

Abstract
Despite our long familiarity with how the genetic code specifies the amino acid sequence, we still know little about why it is organized in the way that it is. Contrary to the view that the organization of the genetic code is a “frozen accident” of evolution, recent studies have demonstrated that it is highly nonrandom, with implications for both codon assignment and usage. We hypothesize that this inherent nonrandomness may facilitate the coexistence of both sequence and structural information in DNA. Here, we take advantage of a simple metric of intrinsic DNA flexibility to analyze mutational effects on the four phosphate linkages present in any given codon. Application of a simple evolutionary neutral model of substitution to random sequences, translated with alternative genetic codes, reveals that the standard code is highly optimized to favor synonymous substitutions that maximize DNA polymer flexibility, potentially counteracting neutral evolutionary drift toward stiffer DNA caused by spontaneous deamination. Comparison to existing mutational patterns in yeast also demonstrates evidence of strong selective constraint on DNA flexibility, especially at so-called “silent” sites. We also report a fundamental relationship between DNA flexibility, codon usage bias, and several important evolutionary descriptors of comparative genomics (e.g., base composition, transition/transversion ratio, and nonsynonymous vs. synonymous substitution rate). Recent advances in structural genomics have emphasized the role of the DNA polymer's flexibility in both gene function and whole genome folding, thereby implicating possible reasons for codons to facilitate the multiplexing of both genetic and structural information within the same molecular context.
Keywords: genetic code, codon, codon bias, DNA flexibility, minor groove width, silent mutation
Introduction
Upon the discovery of the structure of DNA, and the subsequent illumination of the genetic code over a decade later, the hereditary information contained in the genetic code was presumed to be largely abstracted from details of molecular structure, excepting of course, for the base sequence itself. As a consequence, it was postulated that both codon assignment and degeneracy in the genetic code may be quite arbitrary, having perhaps evolved accidentally with respect to specific base sequences (Crick 1966, 1968), and thus existing as a truly abstract “cipher” or code. However, more recently, it has been demonstrated that the genetic code is quite nonrandom with respect to its ability to minimize the effects of single-base substitutions on proteins, especially at the first and third positions of the codon (Freeland and Hurst 1998). Both the source and degree of this nonrandomness are still debated (Zhu et al. 2003; Archetti 2004; Stoltzfus and Yampolsky 2007); however, most would agree that this nonrandomness ultimately implies some level of evolutionary constraint evident in both the overall distribution of codon assignments and local choice of codons (i.e., codon bias) within genes and genomes (Hershberg and Petrov 2008).
The recent discovery that the genetic code is extremely highly optimized to allow for the carrying of additional parallel codes (Itzkovitz and Alon 2007) begs the question as to what actually are the specific mechanism(s) by which this additional information can be multiplexed over the genetic code. Supported by a growing body of recent research is a view that this additional information is fundamentally structural in character, somehow comprising additional molecular biophysical properties of the DNA polymer itself. Most notable is DNA's ability to bend and deform physically to the requirements of specific DNA–protein interactions that influence gene regulation (Olson et al. 1998; Segal et al. 2006; Parker et al. 2009; Rohs et al. 2009; Parker and Tullius 2011). Research now supports the view that transcriptional gene regulation involves collaborative competition for access to cis-regulatory DNA among several classes of proteins that activate gene expression (e.g., transcription factors and chromatin remodelers) and the various histone proteins that package eukaryotic DNA (i.e., forming nucleosomes) (Jiang and Pugh 2009; Segal and Widom 2009; Moyle-Heyrman et al. 2011; Tims et al. 2011; Yosef and Regev 2011). This type of structural information contained in DNA polymer is not likely to be limited to cis-regulatory regions as nucleosome formation is required of all genomic DNA in eukaryotes, and coding regions, in particular, are known to allow for tight stacking of adjacent nucleosomes against strongly positioned nucleosomes occurring at transcription start sites (Mavrich et al. 2008).
It is often traditionally assumed that both cis-regulatory and coding DNA are inert with respect to the information they carry, wholly dependent upon interactions with complex protein-based systems designed to convert sequence information into cell functions. However, in reality, the DNA polymer is a dynamic and potentially flexible partner in probably all protein–DNA interactions. Although the close proximity of negatively charged phosphate groups on the backbone of the double helix lends considerable stiffness to DNA polymer, there is also a tendency for DNA to form mini kinks that can allow for considerable sequence-dependent DNA deformation without the imposition of strong structural constraints; as is well demonstrated in DNA deformation to the structure of the nucleosome core (Ruscio and Onufriev 2006; Tolstorukov et al. 2007). As a consequence of this sequence-associated tendency of DNA to kink, the flexibility of local regions of DNA polymer is strongly associated with both GC content (Heddi et al. 2010) and purine–pyrimidine ground states (Olson et al. 1998). Recently, it has been shown that these biophysical properties of the DNA polymer can play direct and significant roles in governing DNA–protein interactions (Tolstorukov et al. 2004; Rohs et al. 2009; Tullius 2009; Hebert and Crollius 2010) and their evolution (Tirosh et al. 2007; Parker et al. 2009; Babbitt et al. 2010; Kenigsberg et al. 2010; Nikolaou et al. 2010; Dai et al. 2011; Parker and Tullius 2011).
In all forms of life, the genetic code must coexist with structural flexibilities of DNA that can either favor or disfavor interactions with specific proteins. In the genomes of almost all Eukarya and some Archaea, where packaging of DNA involves considerable compaction by chromatin architectural proteins, the problem of multiplexing these fundamentally different types of information into a single molecular structure is paramount. Recent observations that codon biases vary in association with nucleosome occupancy (Warnecke et al. 2008), that a strong codon bias exists toward the avoidance of long A-tracts that are disruptive to nucleosome positioning (Cohanim and Haran 2009), and that genomic mutational frequencies are strongly associated with their affects on the ability of DNA to deform to the structure of the nucleosome core (Babbitt and Cotter 2010) would also imply a universal selective pressure to allow the genetic information contained in codons to freely coexist with additional regulatory information encoded by varying local DNA flexibility. In support of this idea, Parker and Tullius (2011) have noted that the third position of the codon ideally defines the minor groove width of the DNA, which in turn, plays a crucial role in regulating DNA–protein interactions through DNA shape-induced changes in electrostatic potential (Rohs et al. 2009). So, in theory, it would seem that most mutations with strong potential to affect DNA flexibility would be less likely to alter amino acid sequences of proteins. In one of the few articles to ever address this point specifically, Bainsee et al. (2001) actually observed that the genetic code is quite accommodating of potential structural information in DNA, but because the range of structural variation allowed by the genetic code was low compared with some alternative codes, they speculated that the code has probably not evolved under these structural constraints. However, these author’s conclusions are not based on any direct analysis of mutational impacts on DNA shape or flexibility. Beyond this work, there has been little further attempt to explore the evolutionary relationship between DNA flexibility and the functional organization of the genetic code.
Here, we have taken advantage of a recently published metric of intrinsic DNA flexibility, based on a large sequence-dependent nuclear magnetic resonance data set of 31P chemical shifts in solution, representing BI ↔ BII conformation states of phosphate linkages in the DNA backbone (Heddi et al. 2010). We use a metric scale for intrinsic DNA flexibility to assess the mutational effects of single-base substitutions on the flexibility of DNA polymer comprising various codons. Our primary objective is to examine how mutational impacts on DNA flexibility are related to the organization of the genetic code (i.e., codon assignment), the biased usage of codons of the Saccharomyces cerevisiae genome (i.e., codon bias), the relative frequencies of purine and pyrimidine conserving substitutions in four closely related yeast genomes (i.e., transition/transversion or Ts:Tv ratio), and the level of functional constraint acting upon protein evolution (i.e., dN/dS). We hypothesize that the nonrandomness evident in both codon assignment and codon usage may reveal a signature of mutational bias and/or functional constraint regarding the intrinsic flexibility of DNA.
Materials and Methods
The TRX Score and Intrinsic Flexibility of B-DNA
In solution, the P linkages of the B-DNA backbone can reside in one of two molecular states, referred to as BI or BII conformations, at any point in time. The TRX or “twist, roll, and X displacement” scale is a measure of the average percentage of time that a particular P linkage connecting two bases resides in the BII conformation; the higher this percentage, the more flexible the dinucleotide or dimer (Heddi et al. 2010). This TRX scale was derived from a large nuclear magnetic resonance data set. The TRX scale is presented in table 1 in conjunction with the AT content and purine (R)–pyridimine (Y) sequence state of each dimer. Olson et al. (1998) conducted a study of DNA–protein crystal structures demonstrating that YR dimers are generally most flexible, often occupying critical sites of DNA–protein interaction (RY dimers are least flexible). It is now widely accepted that YR dimers accommodate lateral displacements in the long axis of DNA, known as “kink and slide” deformations, which are critical to many protein–DNA interactions including nucleosome formation (Tolstorukov et al. 2007; Wang et al. 2010). As presented in table 1, the TRX scale also confirms the contribution of YR/RY dimers, as well as the contribution of base composition toward DNA flexibility (with higher GC content associated with more flexibility).
Table 1.
The Flexibility of DNA as Measured by TRX Score, AT Composition, and Pyrimidine–Purine (YR) Ordering of a Given Dimer (Dinucleotide)
| Dimer | Purine:Pyrimidine | Frequency of AT | TRX |
|---|---|---|---|
| CpG:CpG | YR:YR | 0 | 43 |
| CpA:TpG | YR:YR | 0.5 | 42 |
| GpG:CpC | RR:YY | 0 | 42 |
| GpC:GpC | RY:RY | 0 | 25 |
| GpA:TpC | RR:YY | 0.5 | 22 |
| TpA:TpA | YR:YR | 1 | 14 |
| ApG:CpT | RR:YY | 0.5 | 9 |
| ApA:TpT | RR:YY | 1 | 5 |
| ApC:GpT | RY:RY | 0.5 | 4 |
| ApT:ApT | RY:RY | 1 | 0 |
Note.—The TRX score appears to capture the function of both AT content and YR ordering. The base stacking of weak-bonded base pairs (A and T) results in narrower minor groove width and less flexible DNA. The YR dimers (green fill) are highly susceptible to lateral displacementsor “kink and slide” deformations in protein–DNA crystal structures, whereas the RY dimers (red fill) are least deformable (Olson 1998).
Correlation of TRX Score to Minor Groove Width Dimensions of DNA
It was recently reported that DNA shape, defined by the minor groove width of the double helix, correlates strongly to DNA flexibility (Rohs et al. 2009). By narrowing minor groove width, negatively charged phosphate groups on the backbone more strongly repel each other causing the subsequent decrease in flexibility. To assess the relationship between DNA shape and TRX score, we investigated the statistical association of minor groove width (in angstroms) of tetramers given in supplementary tables of Rohs et al. (2009) to their sum TRX scores calculated according to method of Heddi et al. (2010). These data include average minor groove widths of both protein bound and free DNA.
Estimation of Mutational Impact on the DNA Flexibility for a Given Codon (dTRX)
DNA flexibility over the extent of a single codon (i.e., codon linkage flexibility) was defined by the total sum conformational state, or sum TRX score, for the four phosphate (P) linkages on the leading strand DNA sugar–phosphate backbone for a given codon (fig. 1A). Two of these P linkages are internal to a given codon, flanking the base in the second position of the codon, thereby contributing to DNA flexibility only in accordance with codon assignment. The other two P linkages are external to a given codon, preceding the first base position and following the third base, and are thereby defined through the combination of codon assignment and usage of neighboring adjacent codons. Therefore, these external P linkages can also potentially influence overall DNA flexibility through patterns of codon usage or “codon bias.” In our comparative genomic analyses, we used the multiple alignments for four extant Saccharomyces spp. (S. cerevisiae, S. paradoxus, S. mikatae, and S. bayanus) previously published by Kellis et al. (2003) implementing PAML software (baseml program; Yang 2007) to assemble ancestral sequence reconstructions on the clade for all the aligned genes. These reconstructions allow us to determine the evolutionary directionality of substitution events in a way that pairwise comparison of extant sequences would not. The mutational effect of single-base substitutions on codon linkage flexibility, or ΔTRXcodon, was defined as
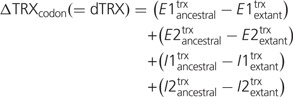 |
or in the case of simulations of neutral evolution on random seuences (described in the next section)
 |
Etrx and Itrx specify the TRX scores for “external” and “internal” P linkages, respectively (note: differences are always 0 for the two linkages unaffected by the single-base substitution at a particular codon position, i.e., where extant and ancestral sequences are identical). In conclusion, a negative value of dTRX indicates a mutational impact whereby the stiffness of the DNA backbone in a given codon is decreased (i.e., flexibility increased), whereas a positive value indicates a stiffening of the DNA.
Fig. 1.—

DNA flexibility as defined by the four phosphate linkages in the codon (TRX). (A) External linkages are defined by neighboring adjacent codons, whereas internal linkages are defined by the codon itself. Adjacent codons are displayed with alternating stick and wireframe display. (B) A strong and highly significant correlation (r = 0.66, P < 0.0001, df = 193) exists between the sum flexibility(TRX score) and the average minor groove width (angstroms) of tetramers in free DNA and protein-bound DNA structures reported in Rohs et al. (2009).
Assessing Effects of Codon Assignment on dTRX: Neutral Simulations on Random Sequences Using Alternative Genetic Codes
To assess the ability of codon assignments in alternative genetic codes to bias average effects on dTRX, we randomly permuted base assignments of the three codon positions according to figure 1 in Itzkovitz and Alon (2007) to create 1,152 (=4!×4!×2) alternate genetic codes. There are 4! (=24) permutations of the four nucleotides, giving 243 (=13,824) alternative codes. The number of permutations can be reduced to 1,152 by consideration of the wobble constraint (i.e., recognizing only the YR state of third position). This permutation scheme retains the overall composition of base assignments in the genetic code and the number of codons assigned to each amino acid; however, it randomizes the positions of the bases within codons specified by the alternative codes. For each alternative code, we generated random amino acid sequences that were 1,000,000 residues in length built with an equal probability of each amino acid at each residue position. Amino acids sequences were reverse translated using an equal probability for choice of codon (i.e., no codon bias), and the resulting DNA sequences were diverged neutrally at 16% of their sites (i.e., 8% divergence on each lineage) using an equal probability of each type of base change (i.e., substitutions were modeled according to Jukes–Cantor or JC69). The random DNA sequences generated in this manner are theoretically 48.3% GC, therefore the neutral simulations were conducted at very near equilibrium regarding GC content. This is important due to the fact that the neutral evolution of TRX scores, being highly dependent upon GC content, can potentially simply reflect nonequilibrium changes in GC content under the neutral model. To ensure that this potential bias was not driving our main result in any way, we also conducted our simulations using DNA sequences randomly generated at 50% GC and compared this to our simulations using randomly generated amino acid sequences. Finally, the average dTRX for each of the four functional classes of mutations (i.e., synonymous Ts, synonymous Tv, nonsynonymous Ts, and nonsynonymous Tv) were calculated under each alternative coding scheme and compared with the average dTRX derived from an identical neutral simulation using the standard genetic code. These averages simulated under neutral evolutionary models with both real and alternative genetic codes were also compared with overall genomic average dTRX observed when comparing the genomes of the four extant yeast spp. to their ancestral states (later).
Comparative Genomic Analysis of dTRX in Four Closely Related Yeast Genomes
Multiple alignments of the four extant yeast species were used to construct ancestral sequence reconstructions (using baseml program in PAML). The directionality of single-base substitutions was determined by comparing extant sequences with their ancestral states. The relative positions of mutations with respect to reading frame (i.e., codon position) were noted as well. We individually averaged the mutational impacts on codon linkage flexibility (dTRX) across the six classes of substitutions (i.e., two types of Ts and four types of Tv) and compared their overall frequencies on both synonymous (i.e., silent = S) and nonsynonymous sites (i.e., AA replacement sites = N). We also averaged dTRX for all 540 possible classes of codon substitutions as well. Note: these are reported as averages for each class of codon substitution involving a base change at first or third positions, necessary due to variance contributed to the flexibility of external P linkages by base pairings of adjacent codons. In this analysis, codon substitutions were classified/color coded according to their effect on purine–pyrimidine ground state of the DNA sequence (i.e., Ts vs. Tv) and their effect on amino acid sequence (N vs. S). Thus four functional types of mutation are defined with respect to codons (synonymous Ts, synonymous Tv, nonsynonymous Ts, and nonsynonymous Tv).
Assessing Effects of Codon Bias on dTRX in S. cerevisiae with Random Synonymous Codon Reassignments
To distinguish the potential mutational effects of codon assignment from codon usage bias, we randomly assigned synonymous codons at observed sites of single-base substitution throughout the four yeast genomes and reanalyzed average dTRX on all 540 types of codon substitutions. We compared this with similar results obtained from unaltered coding and noncoding regions, as well as simulated neutral evolution on random DNA sequences. Here again, neutral simulations of evolutionary diverged 1MB random DNA sequences were constructed using equal probabilities of base substitutions (i.e., the Jukes–Cantor model). However, the random sequences used for the neutral simulations were constructed with the same overall dinucleotide frequencies as the S. cerevisiae genome. Evolutionary divergence was 8% on each branch or lineage (16% total divergence). Where necessary to quantify codon bias at the level of single genes, we chose the weighted relative entropy metric of Suzuki et al. (2004). For any given gene, we report codon bias (as = 1−Suzuki's weighted relative entropy) on a scale of 0 (indicating no bias) to 1 (indicating universal preferences for single codons). This method was preferred over others (e.g., codon adaptation indices; Sharp and Li 1987) as it makes no assumption as to an optimal level of bias as defined by comparisons to reference sets of highly expressed genes. It also has several advantages over simpler metrics of codon evenness in that it accounts for the number of distinct amino acids, their relative frequency in a given gene, and the degree of codon degeneracy. Finally, we also compared respective trends in the associations of average gene-specific flexibility or TRXsum (i.e., average coding region flexibility) and average gene-specific mutational shifts in flexibility or |dTRX| (i.e., average mutational shift in flexibility taken over entire coding region) to several of the most important descriptors of comparative genomics (i.e., Ts:Tv, codon bias, and dN/dS). To illuminate the potential role of codon usage bias in also contributing toward gene-averaged mutational shifts in local DNA flexibility, we also conducted identical analyses where synonymous codons were chosen randomly (i.e., shuffled) at sites of mutation on ancestral sequences. Where possible, we compared trends to noncoding regions as well. Additional analyses were conducted using a randomized TRX scale (i.e., random integers from 0 to 43) to validate the biological signal in our results.
Results
Correlation of TRX Score to Minor Groove Width Dimensions of DNA
As expected, we found a strong and highly significant positive correlation between the minor groove widths (in angstroms) of tetramers observed in both free DNA structures and protein-bound DNA structures and their calculated sums of TRX scores (r = 0.66, P < 0.0001, df = 193; fig. 1B) indicating a strong role of P linkage flexibility in determining DNA shape.
Assessing Effects of Codon Assignment on dTRX: Neutral Simulations on Random Sequences Using Alternative Genetic Codes
We investigated directly the potential role that codon assignments in the standard genetic code may play in biasing mutational impacts on DNA flexibility at the level of codons. We generated 1,152 alternative genetic codes and compared the average random mutational impacts (dTRX) for each alternative code to those of the real code over each of the four functional classes of substitutions (i.e., synonymous Ts, synonymous Tv, nonsynonymous Ts, and nonsynonymous Tv). To assess potential influences of selective constraints on dTRX, we also included average random mutational impacts estimated from the real patterns of mutation in the yeast genomes as well. We found that the standard genetic code appears extremely highly optimized to favor synonymous substitutions that increase DNA flexibility under neutral evolutionary processes, particularly with respect to synonymous Ts (fig. 2 and supplementary fig. SA, Supplementary Material online, top; probabilities under normal distribution are synonymous Ts = 0.016, synonymous Tv = 0.027; nonsynonymous Ts and Tv are 0.980 and 0.776, respectively). A marked signature of selective constraint was also evident, as synonymous substitutions actually observed in the yeast genomes exhibit on average only very slight increases in DNA flexibility. The genetic code also appears somewhat optimized to reduce the effects of nonsynonymous Ts on DNA flexibility; an opposite trend to that observed in synonymous substitutions, with little evidence of selective constraint here (fig. 2 and supplementary fig. SA, Supplementary Material online; bottom, y axis). Conversely, nonsynonymous Tv appear to have very large levels of selective constraint on DNA flexibility with nearly no evident optimization in the code (fig. 2 and supplementary fig. SA, Supplementary Material online; bottom, x axis). Taken as a whole, these patterns in selective constraint support that mutations at so-called silent sites are actually quite evolutionarily constrained with respect to DNA flexibility, whereas nonsilent mutations are far less constrained. In addition, despite the tendency of silent mutations to increase DNA flexibility, most mutations appear to have accumulated in the yeast genomes with little to no effect on codon flexibility (i.e., average dTRX ≈ 0); supporting our hypothesis that although natural selection and genetic drift have been working to alter protein structures over time, the structural information contained in DNA polymer as it affects its flexibility is generally conserved by selection acting on silent mutations and nonsilent Tv and through optimization of the genetic code with respect to nonsilent Ts.
Fig. 2.—

Average mutational impacts on DNA flexibility for evolutionary neutral simulations under alternative genetic codes. A total of 1,152 alternative permutations of the genetic code were computed using the method of Itzkovitz and Alon (2007), and the average mutational impact for four classes of substitutions (synonymous Ts, synonymous Tv, nonsynonymous Ts, and nonsynonymous Tv) were calculated using neutral evolutionary simulations on reverse translated random amino acid sequences with no codon usage bias (48.3% GC content). Values for neutral simulations using the standard genetic code (red) and real mutations in yeast genomes (blue), presumably selectively constrained, are shown for comparison. The level/direction of selective constraint is indicated by the black arrow.
Comparative Genomic Analysis of dTRX in Four Closely Related Yeast Genomes
Not surprisingly, comparative sequence analysis of the multiple alignments clearly indicated that the average mutational impacts of substitutions on the flexibility of codons (dTRX) were far greater, in those four substitution types which alter the local GC content. This was to be expected as TRX scores correlate directly with GC content. Substitutions resulting in an additional G or C increase flexibility on average (or decrease stiffness; = negative dTRX value), whereas substitutions resulting in an additional A or T decrease flexibility on average (or increase stiffness; = positive dTRX). These differences in average genomic mutational impacts on codons (dTRX) were highly significant (F = 646.23, num df = 5, denom df = 265, P < 0.0001; fig. 3A). Comparison of overall genomic frequencies of these substitutions at both synonymous and nonsynonymous sites revealed very high frequencies of purine and pyrimidine conserving mutations (i.e., Ts), especially at synonymous sites (fig. 3B). Therefore, the overall tendency of high frequencies of GC→AT (Ts), often commonly associated with mutational biases imposed by spontaneous deamination, appears compensated to a considerable degree in its overall effect on DNA flexibility by equally high frequencies of AT→GC (Ts). More detailed analyses of the frequencies of all 540 possible codon substitutions also reveals a compensating trend around dTRX in synonymous Ts (fig. 4A). We compared this plot to a similar one derived from noncoding regions of our alignments (fig. 4B) and found that a high rate of Ts still occurs, but relative Tv rates are slightly higher in GC content-preserving or GC content-elevating Tv. As a codon-based substitution analysis is not really appropriate when applied to noncoding sequence, we wondered how this result might compare to coding sequences in which codon bias was controlled or eliminated (fig. 4C) or in which mutations accumulated neutrally on random DNA sequences (fig. 4D). Here, we also find elevation of synonymous Tv rates, and in neutral simulations, the overall effects on dTRX lean markedly toward increasing the flexibility of P linkages in codons (fig. 4D), further supporting our earlier conclusion that a built-in mutational bias may exist regarding synonymous substitutions under the standard genetic code to increase DNA flexibility and therefore through the process of random genetic drift.
Fig. 3.—

Mutational impact on DNA flexibility and frequency of occurrence for different classes of substitutions in the yeast genomes. (A) Average mutational impact of single-base substitutions on the flexibility of codon phosphate linkages (delta TRX) for all classes of substitutions. Negative values indicate mutational shifts toward increased DNA flexibility. (B) Frequencies of the same classes of substitutions for both functional (amino acid replacing) and silent substitution types. High frequency mutations (synonymous Ts) have strong counteracting average effects on DNA flexibility.
Fig. 4.—

Association of genomic frequency and average mutational impact of single-base substitutions on the flexibility of codon phosphate linkages (dTRX) for all 540 possible codon substitutions. Results are shown for (A) yeast coding and (B) noncoding sequences, as well as (C) coding sequences where codons at the sites of mutations were replaced with random synonymous codons (i.e., eliminating codon usage bias). (D) Frequencies of codon substitutions in a simulation of neutral evolution acting on random DNA (using standard genetic code).
Assessing Effects of Codon Bias on dTRX in S. cerevisiae with Random Synonymous Codon Reassignments
To further investigate the importance of mutational impacts on codon flexibility (dTRX) in driving common patterns observed in comparative genomics, we averaged the |dTRX| for the coding region of each gene and compared avg|dTRX| with Ts:Tv ratios and levels of functional conservation on proteins (i.e., dN and dN/dS). We also conducted identical analyses where synonymous codons were chosen randomly (i.e., shuffled) at sites of mutation on ancestral sequences. Where possible, we compared trends to noncoding regions as well. We found that the elevation in Ts:Tv ratio is significantly associated with genes demonstrating large mutational shifts in DNA flexibility at the levels of individual codons and that this trend is maintained almost completely by existing codon usage biases of single genes (with bias, r = 0.26, P < 0.0001; without bias r = 0.06, P = 0.03; fig. 5A). A similar plot is shown in figure 5B using DNA flexibility (TRXcodon) in lieu of dTRX to highlight the profound overall differences in DNA flexibility between coding and noncoding regions in the yeast genome. We also found a significant anticorrelation between gene-specific mutational shifts in |dTRX| and their level of nonsynonymous substitution (dN) and dN/dS, indicating that more highly conserved genes have higher overall shifts in DNA flexibility at the levels of codons (for dN/dS, r = −0.14, P < 0.0001; fig. 5C and for dN, r = −0.29 , P < 0.0001; fig. 5D). Again, this correlative trend is wholly dependent on existing codon bias (for dN/dS without codon bias, r = 0.029, P = 0.328; fig. 5C and for dN without bias r = −0.038, P = 0.197; fig. 5D). These correlative trends support that the evolution of amino acid sequence and phosphate linkage flexibility are only partially decoupled as it would appear that highly conserved coding sequences (low dN or low dN/dS) are associated with higher average mutational shifts in |dTRX|. Such a trade-off in the efficiency of selection acting in the context of the multiplexing of sequence-based and structurally based information might be expected when more than one type of information must share the same molecular structure. Many of these highly conserved genes also appear to have comparably high Ts:Tv ratio, further supporting that higher rates of synonymous AT→GC (Ts) may actually function to conserve DNA flexibility at the level of individual codons without adversely affecting protein function. A very weak but significant anticorrelation between gene-specific mutational shifts in |dTRX| and synonymous substitutions (dS) was also observed to be maintained by codon bias (for dS with codon bias, r = −0.08, P = 0.007 and for dS without bias r = 0.053, P = 0.077; fig. 5D).
Fig. 5.—

Fundamental relationships between codon bias, DNA flexibility, and comparative evolutionary genomics. (A) Across all genes, codon bias maintains a strong correlation between the relative rate of purine and pyrimidine conserving mutations (Ts/Tv) and the average overall mutational impact on the DNA flexibility of codons. (B) Comparative relationships between Ts/Tv and average DNA flexibility of codons. (C) Codon bias also maintains a strong correlation between average mutational impacts on flexibility and elevated levels of functional conservation of a given gene (i.e., low dN/dS). (D) This correlation is driven largely by trend in dN (not shown—correlation with dS is small). In the unbiased groupings (gray), codon usage bias was eliminated through random replacement with synonymous codons at all sites affected by base substitution.
Discussion
Taken as a whole, our results strongly implicate a major role for local variability of DNA polymer flexibility in affecting the molecular evolution of coding regions in yeast genomes. The molecular evolution of this intrinsic structurally encoded information is only possible because different types of substitutions have substantially different effects on DNA flexibility and minor groove width. These mutational effects are largely in accordance with relative impact on local GC content and the formation or dissolution of YR dimers, where lateral displacements of the DNA long axis can occur (i.e., mini kinks or “kink and slide” deformations). Later, we outline two lines of supporting evidence that DNA shape and flexibility are subject to significant evolutionary forces—both from neutral mutational bias imposed by the genetic code itself and from purifying selective pressure to generally minimize mutational impacts on DNA flexibility. Finally, we offer a potential functional explanation for the patterns we observe.
Optimization and Selective Constraints Regarding Codon Assignments
We find that the organization of codon assignment within the genetic code is highly optimized to maximize the tendency of synonymous substitutions to increase the intrinsic flexibility of the DNA polymer under neutral evolution (i.e., genetic drift). This organization of codon assignment in the genetic code is also optimized to minimize the tendencies of nonsynonymous transitions to alter DNA flexibility as well. Over evolutionary timescales, this combination of silent mutations with large potential impacts on DNA flexibility and nonsilent mutations with small impacts on flexibility may have played an important role in establishing many of the common characteristics of coding regions observed in comparative genomics, including elevated GC content, elevated Ts:Tv (Yang 2006), and dense nucleosome stacking (Mavrich et al. 2008). With respect to nonsilent transversions, the organization of codon assignments appears not to be optimized to control their tendency to increase DNA flexibility. Instead, nonsynonymous transversions appear strongly selectively constrained to have little effect on DNA flexibility. Together, these results implicate a combined role for both purifying selection and genetic code optimization, in facilitating the coexistence of structural with genetic information. The potential selective constraint of nonsilent transversion, which would contribute to elevating Ts:Tv across most coding regions, may be particularly indicative of this multiplexing of genetic and structural information into the same molecular context. Additionally, the existence of significant selective constraints on DNA flexibility at synonymous sites seriously jeopardizes the notion of the existence of truly “silent sites” in the genome. The suggestion that non-neutral evolution can act upon so-called silent sites is not new (Chamary et al. 2006), but our work suggests a universal mechanism by which it can occur. Parker and Tullius (2011) have recently recognized that the third position of the codon, where most synonymous substitutions occur, is largely responsible for defining DNA shape by the width of the minor groove. Thus, the reading frames of coding regions have an ability to easily coincide and avoid interfering with the structural characteristics of DNA polymer that determine its local flexibility. In this sense, the existence of a triplet code may be no accident of early evolution.
Codon Bias Supports Correlative Trends between DNA Flexibility and Descriptive Metrics of Comparative Genomics
We also cannot ignore the potential involvement of codon bias in maintaining DNA flexibility in coding regions either. Although, we did not manipulate codon bias when generating random sequences from alternative codes, we were able to observe its significant role in maintaining significant genome-wide associations regarding DNA flexibility, Ts:Tv ratio, dN, and dN/dS averaged over the entire coding regions of individual genes. We conclude that existing codon biases have probably evolved in response to the need to maintain a significant functional level of flexibility in the coding regions of the genome.
This finding is somewhat surprising in light of previous research strongly linking codon usage to the availability of cytosolic tRNA. Translation-optimized codon usage is well documented in many model organisms (Ikemura 1985), most likely due to selection for maintaining translational accuracy (Stoletski and Eyre-Walker 2007). Codon usage bias also correlates strongly with gene expression level within most species (Duret and Mouchiroud 1999; Duret 2002) and follows patterns of gene coregulation and coexpression (Fraser et al. 2004). It was recently proposed that the primary source of selective constraint on coding sequence evolution is the potential misfolding of mistranslated proteins, especially in highly expressed genes where mistakes at the level of translation can more easily overwhelm the coping mechanisms of cells (Drummond and Wilke 2008; Yang et al. 2010). This potentially universal constraint was proposed to account for the general observation that highly expressed proteins evolve quite slowly (Drummond et al. 2005). It also calls into question the utility of nonsynonymous to synonymous substitution ratios (dN/dS or Ka/Ks) in identifying functional constraints on protein evolution that are not potentially confounded by translation-induced variation in gene expression (Drummond and Wilke 2008).
However, a major problem with the view that selective constraints acting on coding sequence evolution are mostly affecting molecular events during translation is the fact that gene expression is regulated in large part through transcription rather than translation (70% transcriptional to 30% translational according to Lu et al. 2007). Although this, in itself does not negate the role of translational optimization in shaping the evolution of coding sequences, it suggests that selection should also act on events occurring during transcription as well. If the evolution of translational optimization explained all aspects of observed codon usage bias, then one would expect that selective constraints of coding and noncoding regions should differ. However, evidence that codon usage biases favored within specific genomes can be actually be predicted by features of the intergenic regions of those genomes (Chen et al. 2004; Hershberg and Petrov 2009) implies that mutational and/or selective biases acting on whole genomes have actively shaped the functional organization of the genetic code as well. Our finding that codon usage biases in yeast support significant trends between mutational impacts on DNA flexibility and several well-known descriptors of molecular evolution (Ts:Tv and dN:dS) is also in accord with this emerging perspective that codon bias may result from selective constraints related to transcriptional processes and translational efficiency.
Mutational Biases May Counteract Negative Effects of Spontaneous Deamination on DNA Flexibility
Although mutationally biased by the genetic code, the high frequency of AT→GC transitions we observe, particularly at third position synonymous sites, also appear to be potentially functional in counteracting the large negative effects of spontaneous deaminations (i.e., GC→AT transitions) upon structurally encoded information in the genome. Recently, a strong mutational bias toward A and T generating mutations was observed in mutation accumulation lines of Caenorhabditis nematodes (Denver et al. 2012). This suggests that genomic DNA has a marked tendency to become “stiffer” over long evolutionary timescales when subjected to genetic drift, particularly in small populations. Indeed, the observation of very high AT content resulting from the massive genomic decay in bacterial endosymbionts of aphids (Burke and Moran 2011) is also potentially indicative of this sort of mutational bias as well. Perhaps, a mutational bias reducing DNA flexibility is universal and requires a highly optimized organization of the genetic code to counteract stiffening of genomic DNA over time. The decreased flexibility commonly observed in almost all noncoding regions of the genome would seem to support this speculation, as noncoding regions are obviously not “protected” against this mutational bias by the presence of the genetic code. However, selection may also certainly play a role in these regions as well, as it is widely accepted that reduced DNA flexibility in gene promoter regions is functional, allowing ease of access for transcription factors, through a reduction of DNA deformability to the nucleosome core (Jiang and Pugh 2009). The recent discovery of strong universal selective pressures to increase GC content in most bacteria (Hildebrand et al. 2010), and perhaps even the existence of GC-biased gene conversion in eukaryotes (Duret and Galtier 2009), perhaps may also suggest an underlying need to counteract a universal mutational pressure of genomic DNA to become stiffer over evolutionary timescales. However, the existence of truly “universal” mutational biases in molecular evolution has been a contentious issue in the past (Keller et al. 2007), perhaps best left for future experimental evidence to resolve (Denver et al. 2012).
Conclusion
The gene products produced by eukaryotic genomes function in a vast variety of ways; however, all gene products originate from information that is universally packaged first into the nucleosome and then into successive levels of higher order chromatin. Ultimately, this packaging and unpackaging of information during transcriptional gene regulation depends in large part upon local variations in the intrinsic flexibility of DNA, which are a direct function of sequence-dependent conformational states of the phosphate linkages in the DNA polymer backbone. In this sense, DNA structurally encodes for its own packaging. In coding regions, this structurally encoded information must always coexist with genetic information, requiring a mechanism of multiplexing two very disparate types of information into the same molecular context. The observation that triplet codons appear scaled precisely to the same repeat frequency of base pairs defining the width of the minor groove, and therefore defining the intrinsic flexibility of DNA, is probably no happy accident. In general, it allows information that is silent in one context to be active in another.
Because biology is increasingly being dominated by vast amounts of sequence data, the field of molecular evolution has traditionally ignored the role of structural variation in the DNA double helix in shaping the genomic spectrum of mutation. In this study, our demonstration of a fundamental connection between DNA flexibility and the organization of codons offers a compelling reason to no longer ignore the simple fact that DNA is not merely a sequence of letters but is also the molecular scaffolding for a complex and dynamic chromatin structure that interacts, through bending and binding, with a large host of other proteins in the cell.
List of Abbreviations
- Ts
= purine to purine or pyrimidine to pyrimidine substitution (transition).
- Tv
= purine to pyrimidine or pyrimidine to purine substitution (transversion).
- dN
= rate of nonsynonymous or amino acid replacing substitutions.
- dS
= rate of synonymous or “silent” substitutions.
- TRX
= intrinsic flexibility score (Heddi et al. 2010).
- TRXsum
= sum of TRX score over codon.
- dTRX
= delta TRX = overall sum of mutational changes in DNA flexibility (TRX score) for the phosphate linkages in a given codon.
- avg |dTRX|
= |dTRX| summed over all codons for a given gene.
Supplementary Material
Supplementary figure SA is available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This research was supported by an internal grant from the Office of Sponsored Research Services, Rochester Institute of Technology. G.A.B. acknowledges both personal correspondence and public lectures of the late Jonathon Widom (Northwestern University) regarding “multiplexing” of both genetic and structural information in the nucleosome, as a primary motivation for this research.
Literature Cited
- Archetti M. Codon usage bias and mutation constraints reduce the level of error minimization of the genetic code. J Mol Evol. 2004;59(2):258–266. doi: 10.1007/s00239-004-2620-0. [DOI] [PubMed] [Google Scholar]
- Babbitt GA, Cotter CR. Functional conservation of nucleosome formation selectively biases presumably neutral molecular variation in yeast genomes. Genome Biol Evol. 2011;3:15–22. doi: 10.1093/gbe/evq081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbitt GA, Tolstorukov MY, Kim Y. The molecular evolution of nucleosome positioning through sequence-dependent deformation of the DNA polymer. Special issue—current perspective in nucleosome positioning. J Biomol Struct Dyn. 2010;27(6):765–780. doi: 10.1080/07391102.2010.10508584. [DOI] [PubMed] [Google Scholar]
- Bainsee P, Baldi P, Brunak S, Pedersen AG. Flexibility of the genetic code with respect to DNA structure. Bioinformatics. 2001;17(3):237–248. doi: 10.1093/bioinformatics/17.3.237. [DOI] [PubMed] [Google Scholar]
- Burke GR, Moran NA. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol Evol. 2011;3:195–208. doi: 10.1093/gbe/evr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at silent sites in mammals. Nat Rev Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH. Codon usage between genomes is constrained by genome-wide mutational processes. Proc Natl Acad Sci U S A. 2004;101(10):3480–3485. doi: 10.1073/pnas.0307827100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohanim AB, Haran TE. The coexistence of the nucleosome positioning code with the genetic code on eukaryotic genomes. Nucleic Acids Res. 2009;37(19):6466–6476. doi: 10.1093/nar/gkp689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FHC. Codon-anticodon pairing: the wobble hypothesis. J Mol Biol. 1966;19:548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- Crick FHC. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- Dai Z, Dai X, Xiang Q. Genome-wide DNA sequence polymorphisms facilitate nucleosome positioning in yeast. Bioinformatics. 2011;27(13):1758–1764. doi: 10.1093/bioinformatics/btr290. [DOI] [PubMed] [Google Scholar]
- Denver DR, et al. Variation in base-substitution mutation in experimental and natural lineages of Caenorhabditis nematodes. Genome Biol Evol. 2012;4(4):513–522. doi: 10.1093/gbe/evs028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2005;102(40):14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L. Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev. 2002;12:640–649. doi: 10.1016/s0959-437x(02)00353-2. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion in the evolution of mammalian landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci U S A. 1999;96:4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Wall DP, Eisen MB. Coevolution of gene expression among interacting proteins. Proc Natl Acad Sci U S A. 2004;101:9033–9038. doi: 10.1073/pnas.0402591101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeland SJ, Hurst LD. The genetic code is one in a million. J Mol Evol. 1998;47(3):238–248. doi: 10.1007/pl00006381. [DOI] [PubMed] [Google Scholar]
- Hebert C, Crollius HR. Nucleosome rotational setting is associated with transcriptional regulation in promoters of tissue-specific genes. Genome Biol. 2010;11:R51. doi: 10.1186/gb-2010-11-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heddi B, Oguey C, Lavelle C, Foloppe N, Hartmann B. Intrinsic flexibility of B-DNA: the experimental TRX scale. Nucleic Acids Res. 2010;38(3):1034–1047. doi: 10.1093/nar/gkp962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. General rules for optimal codon choice. PLOS Genet. 2009;5(7):e1000556. doi: 10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand F, Meyer A, Eyre-Walker A. Evidence of selection upon genomics GC-content in bacteria. PLOS Genet. 2010;6(9):e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Evol Biol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Itzkovitz S, Alon U. The genetic code is nearly optimal for allowing additional information with protein-coding sequences. Genome Res. 2007;17:405–412. doi: 10.1101/gr.5987307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BP. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller I, Bensasson D, Nichols RA. Transition-transversion bias is not universal: a counter example from grasshopper pseudogenes. PLOS Genet. 2007;3(2):e22. doi: 10.1371/journal.pgen.0030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Kenigsberg E, Bar A, Segal E, Tanay A. Widespread compensatory evolution conserves nucleosome organization in yeast. PLOS Comp Biol. 2010;6:e1001039. doi: 10.1371/journal.pcbi.1001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- Mavrich TN, et al. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18(7):1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle-Heyrmann G, Tims HS, Widom J. Structural constraints in collaborative competition of transcription factors against the nucleosome. J Mol Biol. 2011;412(4):634–646. doi: 10.1016/j.jmb.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaou C, Althammer S, Beato M, Guigo R. Structural constraints revealed in consistent nucleosome positions in the genome of S. cerevisiae. Epigenetics Chromatin. 2010;3:20. doi: 10.1186/1756-8935-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95(19):11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SCJ, Hansen L, Abaan HO, Tullius TD, Marguilies EH. Local topography correlates with functional non-coding regions of the human genome. Science. 2009;324:389–392. doi: 10.1126/science.1169050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SCJ, Tullius TD. DNA shape, genetic codes, and evolution. Curr Opin Struct Biol. 2011;21:1–6. doi: 10.1016/j.sbi.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, et al. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscio JZ, Onufriev A. A computational study of nucleosomal DNA flexibility. Biophys J. 2006;91:4121–4132. doi: 10.1529/biophysj.106.082099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. From DNA sequence to transcriptional behavior: a quantitative approach. Nat Rev Genet. 2009;10(7):443–456. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The codon adaptation index—a measure of directional synonymous codon usage, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletski N, Eyre-Walker A. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol Biol Evol. 2007;25:568–579. doi: 10.1093/molbev/msl166. [DOI] [PubMed] [Google Scholar]
- Stoltzfus A, Yampolsky LY. Amino acid exchangeability and the adaptive code hypothesis. J Mol Evol. 2007;65(4):456–462. doi: 10.1007/s00239-007-9026-8. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Saito R, Tomita M. The “weighted sum of relative entropy”: a new index for synonymous codon usage bias. Gene. 2004;335:19–23. doi: 10.1016/j.gene.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Tims HS, Gurunathan K, Levitus M, Widom J. Dynamics of nucleosome invasion by DNA binding proteins. J Mol Biol. 2011;411:430–448. doi: 10.1016/j.jmb.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Berman J, Barkai N. The pattern and evolution of yeast promoter bendability. Trends Genet. 2007;23(7):318–321. doi: 10.1016/j.tig.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Tolstorukov MY, Colasanti AV, McCandlish DM, Olson WK, Zhurkin VB. A novel roll-and-slide mechanism of DNA folding in chromatin: implications for nucleosome positioning. J Mol Biol. 2007;371:725–738. doi: 10.1016/j.jmb.2007.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstorukov MY, Jernigan RL, Zhurkin VB. Protein-DNA hydrophobic recognition in the minor groove is facilitated by sugar switching. J Mol Biol. 2004;337(1):65–76. doi: 10.1016/j.jmb.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Tullius TD. DNA binding shapes up. Nature. 2009;461:1225–1226. doi: 10.1038/4611225a. [DOI] [PubMed] [Google Scholar]
- Wang D, Ulyanov NB, Zhurkin VB. Sequence-dependent kink and slide deformations of nucleosomal DNA facilitated by histone arginines bound in the minor groove. J Biomol Struct Dyn. 2010;27(6):843–859. doi: 10.1080/07391102.2010.10508586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke T, Batada NN, Hurst LD. The impact of the nucleosome code on protein coding sequence evolution in yeast. PLOS Genet. 2008;4:e1000250. doi: 10.1371/journal.pgen.1000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhuang S, Zhang J. Impact of translational error-induced and error-free misfolding on the rate of protein evolution. Mol Syst Biol. 2010;6:421. doi: 10.1038/msb.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. New York: Oxford University Press; 2006. Computational molecular evolution. [Google Scholar]
- Yang Z. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yosef N, Regev A. Impulse control: temporal dynamics in gene transcription. Cell. 2011;144(6):886–896. doi: 10.1016/j.cell.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CT, Zeng XB, Huang WD. Codon usage decreases the error minimization within the genetic code. J Mol Evol. 2003;57(5):533–537. doi: 10.1007/s00239-003-2505-7. [DOI] [PubMed] [Google Scholar]