

The last 15 years have witnessed a significant shift in our understanding of neurodegenerative diseases. We increasingly recognize that the degenerative process in disorders such as Alzheimer disease,1 Huntington disease,2 and Parkinson disease3–5 begins years, if not decades, prior to the appearance of typical clinical manifestations. Whether ALS is also characterized by a prolonged presymptomatic period is unclear, but definitively answering this question is likely to have profound implications for understanding disease biology, uncovering environmental risk factors, developing effective therapies, and even disease prevention.
To effectively study presymptomatic ALS, one must first identify people who are at risk for developing disease. Although advancing age is a risk factor for ALS, the broad range of age at symptom onset limits its utility in identifying at-risk individuals. Absent any known environmental risk factor, it is currently only possible to study individuals genetically predisposed to developing ALS—namely, presymptomatic gene mutation carriers.
We begin this review with the rationale for studying presymptomatic ALS and a review of the relevant literature. We then turn to the many challenges inherent to studying presymptomatic ALS, and use the Pre-symptomatic Familial ALS (Pre-fALS) study to illustrate some of the practical approaches we have employed to overcome these challenges.
RATIONALE
Disease biology.
There is longstanding debate about the onset and progression of ALS.6 Some have suggested that ALS is primarily an upper motor neuron (UMN) disorder with secondary anterograde degeneration (“dying forward”) of the lower motor neuron (LMN)7–9; others have postulated a retrograde (“dying back”) process in which pathology begins in the LMN.10 Within the last few years, 2 related ideas about disease onset and progression have emerged. First, the disease process—namely, the underlying pathologic process that leads to motor neuron (MN) degeneration—begins focally within the axes of the UMN or LMN systems. Second, the disease process spreads via contiguity, with the rate of spread through each axis being central to determining phenotype.11,12 If correct, these hypotheses would have profound implications for how we conceptualize ALS pathogenesis—e.g., providing some support for the theory that misfolded/aggregated proteins recruit neighboring mutant or wild-type proteins in a prion-like fashion.13 It has been difficult to test these hypotheses in a natural history study, however, due to the relatively advanced stage of disease at which patients with ALS emerge over the clinical horizon. The long (median ∼10 months) delay from symptom onset to diagnosis compounds this problem.14–18 Even if the disease process had begun focally, by the time of diagnosis, confluence across multiple affected neighboring anatomic regions would make it almost impossible to dissect out the early stages of disease onset and spread. Testing these hypotheses, therefore, requires studying disease process from inception and through the early stages of progression. This can only realistically be accomplished by studying those at risk for developing disease.
Studying gene mutation carriers also has the potential to elucidate the time frame over which MN degeneration proceeds in ALS. One possibility is that ALS emerges explosively against a background of an otherwise healthy nervous system, with catastrophic loss of MNs and emergence of progressive weakness. Alternatively, there may be slow attrition of MNs, with compensatory mechanisms temporarily warding off symptoms, and progressive weakness eventually developing once the degenerative process overwhelms compensatory mechanisms.19 Under either scenario, genetic susceptibility might produce an endophenotype of structural or functional vulnerability to MN degeneration that could be elucidated through careful evaluation of presymptomatic gene mutation carriers.
Environmental risk factors.
Studying the full course of disease, including the presymptomatic stage, allows better definition of the role that environmental exposures might play. The temporal relationship between environmental exposures and disease onset, for example, may only be examined once we are able to define the earliest onset of disease (figure 1).
Figure 1. Schematic illustration of the full temporal course of the disease process in amyotrophic lateral sclerosis, showing different time periods during which environmental factors might be relevant.

During period A, prior to the onset of disease, environmental exposures might act, alone or in conjunction with other factors, either to cause disease or to modify the risk and timing of disease onset. During period B, between the onset of disease and the appearance of symptoms, environmental exposures might modify the course of the disease process, hastening or slowing the rate of progression and the latency to onset of symptoms, for example. Environmental exposures during this period cannot, by definition, cause disease since they postdate the onset of disease. Presymptomatic studies in amyotrophic lateral sclerosis (e.g., Pre-fALS study) offer an opportunity to delineate the distinction between periods A and B. During period C, after symptom onset, environmental exposures might also modify the course of disease, either for better or for worse.
Early therapeutic intervention.
Studying a population at risk for ALS creates an opportunity for early therapeutic intervention. Regular and careful follow-up with greater attention to early symptoms by both the individual and the study team is likely to expedite diagnosis, thereby allowing very early therapeutic intervention (e.g., with riluzole) or enrollment in clinical trials. The motivations for early rather than late therapeutic intervention are many. First, potential therapies may be less effective if administered late, once the disease process has become established. Second, preventing neuronal loss may be more realistic than regeneration of diseased or dead MNs; if the disease process truly begins focally and spreads by contiguity, early intervention might preserve neuronal number/function in unaffected or minimally affected regions. Moreover, it is preferable to slow or halt progression during the early stages of disease when disability is minimal; halting progression may be less appealing to patients once severe disability has already developed.
Disease prevention.
For a rare disease such as ALS, prevention can only practically be considered for individuals who are at risk for disease. Until very recently, the only known such population were gene mutation carriers from familial ALS (fALS) pedigrees. With the recent recognition that reduced penetrant hexanucleotide repeat expansions in C9ORF72 may be responsible for 5%–7% of apparently sporadic ALS,20,21 it may be possible to identify—e.g., by screening their first-degree relatives—an even larger population of people genetically predisposed to developing ALS. Studying these gene carriers and defining the presymptomatic phase of the disease is the only way to acquire necessary insights into the feasibility and design of a preventive trial,22 including how to identify and recruit at-risk individuals, quantify their long- and short-term risks of developing ALS, operationally define disease onset with high confidence and good reliability, and select biological markers of early disease.
Early evidence for presymptomatic disease.
Studies of presymptomatic SOD1+ carriers (i.e., those with an SOD1 gene mutation) have employed electrophysiologic methods such as motor unit number estimation (MUNE) and cortical excitability, as well as MRI techniques such as diffusion tensor imaging (DTI) and magnetic resonance spectroscopy (MRS) (table). Cross-sectional studies have suggested no differences between SOD1+ carriers and various control groups in electrophysiologic parameters,19,23 but longitudinal studies have reported reduced MUNE and increased cortical excitability 3–10 months in advance of symptom onset.24,25 MRI studies (all cross-sectional) have yielded conflicting results with 1 brain DTI study showing differences between SOD1+ carriers and age-matched controls,26 but a second study not confirming these findings.23 Additionally, a recent cross-sectional MRS study of the cervical cord revealed differences in tissue metabolites (e.g., reduced NAA/Cr) in SOD1+ carriers compared to controls.27 The expected heterogeneity of the study population with respect to the anticipated latency to onset of disease may underlie these varying cross-sectional findings.
Table.
Early evidence for presymptomatic disease in ALS
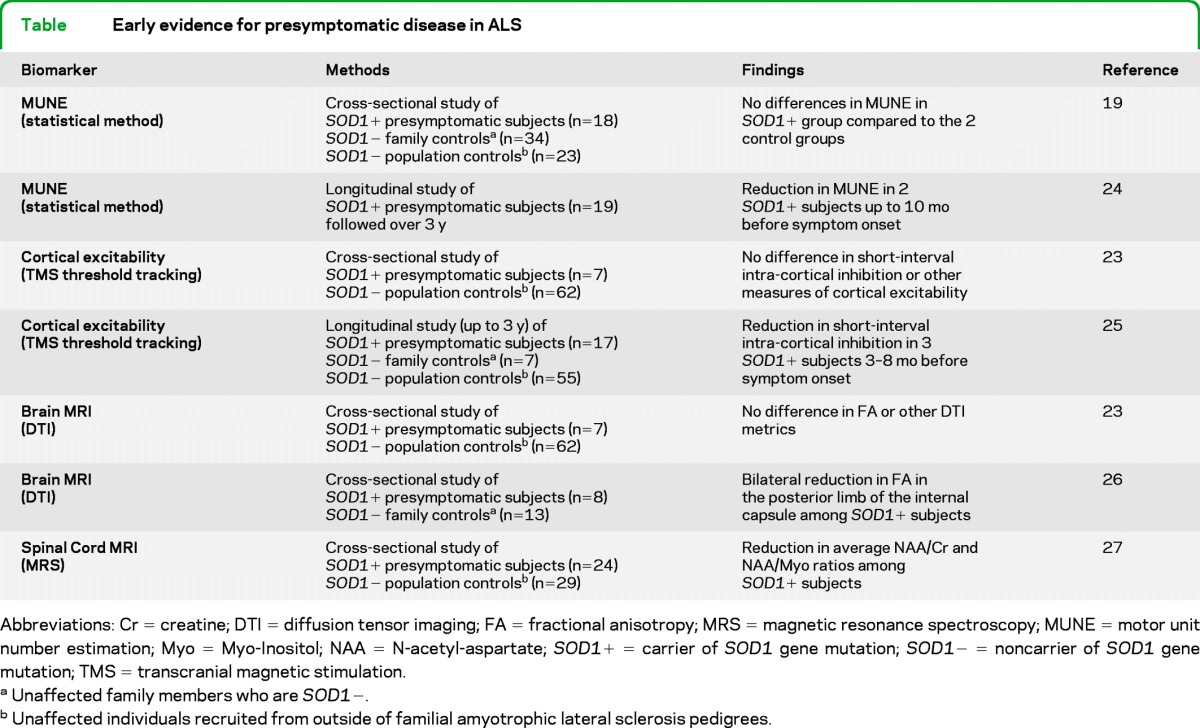
Abbreviations: Cr = creatine; DTI = diffusion tensor imaging; FA = fractional anisotropy; MRS = magnetic resonance spectroscopy; MUNE = motor unit number estimation; Myo = Myo-Inositol; NAA = N-acetyl-aspartate; SOD1+ = carrier of SOD1 gene mutation; SOD1− = noncarrier of SOD1 gene mutation; TMS = transcranial magnetic stimulation.
Unaffected family members who are SOD1−.
Unaffected individuals recruited from outside of familial amyotrophic lateral sclerosis pedigrees.
CHALLENGES
Identifying an at-risk population.
A distinction has traditionally been drawn between fALS (in which at least 2 biologically related family members have had ALS) and sporadic ALS (in which only a single individual within a family was known to be affected). With minimal knowledge of the causes of sporadic ALS (and hence, at least until recently, no ability to identify people at risk), attention has understandably focused on the genetic etiology of fALS, which accounts for 5%–10% of all cases of ALS.28,29 SOD1 mutations, responsible for ∼14%–20% of fALS28,30 (and therefore 1%–2% of all ALS cases), were the first identified. For almost 15 years, SOD1 remained virtually the only known cause of ALS. Within the last several years, however, there has been an explosion in our knowledge of other genetic causes of ALS, including, among others, mutations in TARDBP (TDP-43),31 FUS/TLS,32,33 VCP,34 Ubiquilin-2,35 and most significantly the recently discovered C9ORF72 gene.20,21 Collectively, these genetic mutations account for ∼10%–15% of all ALS. This substantial increase from just a few years ago creates an opportunity to identify many more people at genetic risk for ALS.
Ethical, legal, and psychosocial considerations.
The question of whether or not presymptomatic genetic testing should be offered for a fatal and untreatable neurodegenerative disease is not new. Presymptomatic genetic testing for Huntington disease, for example, has been available since 1986 and driven by respect for patient autonomy and the right to know.36 Genetic knowledge also empowers individuals and facilitates informed reproductive decisions. There are potential risks, but these can be mitigated by ensuring that consent is adequately informed and by probing each individual's psychosocial readiness to undergo presymptomatic testing.37,38 Other issues to consider are whether the results of presymptomatic testing should be made available to research participants, how to provide adequate pretest and post-test counseling for those who elect to learn their test results (including discussing issues such as inheritance patterns, incomplete penetrance, and discovery of polymorphisms of uncertain pathogenicity), and what follow-up is needed for those found not to harbor a genetic mutation. Equally important is the confidentiality and security of this genetic information, with implementation of the necessary safeguards to ensure that genetic knowledge does not compromise individuals' ability to secure health or life insurance, for example.
Logistical complexity.
Due to the rarity of ALS and the relative infrequency with which the genetic cause of disease is known, individuals potentially at genetic risk for ALS need to be recruited from across a broad geographic area. Even with multiple study sites, potential participants will likely reside some distance from a study center, imposing significant logistical complexity and cost. Initial screening for eligibility, obtaining consent, providing genetic counseling, and collecting blood for genetic testing may therefore be necessary before the study participant is seen in person at a study center. Once eligibility is confirmed, arrangements must then be made for the participant to travel to a study center. Moreover, in the absence of foreknowledge of the duration of the presymptomatic period, it is difficult to know how frequently gene mutation carriers should be evaluated. Ideally it should be sufficiently frequent that at least 1 evaluation takes place between disease onset and symptom onset, but not so frequently as to overburden participants and research personnel. Maintaining contact with participants between in-person evaluations is critical to minimizing loss to follow-up, especially if in-person study visits are relatively infrequent.
Methods for detecting presymptomatic disease.
A significant challenge to detecting presymptomatic disease is that the earliest manifestations of ALS are unknown. Although there is a wealth of investigative tools available—e.g., brain and spinal cord MRI, physiologic assessment of UMN and LMN function, and quantifiable “wet” biomarkers in the CSF or blood—their sensitivity to detect early manifestations of ALS has yet to be established. Moreover, standardizing and demonstrating reproducibility of these investigative modalities will require significant work. Constraints imposed by study participants' time and tolerance also limit the nature and number of procedures that can routinely be used. The growing recognition of the relationship between ALS and frontotemporal dementia (FTD) further complicates definition of presymptomatic disease. Early signs of cognitive/behavioral impairment in the absence of motor manifestations may reflect early FTD, but the individual should nonetheless be considered presymptomatic with respect to motor manifestations of ALS.
APPROACH: THE pre-fALS STUDY
We initiated the Pre-fALS study in 2007 as a systematic attempt to assemble and longitudinally follow a cohort of individuals at genetic risk for ALS. Pre-fALS is approved by the University of Miami institutional review board; all participants have provided informed consent. The explicit goals of Pre-fALS are to 1) characterize the presymptomatic phase of ALS using a wide range of biomarkers; 2) ascertain the incidence rate with which at-risk individuals progress to manifest disease; 3) explore potential genetic and environmental modifiers of the timing of progression to manifest disease; 4) establish a biobank of rigorously collected biological samples for “wet” biomarker discovery; and 5) use the knowledge gained to facilitate design of a preventive/early therapeutic trial. The Pre-fALS protocol has evolved since its inception to allow for adaptation to new scientific advances and development of new biomarkers, as well as input from the scientific and fALS community. The scope of Pre-fALS has also broadened in response to the identification of new genetic causes of ALS. The underlying goals, design, and approach, however, have remained unchanged. Here we use Pre-fALS (NCT00317616) as an example to illustrate how one might approach the aforementioned challenges.
Identifying an at-risk population.
The relative rarity of fALS has led us to identify and enroll pedigrees (figure 2, #1) from across the United States using both passive and active recruitment strategies. Before enrolling unaffected family members, we first determine the genetic cause of ALS in the family by testing (or obtaining results of prior testing from) an affected individual or an obligate carrier (figure 2, #2). When Pre-fALS first began, we only included SOD1 families, as SOD1 was at the time the only known (and relatively common) cause of ALS. We have since expanded Pre-fALS to include also families with more recently identified mutations (e.g., TARDBP, FUS, VCP, C9ORF72). Parenthetically, we use the term Pre-fALS study (or Pre-fALS for short) to encompass the extensive screening process to identify presymptomatic gene carriers; the term Pre-fALS cohort is reserved for the subset of Pre-fALS participants who are subsequently enrolled in longitudinal follow-up.
Figure 2. Pre-Symptomatic Familial ALS study schema showing recruitment of presymptomatic individuals from familial amyotrophic lateral sclerosis pedigrees in which the genetic cause of amyotrophic lateral sclerosis is known.

Family members known not to carry the mutated gene (“known gene−”) are excluded. Presymptomatic individuals known to carry the mutated gene (“known gene+”) are directly enrolled in longitudinal follow-up. Other presymptomatic individuals with a high likelihood of having inherited the mutated gene are offered genetic testing. Those who elect to learn the results of genetic testing (“disclosure”) are offered genetic counseling, and only gene carriers (“gene+”) are enrolled in longitudinal follow-up. For those who elect not to learn the results of genetic testing (“nondisclosure”), both carriers and noncarriers of mutated gene (“gene +/−”) are enrolled in the Pre-symptomatic Familial ALS (Pre-fALS) cohort. fALS = familial amyotrophic lateral sclerosis.
Once the genetic cause of ALS in the family is known, unaffected family members may be considered for Pre-fALS (figure 2, #3). We focus enrollment efforts on these first-degree relatives because they have the highest likelihood of having inherited the mutated gene, and genetic counseling is more readily performed for them. Moreover, this targeted recruitment approach provides an “enriched” study population, thereby maximizing our likelihood of identifying presymptomatic gene carriers for the Pre-fALS cohort. To date, no eligible individuals have declined participation.
After additional screening (e.g., for psychosocial readiness), those who are known to be gene mutation carriers through previous genetic testing are enrolled directly into the Pre-fALS cohort (figure 2, #4). Participants who do not know their genetic status are offered testing and the option of whether or not to learn the results (figure 2, #5); we refer to these as the “disclosure” and “nondisclosure” groups, respectively. Predecision counseling is provided as needed to help participants choose between disclosure and nondisclosure of results. Those who elect disclosure receive both pretest and post-test counseling (figure 2, #6) and are informed of the results; only gene mutation carriers are enrolled in the Pre-fALS cohort (figure 2, #7). Because obligate carriers do not always know that they carry the mutated gene, they too may receive genetic counseling before entering the Pre-fALS cohort. The nondisclosure group comprises a mixture of presymptomatic gene carriers and noncarriers (who may serve as controls in some of our biomarker studies). In order to avoid implicit disclosure of results, all participants who elect nondisclosure are enrolled in the Pre-fALS cohort, irrespective of genetic results (figure 2, #8).
Ethical, legal, and psychosocial considerations.
That Pre-fALS is a single-center study with participants recruited from across the United States raises the question of how best to provide adequate counseling for those who elect to learn their test results. During the first 3 years of Pre-fALS, participants who elected disclosure were randomized to receive counseling either via telephone or in person. The findings38 highlight the strengths and weaknesses of each approach and provided proof-of-principle that genetic counseling could adequately be performed remotely, at a significantly reduced logistical burden and cost. Based on these results, Pre-fALS procedures were modified such that counseling is by default provided telephonically, though participants may choose in-person counseling. Following pretest counseling, Pre-fALS participants may change their minds and elect instead not to learn their results. Similarly, those who initially elect nondisclosure have the option to learn their results at any stage after receiving appropriate genetic counseling.
A series of measures have been adopted to safeguard the confidentiality of genetic test results. Despite the challenges in the era of electronic medical record, research records are strictly separated from medical records. DNA samples are assigned an ID number that requires complex linkage to identifiable information, and all research personnel who have contact with study participants are blinded to the results of genetic testing for nondisclosure participants. In addition, we operate under an NIH certificate of confidentiality, which enables us to provide study participants with a legally enforceable guarantee of research record protection.
As part of the consenting process, participants are counseled that they will be informed if evidence of ALS is detected. The significance of early cognitive/behavioral symptoms is discussed in the context of specific gene mutations. This approach respects participants' right to know and affords the opportunity to initiate early treatment and participation in therapeutic trials.
Logistical complexity.
Telephone counseling has been used to overcome the logistical challenge of counseling those residing great distances from the study center. In addition, completing the array of study procedures performed at each in-person visit (outlined in the next section) requires careful coordination to ensure collection of biological samples in the fasting state, avoidance of lumbar puncture <24 hours prior to air travel, and availability of both equipment (e.g., MRI) and study staff with expertise in a diverse range of biomarker techniques. We currently schedule in-person visits every 12–18 months, and provide quarterly telephone interviews in between visits to ascertain the emergence of symptoms that might suggest the appearance of manifest disease. Participants are also urged to contact the study center at any time if they become aware of worrisome symptoms, which in turn prompt an ad hoc in-person visit for more detailed evaluation. In addition, we maintain contact with participants through telephone calls and a regular newsletter.
Methods for detecting presymptomatic disease.
The presymptomatic phase of ALS is anchored by disease onset and the appearance of motor manifestations. We use the revised El Escorial diagnostic criteria for fALS (progressive UMN or LMN signs in an individual with an ALS susceptibility gene mutation) to define clinically manifest disease based on careful examination by the same ALS specialist (M.B.) at each in-person visit. During the first 3 years of Pre-fALS, a second neurologist also performed independent evaluations, with complete agreement between the 2 neurologists in determining the presence/absence of clinically manifest disease. In light of the emerging connection between ALS and FTD, especially in families with FUS, TDP-43, VCP, and C9ORF72 mutations, detailed cognitive/behavioral testing has been added to the battery of evaluations. Supine and erect vital capacities are compared to detect subtle diaphragmatic weakness. Electromyographic examination of cranial, thoracic paraspinal, and limb muscles bilaterally is used to test for subclinical LMN dysfunction. Recognizing the need to explore a broad range of investigative modalities potentially sensitive to early manifestations of the disease process, this core set of evaluations is currently supplemented by biomarkers that aim to quantify UMN and LMN function: LMN numbers innervating hand and foot muscles are estimated using the modified incremental MUNE technique;39 multi-frequency electrical impedance myography, which quantifies the biomechanical properties of muscle,40 is employed to study muscles in both arms and legs; evidence of UMN pathology is sought using whole-brain and spinal cord MRS and DTI; fine quantitative motor testing41 provides an integrated view of UMN and LMN function. Finally, a range of biological samples (serum, plasma, DNA, RNA, buffy coat, urine, and, when possible, CSF and cell lines) are rigorously collected, processed, and stored for future “wet” biomarker discovery.
PROGRESS TO DATE AND THE WAY FORWARD
Through Pre-fALS we have found ways to overcome many of the obstacles that hinder the study of presymptomatic ALS. With >430 fALS pedigrees ascertained and the genetic cause identified in ∼100 of these families thus far, we have been inundated by the interest and willingness of unaffected family members to participate in Pre-fALS. By the end of May 2012, >160 subjects have been consented. Approximately 15% of them were known gene+ (figure 2), 15% chose nondisclosure, and 70%—a surprisingly high proportion—chose disclosure. A total of 60 participants from across the United States have been enrolled in the Pre-fALS cohort, with ∼95 person-years of follow-up accumulated by May 2012. Notably, 5 participants have converted from presymptomatic to manifest disease, yielding an estimated incidence rate of ∼6%/year. We have developed and continue to refine the aforementioned multimodal biomarker approach for detection of presymptomatic disease. Significant progress has also been made toward establishing a repository of longitudinally and rigorously collected biological samples from each subject at every visit.
Pre-fALS has benefited tremendously from scientific progress over the last few years, notably in the genetic and biomarker arenas. Scientific advances and growth of the Pre-fALS cohort, however, also bring new challenges. The increasingly diverse array of genetic causes of ALS, for example, adds to the complexity of genetic testing and counseling. The expanding array of potential biomarkers requires flexibility to adopt new techniques as they emerge and to triage methods that prove to be less promising. Moreover, the growing size of the Pre-fALS cohort may require a multicenter platform with the inevitable attendant logistical complexity.
Nevertheless, now that the cohort has been substantially seeded, we are poised to witness exponential growth over the coming years. With a modestly estimated ∼10 new Pre-fALS cohort recruits each year and continued follow-up of those already enrolled, we expect to accumulate >450 person-years of follow-up with a further ∼20 converters to manifest disease over the next 5 years. Over time, therefore, we are increasingly able to look back at the biomarker data collected prior to the emergence of clinically manifest disease. Such analyses offer the best opportunity to identify early markers of the disease process, and move us closer toward our goal of an early therapeutic or even preventive trial.
ACKNOWLEDGMENT
The authors thank their current and past research staff (Eliana Reyes, Natasha Garcia, Catalina Fernandez, Vanessa Vazquez, Julie Steele, Margaret Walker, Meraida Polak, Sharon Usher, Debbie Lu, and Sue Gronka) for their help with participant recruitment and evaluation; Dr. Taylor Harrison for performing the second neurological examination; Christine Stanislaw for providing genetic counseling; Drs. Peter Andersen, Bryan Traynor, and J. Paul Taylor for performing genetic testing; Dr. Merit Cudkowicz and the Northeast ALS (NEALS) consortium for early assistance with data management; colleagues around the country who have referred potential study participants; and, most of all, study participants as well as their family members for their commitment to and engagement in the research process.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- DTI
diffusion tensor imaging
- fALS
familial amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- LMN
lower motor neuron
- MN
motor neuron
- MRS
magnetic resonance spectroscopy
- MUNE
motor unit number estimation
- Pre-fALS
Pre-symptomatic Familial ALS study
- UMN
upper motor neuron
AUTHOR CONTRIBUTIONS
Michael Benatar contributed to all aspects of the work described in this article including study concept and design, obtaining funding, acquiring data, study supervision, and drafting/revising the manuscript. Joanne Wuu contributed to all aspects of the work described in this article including study design, obtaining funding, acquiring data, study supervision, statistical analysis, and drafting/revising the manuscript.
DISCLOSURE
M. Benatar receives research funding from the NIH, the Food and Drug Administration, the Muscular Dystrophy Association, the ALS Association, and TKCIS (subcontract from CDC/ATSDR); served as a site investigator in a multicenter study funded by Alexion Pharmaceuticals and currently serves on the safety monitoring committee of a clinical trial in myasthenia gravis that is funded by Cytokinetics Inc; and has received research support from CytRx Corporation and the Woodruff Foundation. J. Wuu has received research support from the NIH/NIA, the Food and Drug Administration, TKCIS (subcontract from CDC/ATSDR), the Muscular Dystrophy Association, the ALS Association, the Woodruff Foundation, Consolidated Anti-Aging Foundation, and the ALS Recovery Fund. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010; 9: 119– 128 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008; 79: 874– 880 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson's disease with [18F]dopa PET. J Neurol Neurosurg Psychiatry 1998; 64: 314– 319 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ross GW, Petrovitch H, Abbott RD, et al. Association of olfactory dysfunction with risk for future Parkinson's disease. Ann Neurol 2008; 63: 167– 173 . [DOI] [PubMed] [Google Scholar]
- 5. Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology 1996; 46: 388– 393 . [DOI] [PubMed] [Google Scholar]
- 6. Gowers W. Manual of Diseases of the Nervous System. London: J.A. Churchill; 1886 [Google Scholar]
- 7. Charcot J. Sclérose latérale amyotrophique. Oeuvres Complétes 1897; 2: 249– 266 . [Google Scholar]
- 8. Eisen A, Kim S, Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve 1992; 15: 219– 224 . [DOI] [PubMed] [Google Scholar]
- 9. Eisen A, Weber M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001; 24: 564– 573 . [DOI] [PubMed] [Google Scholar]
- 10. Chou SM, Norris FH. Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve 1993; 16: 864– 869 . [DOI] [PubMed] [Google Scholar]
- 11. Ravits J, Laurie P, Fan Y, Moore DH. Implications of ALS focality: rostral-caudal distribution of lower motor neuron loss postmortem. Neurology 2007; 68: 1576– 1582 . [DOI] [PubMed] [Google Scholar]
- 12. Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007; 68: 1571– 1575 . [DOI] [PubMed] [Google Scholar]
- 13. Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 2009; 73: 805– 811 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chio A, Mora G, Calvo A, Mazzini L, Bottacchi E, Mutani R. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology 2009; 72: 725– 731 . [DOI] [PubMed] [Google Scholar]
- 15. Murphy M, Quinn S, Young J, Parkin P, Taylor B. Increasing incidence of ALS in Canterbury, New Zealand: a 22-year study. Neurology 2008; 71: 1889– 1895 . [DOI] [PubMed] [Google Scholar]
- 16. Logroscino G, Traynor BJ, Hardiman O, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry 2010; 81: 385– 390 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Toole O, Traynor BJ, Brennan P, et al. Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J Neurol Neurosurg Psychiatry 2008; 79: 30– 32 . [DOI] [PubMed] [Google Scholar]
- 18. Beghi E, Millul A, Micheli A, Vitelli E, Logroscino G. Incidence of ALS in Lombardy, Italy. Neurology 2007; 68: 141– 145 . [DOI] [PubMed] [Google Scholar]
- 19. Aggarwal A, Nicholson G. Normal complement of motor units in asymptomatic familial (SOD1 mutation) amyotrophic lateral sclerosis carriers. J Neurol Neurosurg Psychiatry 2001; 71: 478– 481 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245– 256 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72: 257– 268 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benatar M, Polak M, Kaplan S, Glass J. Preventing familial amyotrophic lateral sclerosis: Is a clinical trial feasible? J Neurol Sci 2006; 251: 3– 9 . [DOI] [PubMed] [Google Scholar]
- 23. Vucic S, Winhammar JM, Rowe DB, Kiernan MC. Corticomotoneuronal function in asymptomatic SOD-1 mutation carriers. Clin Neurophysiol 2010; 121: 1781– 1785 . [DOI] [PubMed] [Google Scholar]
- 24. Aggarwal A, Nicholson G. Detection of preclinical motor neurone loss in SOD1 mutation carriers using motor unit number estimation. J Neurol Neurosurg Psychiatry 2002; 73: 199– 201 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008; 131: 1540– 1550 . [DOI] [PubMed] [Google Scholar]
- 26. Ng MC, Ho JT, Ho SL, et al. Abnormal diffusion tensor in nonsymptomatic familial amyotrophic lateral sclerosis with a causative superoxide dismutase 1 mutation. J Magn Reson Imaging 2008; 27: 8– 13 . [DOI] [PubMed] [Google Scholar]
- 27. Carew JD, Nair G, Andersen PM, et al. Presymptomatic spinal cord neurometabolic findings in SOD1-positive people at risk for familial ALS. Neurology 2011; 77: 1370– 1375 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chio A, Traynor BJ, Lombardo F, et al. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 2008; 70: 533– 537 . [DOI] [PubMed] [Google Scholar]
- 29. Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2011; 82: 623– 627 . [DOI] [PubMed] [Google Scholar]
- 30. Rosen D, Siddique T, Patterson D. Mutations in Cu. Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362: 59– 62 . [DOI] [PubMed] [Google Scholar]
- 31. Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 2008; 7: 409– 416 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323: 1208– 1211 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009; 323: 1205– 1208 . [DOI] [PubMed] [Google Scholar]
- 34. Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010; 68: 857– 864 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011; 477: 211– 215 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. World Federation of Neurology: Research Committee: Research Group on Huntington's Chorea. Ethical issues policy statement on Huntington's disease molecular genetics predictive test. J Neurol Sci 1989; 94: 327– 332 . [DOI] [PubMed] [Google Scholar]
- 37. Fanos JH, Gelinas DF, Miller RG. “You have shown me my end”: attitudes towards presymptomatic testing for familial amyotrophic lateral sclerosis. Am J Med Genet 2004; 129 A:248– 253 . [DOI] [PubMed] [Google Scholar]
- 38. Fanos JH, Gronka S, Wuu J, Stanislaw C, Andersen PM, Benatar M. Impact of presymptomatic genetic testing for familial amyotrophic lateral sclerosis. Genet Med 2011; 13: 342– 348 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shefner JM, Watson ML, Simionescu L, et al. Multipoint incremental motor unit number estimation as an outcome measure in ALS. Neurology 2011; 77: 235– 241 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rutkove SB. Electrical impedance myography: background, current state, and future directions. Muscle Nerve 2009; 40: 936– 946 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bechtel N, Scahill RI, Rosas HD, et al. Tapping linked to function and structure in premanifest and symptomatic Huntington disease. Neurology 2010; 75: 2150– 2160 . [DOI] [PMC free article] [PubMed] [Google Scholar]