

Abstract
Sphingomonas wittichii RW1 is a bacterium of interest due to its ability to degrade polychlorinated dioxins, which represent priority pollutants in the USA and worldwide. Although its genome has been fully sequenced, many questions exist regarding changes in protein expression of S. wittichii RW1 in response to dioxin metabolism. We used difference gel electrophoresis (DIGE) and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) to identify proteomic changes induced by growth on dibenzofuran, a surrogate for dioxin, as compared to acetate. Approximately 10% of the entire putative proteome of RW1 could be observed. Several components of the dioxin and dibenzofuran degradation pathway were shown to be upregulated, thereby highlighting the utility of using proteomic analyses for studying bioremediation agents. This is the first global protein analysis of a microorganism capable of utilizing the carbon backbone of both polychlorinated dioxins and dibenzofurans as the sole source for carbon and energy.
1. Introduction
Dioxins are some of the most widely studied and prevalent anthropogenic environmental pollutants. Common sources include backyard burning, incineration of plastics, and chlorine bleaching of pulp in paper mills [1]. Documented health effects include acute and chronic effects, including chloracne, various types of cancers, reproductive diseases, circulatory and respiratory diseases, and diabetes [2]. The traditional approach to environmental remediation includes a host of physical and chemical methods, depending on the characteristics of the polluted site and the extent of contamination present [3]. Bioremediation, which is the use of biogenic materials and organisms for environmental cleanup, has also been proposed, including phytoremediation using plants [4] and microbial degradation using primarily bacteria and fungi [5]. Bioremediation is an attractive strategy, as it can destroy the pollutant rather than transferring it from one environmental compartment to another. It also can be less expensive than physical strategies [6]. Common bioremediation strategies include the addition of nutrients, degradative microorganisms, or both.
Sphingomonas wittichii RW1 is a microorganism of great interest to the bioremediation community for its ability to biotransform a large number of toxic polychlorinated dioxins and to utilize both nonchlorinated dibenzo-p-dioxin and nonchlorinated dibenzofuran as a growth substrate and sole source of carbon and energy [5].
One of the major challenges in bioaugmentation strategies relying on the addition of nonnative microbes is to ensure their viability and degradative activity toward the target compounds. Monitoring bioremediation is critical to ensure the efficacy of the process and the reduction of contaminant mass to acceptable levels. Traditionally, the most important characteristics investigated for microorganisms used in bioremediation were their ability to transform the substrate, the rate of substrate removal, and the resulting metabolites [6–9]. To optimize the bacterial degradation of pollutants, it is important to understand how these organisms function during growth on recalcitrant substrates and which factors influence their degradative abilities. This includes analyzing not only the degradative pathways [10–12], but also the peripheral processes and mechanisms that are involved in taxis (i.e., directed motion in a chemical gradient), uptake, and transport during exposures to specific substrates. Analysis of DNA and RNA [13] can shed light on an organism's metabolic potential; however, these measurements poorly correlate to actual protein expression profiles [14]. Therefore, global analyses of protein expression profiles may be a more informative tool for understanding the physiological mechanisms of biodegradation. In addition to identifying important degradative enzymes in a variety of important microbes [15–17], proteomic studies have opened the door to a better understanding of system-wide changes in response to differing substrates [18].
The imperative to perform proteomic analyses is particularly true for S. wittichii RW1 because the enzymes in the dioxin degradation pathway are encoded on different loci throughout the genome [19], certain elements in the pathway are located on a plasmid [20], and there may be alternative pathways at work [21]. The present study builds on previous work [22] and utilized difference gel eletrophoresis (DIGE) coupled with mass spectrometry (MS) to exploit recently gathered RW1 genome data [23]. When used together, these tools yield information on the response of cells of S. wittichii RW1 to dioxin exposure and the bacterium's degradative activity toward this recalcitrant compound.
The aim of this study was to investigate system-wide changes in protein expression during growth on dibenzofuran, a nontoxic surrogate for dibenzo-p-dioxin, as compared to nonselective growth media. Acetate was selected as the nonselective alternate substrate, as growth on this compound was observed to influence expression of select proteins, including the dioxin dioxygenase [24]. Thus, any changes measured in response to cells grown on dibenzofuran should represent cell-wide effects related specifically to the growth substrate and not to unanticipated extraneous effects. This work constitutes the first global assessment of protein expression by S. wittichii RW1.
2. Materials and Methods
2.1. Culture Maintenance
Cultures of S. wittichii strain RW1 (100 mL to 1.0 L) were grown to mid log phase at 30°C in M9 phosphate-buffered minimal medium (pH 7.05) supplemented with either dibenzofuran crystals or 50 mM acetate as growth substrates. Saturated dibenzofuran medium contained approximately 3–5 mg/L of the selective growth substrate in the dissolved phase. Cells were grown overnight on dibenzofuran and acetate as sole carbon sources to an optical density of 0.4–0.6 absorbance units (λ560 nm). Following biomass processing, protein levels in the samples were on the order of 75–200 μg/mL. Protein concentrations were normalized prior to analysis by concentration and resuspension in DIGE sample preparation buffer. Culture purity was confirmed by the streak plate method using Luria Bertani medium supplemented with 1.5% agar.
2.2. Protein Extraction and Cleanup
Cultures were centrifuged at 3,000–5,000 xg for 10 minutes at 4°C. Harvested biomass was washed, spun again, and the resultant pellet suspended in a small volume of 100 mM ammonium bicarbonate (pH ~7.0). This microbial suspension was then sonicated under cooling with ice, using a microtip sonicator (Fisher Scientific, Pittsburgh, PA, USA) in a sequence of three 10-second bursts delivered in thirty-second intervals. The sonicated cells were then immediately centrifuged at 10,000 xg for 10 minutes at 4°C. The supernatant was collected and the protein purified by trichloroacetic acid (TCA)/acetone precipitation. Briefly, 8 parts of 10% TCA in acetone (−20°C) were added per volume of supernatant and, following mixing on a vortex, the resultant dilution was incubated at −20°C overnight. Following centrifugation (10,000 xg, 10 minutes, 4°C), harvested biomass was washed in cold acetone for 10 minutes at −20°C. Following a subsequent centrifugation, the pellet was resuspended in sample preparation buffer (7 M urea, 2 M thiourea, 2% CHAPS, 0.2% DTT, 0.02% bromophenol blue) and stored at −20°C until analyzed. Protein concentrations were measured using the bicinchoninic acid assay (Pierce, Rockford, IL, USA) following dilution to reduce the concentration of interfering agents.
2.3. DIGE Labeling
Twenty-five μg of crude cell lysates of RW1 biological replicates grown on dibenzofuran (n = 3) and acetate (n = 3) were labeled using Cy dyes (GE Healthcare) as described elsewhere [25]. Briefly, samples were adjusted to 1 μg/μL using sample preparation buffer and the pH checked. Subsequently, 0.25 μL of 1 pmol/μL Cy dyes were added to samples for 30 minutes in the dark on ice. To stop the labeling reaction, 0.5 μL of 10 mM lysine was added to the samples, which were mixed and incubated on ice for 10 minutes prior to storage at −20°C until analysis.
2.4. 2D-DIGE
Unless stated otherwise, all procedures were carried out in the dark or minimal light to protect the integrity of the fluorescent dyes. Samples were randomized to reduce the effect of dye bias and in-gel variations. A global pool consisting of fractions of each sample was labeled as outlined above using Cy 2 and added to each sample as an internal standard. One Cy-3- and one Cy-5-labeled sample were added to each gel, as defined by the experimental randomizing procedures. To each sample, an additional 175 μg of unlabeled sample were added; the volume was increased to a total of 450 μL using sample preparation buffer. The reducing agent DTT (dithiothreitol; 1.3 mg per tube) and IPG (immobilized pH gradient) buffer (0.5%) were added, and the samples incubated and mixed in the dark at room temperature for approximately 1 h. Samples were then applied to 24 cm pH 4–7 IPG strips (GE Healthcare) and focused for 60 kVh using the following protocol: 12 h rehydration at 30 V; 1 h step and hold at 500 V; 7 hour gradient to 1,000 V; step and hold at 1,000 V for 1 hour; gradient to 8,000 V for 3 h; step and hold at 8,000 V until 60 kVh. Strips were then reduced and equilibrated using 10 mg/mL DTT (15 min) followed by 25 mg/mL iodoacetamide (15 min). The IPG strips were overlaid on 24 × 26 cm 8–16% gradient Tris-HCl pH 8.8 precast gels (NextGen Sciences, Ann Arbor, MI, USA) cast between low-fluorescing glass plates. Approximately 1 mL of agarose was applied to fix the gels and a Cy-2-labeled molecular weight marker was applied adjacent to the acidic side of the strip. The gels were then run 1-2 W per gel overnight (~22–24 hours) at 20°C until the marker dye ran off the gel. Gels were then imaged with a Typhoon 9400 scanner and processed using DeCyder v6.5 (GE Healthcare) BVA batch processor tool. Gels were poststained using a silver stain as described previously [26]. Images were uploaded to DeCyder (version 6.5) and spurious image objects (water spots, streaks, and mismatches) were identified and excluded from further analysis. Following allocation of changed proteins, individual spots were manually inspected and excluded from analysis if they fell outside acceptable parameters for peak height, area, and slope.
2.5. Gel Picking and Protein Digestion
Pick lists were generated by selecting proteins whose expression was statistically changed in the two growth conditions (P < 0.05) following digital image analysis using DeCyder, and the corresponding spots were automatically picked using an Ettan Spot Picker (GE Healthcare) with Ettan Spot Pick Software v.1.1. Spots were delivered in 100 mM ammonium bicarbonate to a 96-well plate and digested using established protocols. Briefly, gel pieces were sequentially dried using three exchanges of 100% acetonitrile followed by a 10-minute SpeedVac (Savant) drying. Gel pieces were rehydrated in 40 μL of 10 ng/μL trypsin in 100 mM NH4HCO3 on ice for 45 minutes. The supernatant was removed and replaced with 100 mM NH4HCO3 and digested at 37°C overnight. Peptides were then extracted using 50% acetonitrile/0.1% TFA (trifluoroacetic acid) for 30 minutes at 37°C. The peptides were microextracted using Omix C18 tips (Varian, Palo Alto, CA, USA) following the manufacturer's instructions and then deposited on a stainless-steel target plate in a matrix consisting of 10 mg/mL 2,5-dihydroxybenzoic acid.
2.6. MS and Database Searching
Mass spectra were acquired using a Voyager DE-STR matrix-assisted laser desorption/ionization time-of-flight MS (Applied Biosystems, Foster City, CA, USA) in positive reflector mode with delayed extraction using the following parameters: laser energy, 1400 arbitrary units; mass range, 500–5,000 Da; 120 nsec delay, 100 laser shots per spectrum. External calibration was conducted using a standard peptide mixture (bradykinin, insulin B chain, P14R, and ACTH), and internal calibration was carried out using trypsin autolysis peaks. Data were processed in Data Explorer v1.1 (Applied Biosystems, Foster City, CA, USA) using noise reduction (2 standard deviations) and peak deisotoping. Peak masses were searched using the Mascot online search engine (http://www.matrixscience.com/) with the following settings: database, NCBI entire database (5.6 million entries); no missed cleavages; monoisotopic peaks; no fixed modifications; variable modification of methionine oxidation; error tolerance of 150 ppm. Protein identifications were mapped back to the gel using DeCyder v6.5. Database and literature searches were used to further characterize and classify the proteins identified by MALDI-TOF MS. Where ambiguous names were encountered, BLASTp searches [27] were used to identify homologous proteins from orthologous species.
3. Results
Image analysis of 24 cm 2D-DIGE gels loaded with protein of S. wittichii RW1 cells grown on either dibenzofuran or acetate revealed 937 unique spots. Differential in-gel analysis of individual gels determined gel-specific parameters for selection criteria and allowed visual examination of changes between growth on the two substrates (Figure 1). Of the 937 identified spots, 595 were matched between all the gels used to statistically compare the quantitative abundance of proteins. Statistical analysis compared triplicate biological observations for each condition, normalized to the internal pooled standard (Figure 2).
Figure 1.

A representative 2D-DIGE gel of RW1 showing the contrast in protein expression between cells grown on acetate and dibenzofuran. Proteins that are expressed equally for the two conditions are marked with an orange arrow; differentially expressed proteins are shown with green (higher on acetate) or red (higher on dibenzofuran) arrows.
Figure 2.
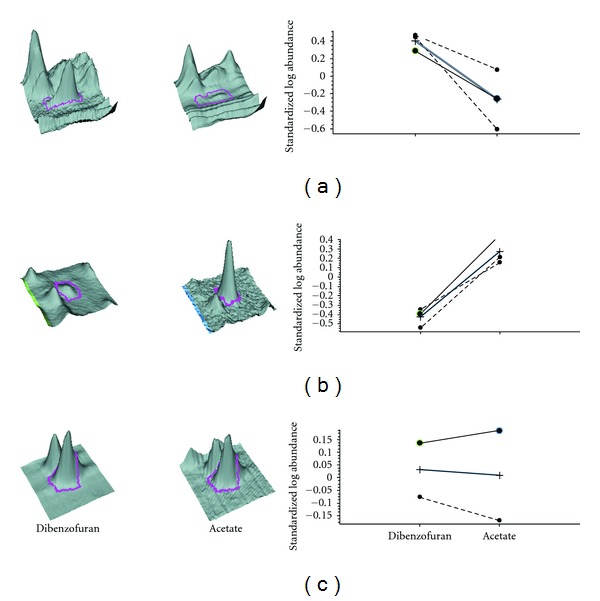
Statistics and spot information for three representative spots from DIGE analysis of the RW1 crude cell proteome. Following visual examination of the spot characteristics and matching parameters, spots were identified as increased ((a), gi∣148555952 glyoxalase/bleomycin resistance protein/dioxygenase), decreased ((b), unidentified protein), or unchanged ((c), gi∣148553776 OmpA/MotB domain protein).
Crude cell lysates from S. wittichii RW1 grown on dibenzofuran showed that, of all proteins observed, 22 proteins were modulated in response to changes in culture conditions. These candidate biomarkers of metabolic activity and phenotype were observed in at least 6 of 9 DIGE images and were modulated as follows: 16 showed an apparent increase and 6 an apparent decrease (Figure 2). These proteins, along with 22 proteins selected due to their high abundance in both growth conditions, were further analyzed and identified using mass spectrometry (Figure 3). A Mascot search of the entire NCBI database using mass spectral data generated by peptide mass fingerprinting identified 23 of the 44 proteins (52%). All protein identifications corresponded to the genome of S. wittichii strain RW1. Among the 16 proteins upregulated during growth on dibenzofuran, 7 were successfully identified (Table 1). Among the 6 proteins downregulated during growth on dibenzofuran, 3 were successfully identified (Table 2). An additional 13 proteins were identified whose expression level remained unchanged regardless of culture conditions (Table 3).
Figure 3.

A scanned and cropped 24 cm gel showing annotations for spots selected for further analysis. The spots were selected due to (i) their relative increase or decrease during growth on the selected substrate or (ii) their high abundance in both samples. The spot numbers are identifiers for the quantitative information and identity; identified spots are marked with an orange flag (see Tables 1–3). The approximate pH range (horizontal) is 4–7, and the approximate molecular weight (vertical) is 110 kDa to 10 kDa.
Table 1.
Proteins identified as being increased in S. wittichii RW1 during growth on dibenzofuran when compared to growth on acetate. The spots were identified on a minimum of 6 gel images.
| Master numbera | Protein accessionb | Gene locus | Protein name | Average ratioc | t-testd |
|---|---|---|---|---|---|
| 919 | 148556489 | Swit_3587 | Alkyl hydroperoxide reductase/thio-specific antioxidant/Mal allergen | 1.52 | 0.099 |
| 922 | 148553900 | Swit_0977 | Catechol 1,2-dioxygenase | 1.8 | 0.078 |
| 921 | 148555809 | Swit_2901 | Putative cold shock DNA-binding domain | 1.89 | 0.078 |
| 539 | 148555586 | Swit_2674 | Adenosylhomocysteinase | 1.97 | 0.062 |
| 418 | 148553385 | Swit_0461 | Elongation factor Ts | 2.10 | 0.05 |
| 934 | 115279619 | Swit_3055 | Meta-cleavage pathway hydrolase | 2.54 | 0.055 |
| 603 | 148555952 | Swit_3046 | Glyoxalase/bleomycin resistance protein/ dioxygenase | 3.88 | 0.031 |
aArbitrary identifier for spot location (see Figure 2).
bNCBI gi∣ number.
cUsing internal standard as 1.0, >1 is an increase and <1 is a decrease in abundance.
dStudent's t-test comparing spot intensity for acetate versus dibenzofuran grown cells.
Table 2.
Proteins identified as decreased in S. wittichii RW1 during growth on dibenzofuran as compared to acetate. The spots were identified on a minimum of 6 gel images.
| Master numbera | Protein accessionb | Gene locus | Protein name | Average ratioc | t-test d |
|---|---|---|---|---|---|
| 821 | 148551036 | Swit_5089 | Fumarylacetoacetate hydrolase | −1.99 | 0.063 |
| 470 | 148553574 | Swit_0650 | Acyl-CoA dehydrogenase domain | −1.74 | 0.031 |
| 161 | 148550568 | Swit_5129 | TonB-dependent receptor | −1.59 | 0.086 |
a Arbitrary identifier for spot location (see Figure 2).
bNCBI gi∣ number.
cUsing internal standard as 1.0, >1 is an increase and <1 is a decrease in abundance.
dStudent's t-test comparing spot intensity for acetate versus dibenzofuran grown cells.
Table 3.
Highly abundant proteins identified as unchanged in S. wittichii RW1 during growth on dibenzofuran or acetate. The spots were identified on a minimum of 6 gel images.
| Master numbera | Protein accessionb | Gene locus | Protein name | Average ratioc | t-testd |
|---|---|---|---|---|---|
| 916 | 148556048 | Swit_3144 | TonB-dependent receptor | −1.33 | 0.23 |
| 929 | 148553835 | Swit_0912 | 4-Oxalocrotonate decarboxylase | −1.26 | 0.25 |
| 256 | 148556278 | Swit_3376 | Chaperonin GroEL | −1.21 | 0.5 |
| 453 | 148553821 | Swit_0898 | Phenylpropionate dioxygenase, ferredoxin reductase subunit | −1.17 | 0.5 |
| 741 | 148553833 | Swit_0910 | Alpha/beta hydrolase fold | −1.08 | 0.9 |
| 933 | 148555961 | Swit_3055 | Meta-cleavage product hydrolase | −1.03 | 0.85 |
| 932 | 148553776 | Swit_0853 | OmpA/MotB domain | −1.00 | 0.93 |
| 749 | 148553834 | Swit_0911 | 4-Oxalocrotonate decarboxylase | 1.09 | 0.3 |
| 927 | 148555704 | Swit_2794 | Opacity protein and related surface antigen-like protein | 1.09 | 0.82 |
| 648 | 148550878 | Swit_4921 | 3-Keto-5-aminohexanoate cleavage enzyme | 1.15 | 0.45 |
| 177 | 148555643 | Swit_2731 | Aconitate hydratase 1 | 1.23 | 0.44 |
| 479 | 148550877 | Swit_4920 | FAD-dependent pyridine nucleotide-disulphide oxidoreductase | 1.57 | 0.5 |
| 937 | 148550856 | Swit_4897 | Dioxin dioxygenase, alpha subunit | 1.59 | 0.38 |
a Arbitrary identifier for spot location (see Figure 2).
bNCBI gi∣ number.
cUsing internal standard as 1.0, >1 is an increase and <1 is a decrease in abundance.
dStudent's t-test comparing spot intensity for acetate versus dibenzofuran grown cells.
Of the 7 identified proteins increased during growth on dibenzofuran, 2 were directly related to the dibenzofuran degradation pathway (Figure 4); the others were involved in downstream metabolic processes (catechol 1,2-dioxygenase, adenosylhomocysteinase), cell growth (elongation factor Ts), and cell protection (cold shock DNA-binding domain protein, alkyl hydroperoxide reductase). The three identified proteins whose expression was decreased (fumarylacetoacetate hydrolase, TonB-dependent receptor, and acyl-CoA dehydrogenase) are involved in biosynthesis, catabolism, and transport. The unchanged proteins represented basic cell functions, although biosynthesis, catabolism, and transport proteins dominated the identities. The alpha subunit of the dioxin dioxygenase, the first step in the dioxin degradation pathway, was also identified as unchanged.
Figure 4.
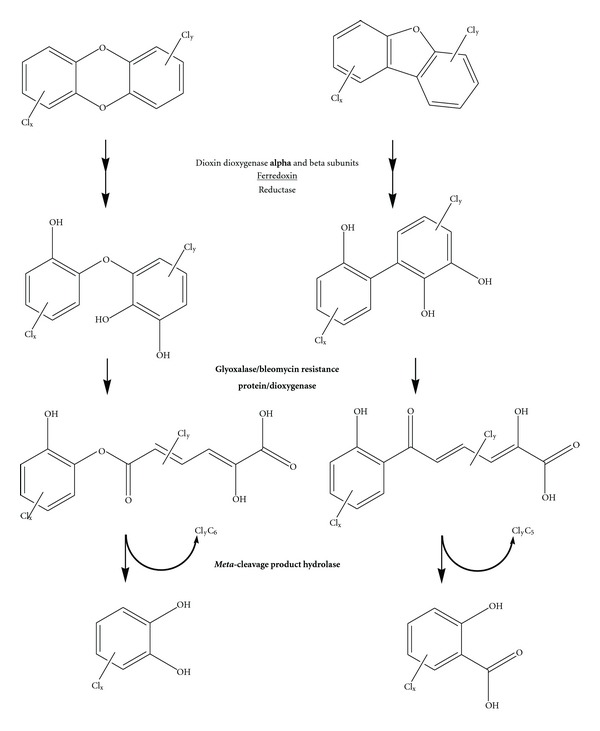
The dioxin (left) and dibenzofuran (right) degradation pathways. Enzymes catalyzing individual reactions are written in the center. Bold typeface indicates a protein that was found to be increased in expression. The ferredoxin (underlined) is located in an operon whose expression was found to be decreased. ClyC5 and ClyC6 represent potentially chlorinated aliphatic moieties.
4. Discussion
DIGE and 2D electrophoresis are an accepted strategy for mining microbial proteomes for biomarkers related to a number of processes [28–30]. The complete protein content of S. wittichii RW1 consists of approximately 5,000 putative proteins from the bacterial chromosome and two megaplasmids. Using simple extraction and purification techniques followed by DIGE, over 500 protein spots were resolved on a large (24 cm) 2-dimensional gel and matched between the three biological replicates, representing approximately 10% of the entire protein content. Of these 500 proteins, 22 were found to be regulated in response to growth condition changes.
Only one study has examined transcriptomic data from S. wittichii RW1 [31]. This study examined the effect of solute perturbation on RW1 grown on salicylate. Of the genes they identified as being changed in their experiments, only two overlapped with the proteins identified in our study: Swit_3144 (TonB-dependent receptor, downregulated) and Swit_3376 (Chaperonin GroEL, upregulated). Both of these gene levels responded to short-term perturbation with PEG8000 but not sodium chloride in the transcriptomic study and were unchanged in our study.
In our study, we also identified a number of proteins that are related to dioxin/dibenzofuran degradation (e.g., dioxin dioxygenase, meta-cleavage product hydrolase, and 2,3-dihydroxybiphenyl 1,2-dioxygenase). Other proteins were identified that showed increases in abundance but whose role was not directly related to the dibenzofuran degradation pathway. The increase in the presence of antioxidants such as alkyl hydroperoxide reductase suggests that there is an increasing stress upon the bacterial cell during growth on dibenzofuran, perhaps due to a change in catabolism resulting in an increase in endogenous peroxide generation [32]. Increases in a cold-shock DNA-binding protein may be further evidence of an increased cellular stress [33]. However, proteins of the cold shock family and related ones are also known to have transport and protein processing roles [34].
Among the proteins in the dioxin degradation pathway, the most prominent on the gel was the meta-cleavage product hydrolase. This identification was produced from two adjacent spots, likely representing an artifact due to the protein's extremely high expression or a reflection of the presence of multiple isoforms or a modified enzyme. S. wittichii RW1 has three known isoforms of this meta-cleavage product hydrolase [21]. The one identified in the present study is the product of the Swit_3055 locus, a gene also known as DxnB2 [21]. Its identification in this study corroborates previous findings [21]. Unlike the dioxin dioxygenase, this gene is found on the chromosome.
The glyoxalase/bleomycin resistance protein/dioxygenase identified in this study is also annotated as a 2,3-dihydroxybiphenyl-1,2-dioxygenase located on the chromosome at the Swit_3046 locus. Again, there are multiple isoforms of this enzyme found both on the chromosome and the megaplasmids. The KEGG dioxin degradation pathway identifies Swit_4182 as the dihydroxybiphenyl dioxygenase involved in biphenyl metabolism and Swit_4902 as the trihydroxybiphenyl dioxygenase in both dioxin and dibenzofuran metabolism. These genes have all been annotated as glyoxalase/bleomycin resistance protein/dioxygenase. The increased expression in response to dibenzofuran suggests that the Swit_3046 dioxygenase plays a more important role in dibenzofuran degradation in vivo.
The high degree of redundancy in the dioxin and dibenzofuran degradation pathways, that is, the presence of multiple ring-hydroxylating alpha and beta subunits, glyoxalase/bleomycin resistance protein/dioxygenases, and meta-cleavage product hydrolases, remains to be explained. One possibility is that the various isoforms have different affinities for chlorinated metabolites that would result from chlorinated dioxins and furans. Further experiments are needed to fully distinguish the roles of these enzymes in S. wittichii RW1 degradation pathways.
Although not directly implicated in dioxin degradation, the fumarylacetoacetate hydrolase is also of interest because the gene encoding this protein (Swit_5089) flanks the ferredoxin Fdx1 (Swit_5088) that has been identified as part of the electron supply chain supporting dioxin dioxygenase activity. Of the multiple isoforms of this enzyme, Fdx1 was found to function in vitro with the dioxin dioxygenase [35]. The electron supply chain also contains two isofunctional reductases [36]. Neither the ferredoxin itself nor the reductases could be identified. In previous studies, the reductase was present as a much smaller fraction of the soluble cell proteome than either the ferredoxin or the dioxin dioxygenase [36], so gel-based methods may not be sensitive enough to detect this protein. If transcription of the ferredoxin is linked to the other genes at that locus, as is predicted [35], the decreased expression in response to dibenzofuran suggests that another ferredoxin is more important in the dioxin degradation pathway in vivo. Further studies are needed to confirm this hypothesis.
The detection of the dioxin dioxygenase alpha subunit and related enzymes in both acetate- and dibenzofuran-grown cells is potentially of importance for the field of bioremediation because it suggests an avenue of biostimulation. When utilizing S. wittichii RW1 as a bioremediation agent, it may be possible to induce the expression of the dioxin degradation pathway using acetate. Induction of the dioxin degradation pathway has not been observed when S. wittichii RW1 is grown on glucose or rich medium [22], and growth in a complex environmental medium (landfill leachate) was correlated with a decrease in copy number of the gene encoding the dioxin dioxygenase alpha subunit [37]. Previous studies using S. wittichii RW1 to transform chlorinated dioxins in soil or fly ash have observed a progressive decrease in degradative activity [5] or viable cells [38, 39], respectively. The addition of acetate may generate sufficient relevant protein biomass to catalyze the successful degradation of dioxin and dioxin-like compounds in environments bioaugmented with S. wittichii RW1.
Proteomic technology has emerged in microbiology more rapidly than in other fields for several reasons. The relatively small genomes code for relatively limited proteomes featuring no or very limited posttranslational modifications compared to higher organisms [40]. Furthermore, microbes are easily controlled and manipulated in the laboratory, both during growth and gene expression. These factors will continue to drive biomarker discovery in microbial proteomes, including phenotypic biomarkers informing on the degradative activity of biomass produced for bioaugmentation of contaminated environments. Furthermore, the field of bioremediation can benefit from methods suitable for monitoring microbial biomarkers in field samples to inform on progress in site bioremediation. This study highlights a number of proteins that were changed in response to dibenzofuran exposure, opens the door to a greater understanding of how S. wittichii RW1 performs and regulates the degradation of dioxins, and suggests ways to enhance the biodegradation of dioxins.
Authors' Contribution
These authors contributed equally to this work.
Acknowledgments
This project was supported in part by Award Number R01ES015445 from the National Institute of Environmental Health Sciences (NIEHS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS or the National Institutes of Health (NIH).
References
- 1.Steenland K, Deddens J. Dioxin: exposure-response analyses and risk assessment. Industrial Health. 2003;41(3):175–180. doi: 10.2486/indhealth.41.175. [DOI] [PubMed] [Google Scholar]
- 2.Bertazzi PA, Consonni D, Bachetti S, et al. Health effects of dioxin exposure: a 20-year mortality study. American Journal of Epidemiology. 2001;153(11):1031–1044. doi: 10.1093/aje/153.11.1031. [DOI] [PubMed] [Google Scholar]
- 3.Kulkarni PS, Crespo JG, Afonso CAM. Dioxins sources and current remediation technologies: a review. Environment International. 2008;34(1):139–153. doi: 10.1016/j.envint.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Campanella BF, Bock C, Schröder P. Phytoremediation to increase the degradation of PCBs and PCDD/Fs: potential and limitations. Environmental Science and Pollution Research. 2002;9(1):73–85. doi: 10.1007/BF02987318. [DOI] [PubMed] [Google Scholar]
- 5.Halden RU, Halden BG, Dwyer DF. Removal of dibenzofuran, dibenzo-p-dioxin, and 2-chlorodibenzo-p-dioxin from soils inoculated with Sphingomonas sp. strain RW1. Applied and Environmental Microbiology. 1999;65(5):2246–2249. doi: 10.1128/aem.65.5.2246-2249.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wackett LP, Hershberger CD. Biocatalysis and Biodegradation: Microbial Transformation of Organic Compounds. 1st edition. Washington, DC, USA: ASM Press; 2001. [Google Scholar]
- 7.Wittich RM, Wilkes H, Sinnwell V, Francke W, Fortnagel P. Metabolism of dibenzo-p-dioxin by Sphingomonas sp. strain RW1. Applied and Environmental Microbiology. 1992;58(3):1005–1010. doi: 10.1128/aem.58.3.1005-1010.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halden RU, Dwyer DF. Biodegradation of dioxin-related compounds: a review. Bioremediation Journal. 1997;1(1):11–25. [Google Scholar]
- 9.Halden RU, Tepp SM, Halden BG, Dwyer DF. Degradation of 3-phenoxybenzoic acid in soil by Pseudomonas pseudoalcaligenes POB310(pPOB) and two modified Pseudomonas strains. Applied and Environmental Microbiology. 1999;65(8):3354–3359. doi: 10.1128/aem.65.8.3354-3359.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basta T, Keck A, Klein J, Stolz A. Detection and characterization of conjugative degradative plasmids in xenobiotic-degrading Sphingomonas strains. Journal of Bacteriology. 2004;186(12):3862–3872. doi: 10.1128/JB.186.12.3862-3872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bünz PV, Schmidt S. The microbial degradation of halogenated diaryl ethers. Biotechnology Advances. 1997;15(3-4):621–632. doi: 10.1016/s0734-9750(97)00040-2. [DOI] [PubMed] [Google Scholar]
- 12.Halden RU, Halden BG, Dwyer DF. Transformation of mono- and dichlorinated phenoxybenzoates by phenoxybenzoate-dioxygenase in pseudomonas pseudoalcaligenes POB310 and a modified diarylether-metabolizing bacterium. Biotechnology and Bioengineering. 2000;69(1):107–112. [PubMed] [Google Scholar]
- 13.Gonçalves ER, Hara H, Miyazawa D, Davies JE, Eltis LD, Mohn WW. Transcriptomic assessment of isozymes in the biphenyl pathway of Rhodococcus sp. strain RHA1. Applied and Environmental Microbiology. 2006;72(9):6183–6193. doi: 10.1128/AEM.00947-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nie L, Wu G, Culley DE, Scholten JCM, Zhang W. Integrative analysis of transcriptomic and proteomic data: Challenges, solutions and applications. Critical Reviews in Biotechnology. 2007;27(2):63–75. doi: 10.1080/07388550701334212. [DOI] [PubMed] [Google Scholar]
- 15.Nelson KE, Weinel C, Paulsen IT, et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environmental Microbiology. 2002;4(12):799–808. doi: 10.1046/j.1462-2920.2002.00366.x. [DOI] [PubMed] [Google Scholar]
- 16.Kim SI, Song SY, Kim KW, Ho EM, Oh KH. Proteomic analysis of the benzoate degradation pathway in Acinetobacter sp. KS-1. Research in Microbiology. 2003;154(10):697–703. doi: 10.1016/j.resmic.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Singh OV. Proteomics and metabolomics: the molecular make-up of toxic aromatic pollutant bioremediation. Proteomics. 2006;6(20):5481–5492. doi: 10.1002/pmic.200600200. [DOI] [PubMed] [Google Scholar]
- 18.Singh OV, Nagaraj NS. Transcriptomics, proteomics and interactomics: unique approaches to track the insights of bioremediation. Briefings in Functional Genomics and Proteomics. 2006;4(4):355–362. doi: 10.1093/bfgp/eli006. [DOI] [PubMed] [Google Scholar]
- 19.Armengaud J, Happe B, Timmis KN. Genetic analysis of dioxin dioxygenase of Sphingomonas sp. strain RW1: catabolic genes dispersed on the genome. Journal of Bacteriology. 1998;180(15):3954–3966. doi: 10.1128/jb.180.15.3954-3966.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basta T, Keck A, Klein J, Stolz A. Detection and characterization of conjugative degradative plasmids in xenobiotic-degrading Sphingomonas strains. Journal of Bacteriology. 2004;186(12):3862–3872. doi: 10.1128/JB.186.12.3862-3872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seah SYK, Ke J, Denis G, et al. Characterization of a C-C bond hydrolase from Sphingomonas wittichii RW1 with novel specificities towards polychlorinated biphenyl metabolites. Journal of Bacteriology. 2007;189(11):4038–4045. doi: 10.1128/JB.01950-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halden RU, Colquhoun DR, Wisniewski ES. Identification and phenotypic characterization of Sphingomonas wittichii strain RW1 by peptide mass fingerprinting using matrix-assisted laser desorption ionization-time of flight mass spectrometry. Applied and Environmental Microbiology. 2005;71(5):2442–2451. doi: 10.1128/AEM.71.5.2442-2451.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller TR, Delcher AL, Salzberg SL, Saunders E, Detter JC, Halden RU. Genome sequence of the dioxin-mineralizing bacterium Sphingomonas wittichii RW1. Journal of Bacteriology. 2010;192(22):6101–6102. doi: 10.1128/JB.01030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bunz PV, Cook AM. Dibenzofuran 4,4a-dioxygenase from Sphingomonas sp. strain RW1: angular dioxygenation by a three-component enzyme system. Journal of Bacteriology. 1993;175(20):6467–6475. doi: 10.1128/jb.175.20.6467-6475.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18(11):2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 26.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Analytical Chemistry. 1996;68(5):850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 27.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hufnagel P, Rabus R. Mass spectrometric identification of proteins in complex post-genomic projects: Soluble proteins of the metabolically versatile, denitrifying Aromatoleum sp. strain EbN1. Journal of Molecular Microbiology and Biotechnology. 2006;11(1-2):53–81. doi: 10.1159/000092819. [DOI] [PubMed] [Google Scholar]
- 29.Giometti CS. Tale of two metal reducers: comparative proteome analysis of Geobacter sulferreducens PCA and Shewanella oneidensis MR-1. Methods of Biochemical Analysis. 2006;49:97–111. [PubMed] [Google Scholar]
- 30.Mazzoli R, Pessione E, Giuffrida MG, et al. Degradation of aromatic compounds by Acinetobacter radioresistens S13: growth characteristics on single substrates and mixtures. Archives of Microbiology. 2007;188(1):55–68. doi: 10.1007/s00203-007-0223-z. [DOI] [PubMed] [Google Scholar]
- 31.Johnson DR, Coronado E, Moreno-Forero SK, Heipieper HJ, Van Der Meer J. Transcriptome and membrane fatty acid analyses reveal different strategies for responding to permeating and non-permeating solutes in the bacterium Sphingomonas wittichii. BMC Microbiology. 2011;11 doi: 10.1186/1471-2180-11-250.Article 250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. Journal of Bacteriology. 2001;183(24):7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Susin MF, Baldini RL, Gueiros-Filho F, Gomes SL. GroES/GroEL and DnaK/DnaJ have distinct roles in stress responses and during cell cycle progression in Caulobacter crescentus. Journal of Bacteriology. 2006;188(23):8044–8053. doi: 10.1128/JB.00824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schiene-Fischer C, Habazettl J, Schmid FX, Fischer G. The hsp70 chaperone DnaK is a secondary amide peptide bond cis-trans isomerase. Nature Structural Biology. 2002;9(6):419–424. doi: 10.1038/nsb804. [DOI] [PubMed] [Google Scholar]
- 35.Armengaud J, Timmis KN. Molecular characterization of Fdx1, a putidaredoxin-type [2Fe-2S] ferredoxin able to transfer electrons to the dioxin dioxygenase of Sphingomonas sp. RW1. European Journal of Biochemistry. 1997;247(3):833–842. doi: 10.1111/j.1432-1033.1997.00833.x. [DOI] [PubMed] [Google Scholar]
- 36.Bunz PV, Cook AM. Dibenzofuran 4,4a-dioxygenase from Sphingomonas sp. strain RW1: angular dioxygenation by a three-component enzyme system. Journal of Bacteriology. 1993;175(20):6467–6475. doi: 10.1128/jb.175.20.6467-6475.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmann EM, Badalamenti JP, Krajmalnik-Brown R, Halden RU. Quantitative PCR for tracking the megaplasmid-borne biodegradation potential of a model sphingomonad. Applied and Environmental Microbiology. 2012;78(12):4493–4496. doi: 10.1128/AEM.00715-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halden RU. Comment on “Biological removal of polychlorinated dibenzo-p-dioxins from incinerator Fly Ash by Sphingomonas wittichii RW1” by I.H. Nam, Y.M. Kim, B.H. Kim, K. Murugesan, and Y.S. Chang. Water Research. 2006;40(9):1918–1920. doi: 10.1016/j.watres.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 39.Nam IH, Hong HB, Kim YM, Kim BH, Murugesan K, Chang YS. Biological removal of polychlorinated dibenzo-p-dioxins from incinerator fly ash by Sphingomonas wittichii RW1. Water Research. 2005;39(19):4651–4660. doi: 10.1016/j.watres.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 40.Gupta N, Tanner S, Jaitly N, et al. Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation. Genome Research. 2007;17(9):1362–1377. doi: 10.1101/gr.6427907. [DOI] [PMC free article] [PubMed] [Google Scholar]