 challenges in Glrx1−/− mice, with striking increases occurring 48 h after six challenges (Fig. 6B). Surprisingly, increases in PSSG were no longer apparent in Glrx1−/− mice, 1 wk postcessation of OVA challenges.
Fig. 6.
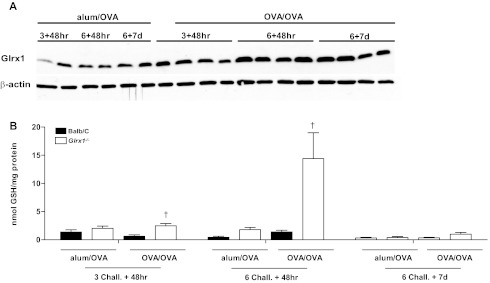
Assessment of protein S-glutathionylation and Glrx1 expression in WT and Glrx1−/− mice following sensitization and challenge with OVA. A: assessment of Glrx1 content in lung tissue from WT mice at different time points following sensitization and challenge with OVA. Lung tissues were homogenized, and Glrx1 content assessed in via Western blot analyses/β-actin is shown as a loading control. Results from 2 independent mice are shown per time point in the alum/OVA groups, whereas 4 independent mice were analyzed per time point in the OVA/OVA groups. B: biochemical analysis of protein S-glutathionylation in homogenized lung tissues of WT (black bars) and Glrx1−/− mice (white bars) 48 h and 7 days following the 6th daily aerosolized OVA challenge. Homogenized lung tissue was prepared, proteins were precipitated, and glutathione was released using Na-borohydride and quantified using the glutathione disulfide reductase recycling assay, with 5,5′-dithiobis-(2-nitrobenzoic acid) as substrate. Na-borohydride-dependent formation of 5′-thio-2-nitrobenzoic acid was calculated and normalized to protein content. Data are expressed as nanomoles tripeptide GSH per milligram protein. Data are expressed as means ± SE from 8 to 10 mice per group. *P < 0.05 (ANOVA) compared with respective alum/OVA control groups. †P < 0.05 (ANOVA) compared with WT OVA/OVA groups.
DISCUSSION
PSSG has emerged as an archetypal, redox-dependent posttranslational modification of target protein cysteines. PSSG not only has the potential to affect protein structure and function, it also protects proteins from irreversible thiol oxidations. The impact of protein S-glutathionylation depends upon the target protein, and activation (1, 2), gain of function (6), and inhibition of various proteins (10, 31, 38) has been observed (21). The biological significance of PSSG is bolstered by the evolution of glutaredoxins, thioltransferases that catalyse reversible S-glutathionylation, and deglutathionylation reactions. Under physiological conditions, cytosolic Glrx1 preferentially catalyses deglutathionylation reactions via the NH2-terminal active site cysteine. This results in S-glutathionylation of Glrx1 itself, which is subsequently reduced by a second GSH molecule (21, 26). Pathophysiological functions for Glrx1 are emerging that include roles in the regulation of apoptosis, transcription, tissue inflammation, and remodeling (26).
In the present study, we addressed the impact of ablation of Glrx1 on OVA-induced allergic airways disease. Our results demonstrate clear increases in the content of PSSG reactivity in lung tissues from Glrx1−/− mice after sensitization and repeated challenges with OVA, compared with WT mice, consistent with its role in catalysis of deglutathionylation reactions in physiological settings. Surprisingly, 7 days after six challenges with antigen, PSSG content decreased in the lungs of Glrx1−/− mice (Fig. 6B). A previous study reported no evidence for compensation of various antioxidant enzymes or Glrx2 content in the lungs of Glrx1−/− mice, which also lacked detectable deglutathionylation activity (19). However, our data indicate the presence of other deglutathionylating events or enzymes in the lung in the setting of allergic inflammation. In this regard, putative roles for the related oxidoreductase, thioredoxin (18) as well as sulfiredoxin (15, 29) have been suggested, which could account for decreases in PSSG content over time in lungs from Glrx1−/− mice. Additional analyses that were beyond the scope of the present study will be required to determine the source of the deglulathionylating activity in lungs of Glrx1−/− mice. In the present study we also observed slight increases in PSSG in lung tissues from mock-sensitized (alum/OVA) Glrx1−/− mice compared WT alum/OVA groups, findings that are in apparent contrast to previous reports indicating that the systemic lack of Glrx1 does not affect baseline PSSG (4, 19). It is plausible that aerosolized antigen, in the absence of previous sensitization, causes a mild perturbation of the redox status, which could be more apparent in the absence of Glrx1.
Our laboratory has previously demonstrated a causal role for activation of the transcription factor NF-κB within the bronchial epithelium in orchestrating allergic inflammation (8, 28, 32). The transcription factor NF-κB has been shown to be inhibited via S-glutathionylation of IKKβ, RelA, and p50 (5, 31, 34, 38). Genetic ablation or siRNA-mediated knockdown of Glrx1 resulted in enhanced S-glutathionylation of IKKβ, and attenuated production of proinflammatory mediators by epithelial cells following stimulation with LPS. Conversely, overexpression of Glrx1 prolonged LPS-induced proinflammatory responses in lung epithelial cells in association with decreases in glutathionylated IKKβ and prolonged activation of NF-κB (5, 38). It is therefore surprising that despite increases in the overall content of PSSG in lung tissue the overall extent of airway inflammation was only transiently diminished in Glrx1−/− mice after sensitization and challenge with OVA compared with WT mice. Nonetheless, expression of proinflammatory mediators was consistently decreased in Glrx1−/− mice compared with WT animals. It is possible that Glrx1 affects the resolution of airway inflammation. Since inflammatory cells in the airways are still prominent 7 days after cessation of the six-challenge regimen, more extended time points will be required to determine the impact of Glrx1 on the resolution of allergic disease. Within the time frame of investigation of the present study we failed to demonstrate clear differences in nuclear content of RelA in lung homogenates from WT and Glrx1−/− mice (data not shown). Future studies will be required to elucidate whether S-glutathionylation of NF-κB family members within the lung epithelium are affected in Glrx1−/− mice. Such analyses were beyond the scope of the present study, and will require dissection of bronchiolar epithelium, the compartment wherein NF-κB activation is predominant. It is also plausible that the systemic ablation of Glrx1 may have obscured the contribution of Glrx1, specifically in airway epithelium towards NF-κB activation, and subsequent inflammatory responses. Therefore, elucidation of the exact role of Glrx1 in orchestrating inflammation awaits additional studies aimed at conditional manipulation of Glrx1 during specific times and specifically within the bronchial epithelium during allergen-driven inflammation.
One of the most striking phenotypes of Glrx1−/− mice in the present study was the enhanced resolution of AHR, compared with WT mice. Despite the sustained presence of OVA-induced inflammatory cells in the lungs at the time of assessment of respiratory mechanics, Glrx1−/− animals exhibited marked reductions in hyperresponsiveness to methacholine compared with WT mice. Notably, the elastance and tissue dampening parameters in Glrx1 −/− mice sensitized to OVA were significantly reduced 7 days following OVA challenge, whereas increases in Newtonian resistance, reflective of conducting airways, still occurred in Glrx1−/− mice, albeit to a slightly lesser extent than WT mice. These findings suggest a more pronounced impact of Glrx1 deficiency in the lung periphery. Additionally, Glrx1−/− mice demonstrated significant decreases in gene expression of MUC5ac and mucus metaplasia 7 days postcessation of OVA challenges, compared with WT mice (Figs. 4 and 5). These findings suggest that the enhanced resolution of AHR in Glrx1−/− mice may be functionally linked to altered regulation of mucin gene expression, mucin glycoprotein processing, and/or secretion. A firm link between oxidants and mucus metaplasia has been established; oxidants can increase expression of Muc5AC mRNA (3, 16, 20, 22, 42) and a role for oxidants in the transcriptional upregulation of the IL-13 gene has been shown (9). In keratinocytes stimulated with IL-4 and IL-13, activation of dual specificity oxidase-1 was reported to be critical in the activation of STAT6, following oxidative inactivation of protein tyrosine phosphatase 1B. These collective studies demonstrate that changes in redox homeostasis are important in the development of mucus metaplasia. However, it is unknown to date whether the signals that regulate mucin gene expression or intracellular processing of mucins is regulated via Glrx1-controlled S-glutathionylation.
S-glutathionylation reflects an oxidative event that can be triggered via multiple biochemical mechanisms, including oxidant production via activation of NADPH oxidases (21, 26). Oxidative events have been strongly implicated in the pathogenesis of allergic airways disease (11, 30, 37). It is therefore perhaps counterintuitive that in mice lacking Glrx1, wherein PSSG (an oxidative event) is increased, AHR, mucus metaplasia, and expression of proinflammatory mediators are decreased. These findings suggest putative beneficial function for S-glutathionylation in the pathophysiology of allergic airways disease. The exact mechanisms, whereby S-glutathionylation exerts protective effects during the pathogenesis of allergic airways disease, awaits additional investigation into the proteins that are targeted via S-glutathionylation, and the impact of S-glutathionylation on the regulation of their structure and function. It is plausible that S-glutathionylation exerts its protective function by preventing irreversible protein oxidations. In this regard, irreversible protein oxidation has been detected in lungs from mice exposed to cigarette smoke in association with loss of S-glutathionylation (23).
In conclusion, the present study demonstrates that a systemic loss of Glrx1 attenuates airways hyperresponsiveness, in association with decreased mucus metaplasia in antigen-induced allergic airways disease. Our results highlight the potential functional importance of the Glrx1/PSSG redox module in the resolution of AHR in allergic asthma, findings that could hold clinical relevance and offer potential therapeutic opportunities.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants T32-HL-076122 and R01-HL-060014.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.M.H. and Y.M.W.J.-H. conception and design of research; S.M.H., J.E.T., K.G.L., J.D.N., M.A., and N.D. performed experiments; S.M.H., J.E.T., V.A., A.S.G., and N.D. analyzed data; S.M.H., L.K.L., and C.G.I. interpreted results of experiments; S.M.H. prepared figures; S.M.H. drafted manuscript; S.M.H., J.E.T., K.G.L., J.D.N., A.S.G., J.L.J.v.d.V., Y.-S.H., C.G.I., and Y.M.W.J.-H. edited and revised manuscript; S.M.H., Y.-S.H., and Y.M.W.J.-H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Ye-Shih Ho for providing us with the Glrx1 knockout mice, the Vermont Lung Center and Charles G. Irvin for providing use of and assistance with the Flexivent, and Jennifer L. Díaz for editorial assistance with this manuscript.
REFERENCES
- 1. Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem 279: 29857–29862, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 10: 1200–1207, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Adler KB, Holden-Stauffer WJ, Repine JE. Oxygen metabolites stimulate release of high-molecular-weight glycoconjugates by cell and organ cultures of rodent respiratory epithelium via an arachidonic acid-dependent mechanism. J Clin Invest 85: 75–85, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aesif SW, Anathy V, Kuipers I, Guala AS, Reiss JN, Ho YS, Janssen-Heininger YM. Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation. Am J Respir Cell Mol Biol 44: 491–499, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aesif SW, Kuipers I, van der Velden J, Tully JE, Guala AS, Anathy V, Sheely JI, Reynaert NL, Wouters EF, van der Vliet A, Janssen-Heininger YM. Activation of the glutaredoxin-1 gene by nuclear factor-κB enhances signaling. Free Radic Biol Med 51: 1249–1257, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, Ho YS, Budd RC, Janssen-Heininger YM. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol 184: 241–252, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Oxidative and nitrosative events in asthma. Free Radic Biol Med 35: 213–225, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Ather JL, Hodgkins SR, Janssen-Heininger YM, Poynter ME. Airway epithelial NF-κB activation promotes allergic sensitization to an innocuous inhaled antigen. Am J Respir Cell Mol Biol 44: 631–638, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bansal G, Wong CM, Liu L, Suzuki YJ. Oxidant signaling for interleukin-13 gene expression in lung smooth muscle cells. Free Radic Biol Med 52: 1552–1559, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 38: 6699–6705, 1999 [DOI] [PubMed] [Google Scholar]
- 11. Ckless K, Hodgkins SR, Ather JL, Martin R, Poynter ME. Epithelial, dendritic, and CD4+ T cell regulation of and by reactive oxygen and nitrogen species in allergic sensitization. Biochim Biophys Acta 1810: 1025–1034, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, Song W, Hazen SL, Bleecker ER, Busse WW, Chung KF, Gaston B, Hastie A, Hew M, Jarjour N, Moore W, Peters S, Teague WG, Wenzel SE, Erzurum SC. Correlation of systemic superoxide dismutase deficiency to airflow obstruction in asthma. Am J Respir Crit Care Med 172: 306–313, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, Calhoun WJ, Janocha AJ, Zheng L, Hazen SL, Erzurum SC. Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am J Pathol 166: 663–674, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Evans CM, Kim K, Tuvim MJ, Dickey BF. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med 15: 4–11, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res 66: 6800–6806, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol 26: 447–452, 2002 [DOI] [PubMed] [Google Scholar]
- 17. Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Airway glutathione homeostasis is altered in children with severe asthma: evidence for oxidant stress. J Allergy Clin Immunol 123: 146–152, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greetham D, Vickerstaff J, Shenton D, Perrone GG, Dawes IW, Grant CM. Thioredoxins function as deglutathionylase enzymes in the yeast Saccharomyces cerevisiae. BMC Biochem 11: 3, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho YS, Xiong Y, Ho DS, Gao J, Chua BH, Pai H, Mieyal JJ. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic Biol Med 43: 1299–1312, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jang MK, Kim SH, Lee KY, Kim TB, Moon KA, Park CS, Bae YJ, Zhu Z, Moon HB, Cho YS. The tyrosine phosphatase, SHP-1, is involved in bronchial mucin production during oxidative stress. Biochem Biophys Res Commun 393: 137–143, 2010 [DOI] [PubMed] [Google Scholar]
- 21. Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45: 1–17, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim HJ, Ryu JH, Kim CH, Lim JW, Moon UY, Lee GH, Lee JG, Baek SJ, Yoon JH. Epicatechin gallate suppresses oxidative stress-induced MUC5AC overexpression by interaction with epidermal growth factor receptor. Am J Respir Cell Mol Biol 43: 349–357, 2010 [DOI] [PubMed] [Google Scholar]
- 23. Kuipers I, Bracke KR, Brusselle GG, Wouters EF, Reynaert NL. Smoke decreases reversible oxidations S-glutathionylation and S-nitrosylation in mice. Free Radic Res 46: 164–173, 2012 [DOI] [PubMed] [Google Scholar]
- 24. Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta 1780: 1304–1317, 2008 [DOI] [PubMed] [Google Scholar]
- 25. Lloyd CM, Saglani S. Asthma and allergy: the emerging epithelium. Nat Med 16: 273–274, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal 10: 1941–1988, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morcillo EJ, Cortijo J. Mucus and MUC in asthma. Curr Opin Pulm Med 12: 1–6, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Pantano C, Ather JL, Alcorn JF, Poynter ME, Brown AL, Guala AS, Beuschel SL, Allen GB, Whittaker LA, Bevelander M, Irvin CG, Janssen-Heininger YM. Nuclear factor-κB activation in airway epithelium induces inflammation and hyperresponsiveness. Am J Respir Crit Care Med 177: 959–969, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem 284: 23364–23374, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peden DB. The role of oxidative stress and innate immunity in O3 and endotoxin-induced human allergic airway disease. Immunol Rev 242: 91–105, 2011 [DOI] [PubMed] [Google Scholar]
- 31. Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-κB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry 40: 14134–14142, 2001 [DOI] [PubMed] [Google Scholar]
- 32. Poynter ME, Cloots R, van Woerkom T, Butnor KJ, Vacek P, Taatjes DJ, Irvin CG, Janssen-Heininger YM. NF-κ B activation in airways modulates allergic inflammation but not hyperresponsiveness. J Immunol 173: 7003–7009, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poynter ME, Irvin CG, Janssen-Heininger YM. Rapid activation of nuclear factor-κB in airway epithelium in a murine model of allergic airway inflammation. Am J Pathol 160: 1325–1334, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qanungo S, Starke DW, Pai HV, Mieyal JJ, Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFκB. J Biol Chem 282: 18427–18436, 2007 [DOI] [PubMed] [Google Scholar]
- 35. Rahman I, Biswas SK, Jimenez LA, Torres M, Forman HJ. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid Redox Signal 7: 42–59, 2005 [DOI] [PubMed] [Google Scholar]
- 36. Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc 1: 3159–3165, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Reynaert NL. Glutathione biochemistry in asthma. Biochim Biophys Acta 1810: 1045–1051, 2011 [DOI] [PubMed] [Google Scholar]
- 38. Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory-κB kinase-β. Proc Natl Acad Sci USA 103: 13086–13091, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol 36: 147–151, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Riesenfeld EP, Sullivan MJ, Thompson-Figueroa JA, Haverkamp HC, Lundblad LK, Bates JH, Irvin CG. Inhaled salmeterol and/or fluticasone alters structure/function in a murine model of allergic airways disease. Respir Res 11: 22, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singer M, Martin LD, Vargaftig BB, Park J, Gruber AD, Li Y, Adler KB. A MARCKS-related peptide blocks mucus hypersecretion in a mouse model of asthma. Nat Med 10: 193–196, 2004 [DOI] [PubMed] [Google Scholar]
- 42. Wright DT, Fischer BM, Li C, Rochelle LG, Akley NJ, Adler KB. Oxidant stress stimulates mucin secretion and PLC in airway epithelium via a nitric oxide-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 271: L854–L861, 1996 [DOI] [PubMed] [Google Scholar]