

Abstract
Sequencing of the complete mitochondrial genome of the soft coral Paraminabea aldersladei (Alcyoniidae) revealed a unique gene order, the fifth mt gene arrangement now known within the cnidarian subclass Octocorallia. At 19,886 bp, the mt genome of P. aldersladei is the second largest known for octocorals; its gene content and nucleotide composition are, however, identical to most other octocorals, and the additional length is due to the presence of two large, noncoding intergenic regions. Relative to the presumed ancestral octocoral gene order, in P. aldersladei a block of three protein-coding genes (nad6–nad3–nad4l) has been translocated and inverted. Mapping the distribution of mt gene arrangements onto a taxonomically comprehensive phylogeny of Octocorallia suggests that all of the known octocoral gene orders have evolved by successive inversions of one or more evolutionarily conserved blocks of protein-coding genes. This mode of genome evolution is unique among Metazoa, and contrasts strongly with that observed in Hexacorallia, in which extreme gene shuffling has occurred among taxonomic orders. Two of the four conserved gene blocks found in Octocorallia are, however, also conserved in the linear mt genomes of Medusozoa and in one group of Demospongiae. We speculate that the rate and mechanism of gene rearrangement in octocorals may be influenced by the presence in their mt genomes of mtMutS, a putatively active DNA mismatch repair protein that may also play a role in mediating intramolecular recombination.
Keywords: mtMutS, 28S rDNA, molecular phylogenetics, soft coral, gene inversion
Introduction
The mitochondrial genomes of the nonbilaterian metazoan phyla have a number of unique features not found in the relatively homogeneous, compact mt genomes of other invertebrate and vertebrate groups (Lavrov 2007). These include additional protein-coding genes (mtMutS: Pont-Kingdon et al. 1995, 1998; atp9: Lavrov et al. 2005; polB: Shao et al. 2006; Haen et al. 2007; Signorovitch et al. 2007; tatC: Wang and Lavrov 2007; Wang and Lavrov 2008) and ORFs of unknown function (Dellaporta et al. 2006; Signorovitch et al. 2007; Kayal et al. 2012), a reduced number of tRNA genes (Beagley et al. 1995; Lavrov 2007; Wang and Lavrov 2008), and unique structural features, such as the linear mt genomes characteristic of the medusozoan cnidarian classes (Bridge et al. 1992; Shao et al. 2006; Kayal and Lavrov 2008; Voigt et al. 2008; Kayal et al. 2012; Smith et al. 2012) and the group I and II introns found in some anthozoan cnidarians, sponges and placozoans (Beagley et al. 1996, 1998; van Oppen et al. 2000; Dellaporta et al. 2006; Medina et al. 2006; Rot et al. 2006; Signorovitch et al. 2007; Chen et al. 2008; Wang and Lavrov 2008; Burger et al. 2009). Anthozoans and some sponges also have unusually slow rates of mitochondrial gene evolution, estimated to be 10−100× slower than other metazoans and up to 5× slower than nuclear genes (Shearer et al. 2002; Hellberg 2006; Huang et al. 2008; Chen et al. 2009).
Among the cnidarian class Anthozoa, members of the subclass Octocorallia (soft corals, gorgonians and sea pens) have a unique protein-coding gene within their mt genome, the mitochondrial MutS (mtMutS) (Pont-Kingdon et al. 1995, 1998). This gene appears to code for a functional homolog of a non-eukaryotic mismatch repair protein related to MutS7 (Bilewitch and Degnan 2011). mtMutS has been found in every octocoral mt genome that has been screened to date (McFadden et al. 2010), but in no other metazoan mt genomes. Among the anthozoans, octocorals have especially slow rates of mitochondrial gene evolution, and the possession of a functional DNA repair mechanism has been hypothesized to explain this difference (Bilewitch and Degnan 2011).
Until recently, it had also been assumed that, concordant with its slow rates of gene sequence evolution, the octocoral mt genome exhibited no gene rearrangements. Although the majority of the octocoral mt genomes that have been sequenced to date do share the same gene order (Park et al. 2012), several recent studies have now documented alternative gene orders in three taxa: the isidid subfamily Keratoisidinae (Brugler and France 2008; van der Hamm et al. 2009), and the species Corallium konojoi and Paracorallium japonicum, both belonging to family Coralliidae (Uda et al. 2011). Each of these alternative gene orders is characterized by a block of five or more genes that has been inverted, with the consequent reversal of the coding strand (Uda et al. 2011). The most commonly documented mechanism of mt genome rearrangement—tandem duplication followed by random loss of genes (Boore 2000) —cannot explain gene inversion, and it has been suggested that octocoral mitochondrial genomes have evolved instead through a process involving intramolecular recombination (Brugler and France 2008; Uda et al. 2011).
Here, we document an additional genus of octocoral with a novel mt gene arrangement. Although the mt gene order in Paraminabea aldersladei (family Alcyoniidae) differs from that known for any other octocoral, it shares several characteristics with Keratoisidinae and Coralliidae, and further supports intramolecular recombination as the predominant mechanism underlying mitochondrial genome evolution in Octocorallia. We map mt gene orders onto a taxonomically comprehensive phylogeny of Octocorallia based on combined mitochondrial and nuclear gene sequences to demonstrate that all known octocoral genome rearrangements can be generated by successive inversions of conserved blocks of protein-coding genes, a mode of mt genome evolution very different from that observed in all other Metazoa, including the other anthozoan sub-class, Hexacorallia.
Materials and Methods
A specimen of Paraminabea aldersladei was collected in 2005 from the Republic of Palau, and preserved in 95% EtOH. The voucher was deposited at the Museum and Art Gallery of the Northern Territory, Darwin, AUS (NTM C14895). DNA was extracted from the EtOH-preserved tissue using Qiagen’s DNEasy Blood and Tissue Kit following the manufacturer’s recommended protocol. Attempts to amplify the genome in several 5−6 kb pieces using a long- polymerase chain reaction (PCR) protocol (Burger et al. 2007) were unsuccessful due to the relatively poor quality of the DNA template. The genome was instead amplified in pieces less than 3 kb in length, using PCR primers designed from complete octocoral mt genomes available in GenBank. Initially, gene order was tested by amplifying and sequencing junctions between genes using forward and reverse primers located in the conserved coding regions flanking each intergenic region (supplementary table S1, Supplementary Material online). Regions of DNA spanning four gene junctions could not be amplified in P. aldersladei (cox1–rns, cob–nad6, nad4l–mtMutS, and nad4–cox3). To test the hypothesis that a gene rearrangement had occurred, primers were paired to anticipate alternative rearrangements, including using unorthodox primer combinations (forward–forward and reverse–reverse pairings) to test for changes in coding strand. Subsequently, the complete mt genome was amplified and sequenced using species-specific primers located within the previously sequenced intergenic regions. All gene regions were PCR-amplified and sequenced using standard, published protocols (e.g., McFadden et al. 2011).
Sequence Analysis
Sequences were trimmed, proofread, and aligned to other octocoral mitochondrial genomes using the LaserGene (DNAStar Inc.) software package. Boundaries of protein-coding genes were determined based on alignment to other annotated octocoral mt genomes (e.g., Park et al. 2012), and locations of the cnidarian start (ATG) and stop (TAA, TAG) codons. tRNAscan-SE (Lowe and Eddy 1997) was used to locate and determine the structure of tRNA genes; ribosomal RNA gene boundaries were determined based only on alignment similarities. Base composition was determined using LaserGene (EditSeq module), and the GraphDNA package (Thomas et al. 2007) was used to analyze strand-specific compositional asymmetry (purine, keto, and GC-skew). To identify features associated with origins of replication, we used the DNA-Walk method of Lobry (1996) implemented in GraphDNA to identify abrupt reversals in compositional bias that are often associated with ORs (Mrazek and Karlin 1998). We also used M-Fold (Zuker 2003), Tandem Repeats Finder (Benson 1999), and Inverted Repeats Finder (Gelfand et al. 2006) to search the entire genome, and in particular the putative control regions, for structures such as stem-loop hairpins, tandem repeats, and inverted repeats. To determine whether any additional genes were present, the entire mt genome was screened for open reading frames with NCBI’s ORF Finder, as well as the prokaryotic genome annotator, RAST-Server (Aziz et al. 2008), using a minimally derived genetic code.
Phylogenetic Analysis
To characterize the phylogenetic distribution of alternative gene orders in Octocorallia, we constructed a phylogenetic tree using combined mitochondrial (mtMutS, cox1) and nuclear (28S rDNA) gene sequences from 145 reference taxa (representing 35 families and 120 genera) for which data are available in GenBank (supplementary table S2, Supplementary Material online). The 16 octocoral species whose complete mt genomes have been published were included in this analysis, as well as representatives of six additional genera for which only gene order has been reported (table 1). Nuclear gene sequences were unavailable for many of the species whose complete mt genomes are known. Whenever possible, therefore, we compensated for missing sequence data by including in our analyses 28S rDNA sequences for other representatives of the same genera. Members of three hexacorallian orders for which cox1 and 28S rDNA sequences were both available were included as outgroup taxa in all analyses: Leiopathes sp. (Antipatharia), Nematostella vectensis (Actiniaria), Metridium senile (Actiniaria), and Montastraea franksi (Scleractinia) (supplementary table S2, Supplementary Material online).
Table 1.
Octocorals for Which Complete Mitochondrial Genomes Have Been Sequenced or for Which mt Gene Order Has Been Reported
| Family | Genus and Species | Gene Order | Total Length (bp) | Total IGR (bp) | GenBank Acc. No. | References |
|---|---|---|---|---|---|---|
| Alcyoniidae | Lobophytum pauciflorum | A | NR | NR | NA | Chen C-T, unpublished MSc thesis |
| Alcyoniidae | Sarcophyton glaucum | A | 18,453 | 438 | AF063191, AF064823 | Pont-Kingdon et al. (1995) and Beaton et al. (1998) |
| Alcyoniidae | Sarcophyton sp. | A | NR | NR | NA | Chen C-T, unpublished MSc thesis |
| Alcyoniidae | Sinularia flexibilis | A | NR | NR | NA | Chen C-T, unpublished MSc thesis |
| Alcyoniidae | S. leptoclados | A | NR | NR | NA | Chen C-T, unpublished MSc thesis |
| Briareidae | Briareum asbestinum | A | 18,632 | 727 | DQ640649 | Medina et al. (2006) |
| Ellisellidae | Junceella fragilis | A | NR | NR | NA | Wu J-H, unpublished PhD dissertation |
| Gorgoniidae | Pseudopterogorgia bipinnata | A | 18,733 | 671 | DQ640646 | Medina et al. (2006) |
| Nephtheidae | Dendronephthya gigantea | A | 18,842 | 804 | FJ372991 | Park et al. (2010) |
| Nephtheidae | D. mollis | A | 18,844 | 806 | HQ694725 | Park et al. (2012) |
| Nephtheidae | D. putteri | A | 18,853 | 817 | HQ694726 | Park et al. (2012) |
| Nephtheidae | D. suensoni | A | 18,885 | 802 | GU047878 | Park et al. (2012) |
| Nephtheidae | D. castanea | A | 18,907 | 804 | GU047877 | Park et al. (2012) |
| Nephtheidae | Nephthea erecta | A | NR | NR | NA | Chen C-T, unpublished MSc thesis |
| Nephtheidae | Scleronephthya gracillimum | A | 18,950 | 908 | GU047879 | Park et al. (2012) |
| Plexauridae | Calicogorgia granulosa | A | 20,246 | 2,249a | GU047880 | Park et al. (2011) |
| Plexauridae | Echinogorgia complexa | A | 19,445 | 1,411 | HQ694727 | Park et al. (2012) |
| Plexauridae | Euplexaura crassa | A | 18,674 | 642 | HQ694728 | Park et al. (2012) |
| Renillidae | Renilla koellikeri | A | 18,911 | 781 | NA | Beagley et al. (1995) |
| Isididae | Acanella eburnea | B | 18,616 | 587 | EF672731 | Van der Hamm et al. (2009) |
| Isididae | Keratoisidinae BAL208-1 | B | 18,923 | 927 | EF622534 | Brugler and France (2008) |
| Coralliidae | Paracorallium japonicum | C | 18,913 | 767 | AB595189 | Uda et al. (2011) |
| Coralliidae | Corallium konojoi | D | 18,969 | 870 | AB595190 | Uda et al. (2011) |
| Paragorgiidae | Paragorgia cf. coralloides | D | 19,016 | NR | NA | Thoma J, Brugler MR, France SC, personal communication |
| Alcyoniidae | Paraminabea aldersladei | E | 19,886 | 1,874 | JX508792 | This study |
Note.—NA, no accession; NR, not reported; Total IGR, sum of non-coding intergenic regions.
aIncludes duplicated ORFs of 672 bp each.
Sequences were aligned using the L-INS-i method and default alignment parameters in MAFFT (Katoh et al. 2005), and Modeltest 3.0 (Posada and Crandall 1998) was used to select appropriate models of evolution. Separate maximum likelihood analyses were run on the 28S rDNA (GTR+I+G model) and combined mitochondrial genes (mtMutS+cox1: TVM+I+G model) datasets using GARLI 2.0 (Zwickl DJ, unpublished PhD dissertation). The resulting phylogenetic trees were highly congruent, so all three gene regions were then concatenated and combined in an analysis with different models of evolution applied to each data partition. A Bayesian analysis of the same combined data set was run using MrBayes v. 3.2.1 (Ronquist et al. 2012); because MrBayes does not support the TVM model, however, a GTR+I+G model was applied to both data partitions. Analyses were run for three million generations (until standard deviation of split partitions <0.01) with a burn-in of 25% and default Metropolis coupling parameters.
Results
Genome Organization
The complete, circular mitochondrial genome of Paraminabea aldersladei (GenBank accession no. JX508792) is 19,886 bp in length, which makes it the second largest mt genome known for octocorals (table 1). Its overall base composition (A+T = 62.44%) is similar to that of other octocorals (range = 62−64%), and the genome contains the typical 14 protein-coding genes (nad1-6, nad4l, cox1-3, atp6, atp8, cob, and the octocoral-specific gene mtMutS), two ribosomal RNA genes (rnl and rns), and one transfer RNA (trnM) (fig. 1 and table 2; trnM structure shown in supplementary fig. S1, Supplementary Material online). The additional length of the P. aldersladei genome is due to the presence of two large intergenic regions (IGRs), one (562 bp) located between nad4 and nad5, and the other (434 bp) between cob and mtMutS. Three other intergenic regions are also relatively large (nad4–nad4l: 106 bp; nad6–trnM: 161 bp; cox1–cox2: 167 bp); two of these IGRs (nad4–nad4l, nad6–trnM) plus the one between cob-mtMutS represent novel gene junctions not found in genomes with the most common octocoral gene arrangement (fig. 2a-A). Although an open reading frame of 540 bp was detected within the nad4–nad5 IGR (positions 13529−14068), the encoded amino acid sequence contains no recognizable conserved domains, making its status as a functional protein questionable.
Fig. 1.—
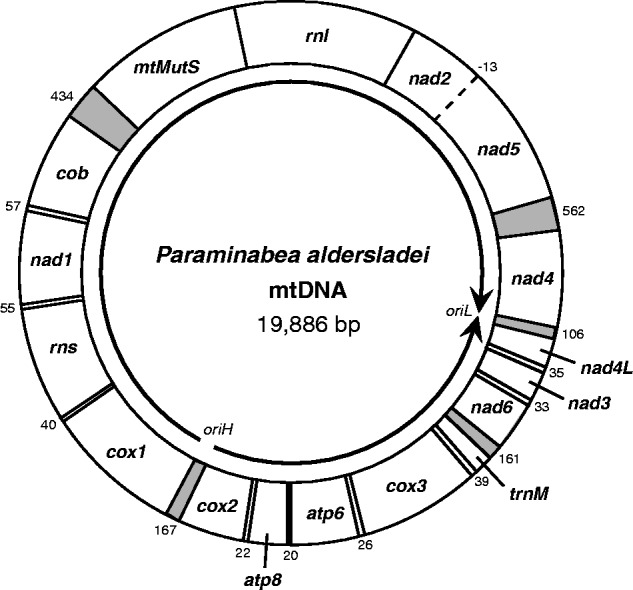
Complete circular mitochondrial genome of Paraminabea aldersladei (GenBank accession no. JX508792). Arrows indicate direction of transcription. Lengths of intergenic regions (IGRs) are indicated by numbers around outside of circle; IGRs > 100 bp are shaded.
Table 2.
Locations and Properties of Genes Encoded by the Paraminabea aldersladei Mitochondrial Genome
| Gene | Position | Length (bp) | Coding Strand | Start Codon | Stop Codon | Intergenic Space |
|---|---|---|---|---|---|---|
| cox1 | 1–1596 | 1,596 | H | ATG | TAA | 40 |
| rns | 1637–2680a | 1,044 | H | — | — | 55 |
| nad1 | 2736–3707 | 972 | H | ATG | TAG | 57 |
| cob | 3765–4931 | 1,167 | H | ATG | TAG | 434 |
| mtMutS | 5366–8359 | 2,994 | H | GTG | TAA | 0 |
| rnl | 8360–10581b | 2,222 | H | — | — | 0 |
| nad2 | 10582–11721c | 1,140 | H | ATG | TAG | −13 |
| nad5 | 11709–13526 | 1,818 | H | ATG | TAG | 562 |
| nad4 | 14089–15537 | 1,449 | H | ATG | TAA | 106 |
| nad4L | 15644–15937 | 294 | L | ATG | TAA | 35 |
| nad3 | 15973–16326 | 354 | L | ATG | TAG | 33 |
| nad6 | 16360–16908 | 549 | L | ATAd | TAA | 161 |
| trnM | 17070–17140 | 71 | L | — | — | 39 |
| cox3 | 17180–17965 | 786 | L | ATG | TAG | 26 |
| atp6 | 17992–18699 | 708 | L | ATG | TAA | 20 |
| atp8 | 18720–18935 | 216 | L | ATG | TAG | 22 |
| cox2 | 18958–19719 | 762 | L | ATG | TAA | 167 |
Note.—Codons in bold are alternative start codons.
aAnnotated by alignment with Park et al. (2012).
bThe 5'-end aligned with Park et al. (2012); 3' terminus unknown.
cStart codon inferred by alignment with McFadden et al. (2004); differs from Park et al. (2012).
dAlternatively, ATG start codon located at position 16872; see text for discussion.
Fig. 2.—

Mitochondrial gene arrangements of Octocorallia. (a) The four previously known octocoral genome arrangements. Blocks of genes that are conserved among species are color-coded: pink: block 1, cox1–rns–nad1–cob; blue: block 2, nad6–nad3–nad4l; yellow: block 3, mtMutS–rnl–nad2–nad5–nad4; green: block 4, trnM–cox3–atp6–atp8–cox2. Gene order A (Briareum asbestinum) is presumed to be ancestral. Arrows and dotted lines show inversions of blocks necessary to generate gene orders B (Keratoisidinae), C (Paracorallium japonicum), and D (Corallium konojoi) from A. a, b, locations of inverted repeats in genomes C, D. Heavy line below gene names indicates genes encoded on light strand. (b) Gene order E (Paraminabea aldersladei) hypothesized to have arisen from ancestral gene order A by long-range inversion of block 2.
Most notably, the gene order of the P. aldersladei mt genome differs from that of all other octocorals. Relative to the most common gene order found to date in the majority of species (fig. 2a-A), P. aldersladei has undergone a translocation and inversion of a block of three genes [nad6–nad3–nad4l] that has been inserted between nad4 and trnM with the direction of transcription reversed (figs. 1 and 2b-E). Nine genes are thus encoded on the heavy strand (cox1–rns–nad1–cob–mtMutS–rnl–nad2–nad5–nad4) with the remaining eight genes encoded on the light strand (nad4l–nad3–nad6–trnM–cox3–atp6–atp8–cox2) (table 2). Analysis of base composition bias using Lobry’s (1996) DNA-Walk method reveals two abrupt reversals in direction (supplementary fig. S2, Supplementary Material online), one of which falls within the cox1–cox2 IGR that has been inferred to be the origin of replication (oriH) in other octocoral mt genomes (Brugler and France 2008; Uda et al. 2011), and corresponds to the reversal in transcriptional orientation. Several stable stem-loop structures were found within the cox1–cox2 IGR, one of which (at positions 19857−19882) (supplementary fig. S3, Supplementary Material online) has a nearly identical sequence to a stem-loop structure found in Briareum that has been inferred to be associated with the replication origin (Brugler and France 2008). The second base composition bias reversal occurs within the nad4–nad4l IGR, also coincident with the point at which the direction of transcription is reversed. One stable stem-loop structure was found within this IGRs, although it did not have a T-rich loop characteristic of mtDNA primase recognition sites. Brugler and France (2008) suggested that oriL was located within the nad4–nad4l IGRs of Keratoisidinae BAL208-1; in that species too, the nad4–nad4l IGR represents the point of transition between genes encoded on the heavy versus light chains. No inverted repeats were detected within the P. aldersladei genome, and no tandem repeats were found within either putative ori; a 15-bp tandem repeat was, however, found within the cob-mtMutS IGR (positions 4968−4997).
Protein-Coding Genes
All of the protein-coding genes found in the P. aldersladei mt genome are of similar or the exact same size as those found in other octocorals (Park et al. 2012), and all terminate with either TAG or TAA (table 2). Unlike other octocorals, however, in which cox1 has been inferred to terminate at CTTT with the addition of two 3' adenines, P. aldersladei has a TAA stop codon at position 1594−1596. Although the majority of protein-coding sequences in P. aldersladei initiate with ATG, an alternative start codon was inferred for mtMutS and also for nad6. P. aldersladei has GTG at the position homologous to the ATG start codon of mtMutS; GTG is commonly used as an alternative start codon in other cnidarian mt genomes (e.g., Kayal et al. 2012). The start codon for nad6 is unclear. An ATG at positions 16870−16872 is 17 amino acids downstream of the location of the ATG inferred to represent the start codon in other octocorals. Conservation of all residues immediately following an ATA at positions 16906−16908, however, suggests that as a possible alternative start codon that is only five amino acids downstream. Use of ATA as a start codon has been suggested previously in several other cnidarian mitochondrial genomes (Beaton et al. 1998; van Oppen et al. 2000), but has yet to be confirmed by protein expression studies.
Phylogenetic Analysis
The combined analysis of mtMutS, cox1 and 28S rDNA sequences (total alignment length 2535 bp) yielded a phylogenetic tree that is similar in most respects to that obtained previously using only mitochondrial gene sequences (mtMutS+nad2) and a smaller set of taxa (McFadden et al. 2006). Maximum likelihood and Bayesian analyses generated identical tree topologies, and differed only in degrees of support for some of the deeper nodes within the tree (fig. 3); in general, Bayesian analysis was less conservative, supporting some nodes that were not well supported by maximum likelihood.
Fig. 3.—

Phylogenetic tree of Octocorallia based on maximum likelihood analysis of combined sequence data for cox1, mtMutS, and 28S rDNA. Taxa for which mitochondrial gene order is known are shown in color: red: gene order A; green: B; dark blue: C; light blue: D; purple: E. Circles on nodes indicate support values: solid circle: ML bootstrap value > 70%, Bayesian pp > 95; open circle: ML bootstrap value < 70%, Bayesian pp > 95. Some clades with strong support (Bayesian pp > 95) have been collapsed to triangles to improve readability. Large circles denote major clades defined in McFadden et al. (2006). CP, Calcaxonia-Pennatulacea; HA, Holaxonia-Alcyoniina; AC, Anthomastus-Corallium. Tree is rooted to Hexacorallia, outgroup taxa not shown.
The phylogeny recovers the same major clades found in previous molecular phylogenetic analyses of Octocorallia (Berntson et al. 2001; McFadden et al. 2006). Two large clades are recovered with moderate to strong support: Holaxonia-Alcyoniina (ML bootstrap = 95%, Bayesian pp = 0.99), comprising the majority of families and genera of order Alcyonacea belonging to the sub-ordinal groups Holaxonia (gorgonians), Alcyoniina (soft corals), and Stolonifera (stoloniferous octocorals); and Calcaxonia-Pennatulacea (ML bootstrap = 63%, Bayesian pp = 0.99), comprising the orders Pennatulacea (sea pens), Helioporacea (blue coral), and the alcyonacean suborder Calcaxonia (mostly deep-water gorgonians). Two smaller clades, Anthomastus-Corallium (ML bootstrap = 94%, Bayesian pp = 1.0) and a clade of alcyonaceans belonging primarily to the sub-ordinal group Scleraxonia (ML bootstrap = 74%, Bayesian pp = 0.99) fall outside of Holaxonia-Alcyoniina, but their relationships to one another and to Calcaxonia-Pennatulacea remain unresolved (fig. 3). The stoloniferan family Cornulariidae, not included in previous analyses, is the sister group to all other Octocorallia.
Evolution of Mitochondrial Gene Order in Octocorallia
Species with the most common mitochondrial gene order (fig. 2a-A) are found within both major clades of octocorals as well as the clade of Scleraxonia at the base of the tree (fig. 3). To date, gene order A is the only gene order that has been found within the large Holaxonia-Alcyoniina clade. Gene order A also occurs in Briareum (Scleraxonia), Renilla (Pennatulacea) and Junceella (Calcaxonia: Ellisellidae).
The four alternative gene orders that have been documented to date (fig. 2, B–E) all fall within either the Calcaxonia (subfamily Keratoisidinae) or Anthomastus-Corallium clades (Corallium konojoi, Paracorallium japonicum, and Paraminabea aldersladei) (fig. 3). It is particularly noteworthy that three novel gene orders are found among the approximately nine genera that are known to belong to the small Anthomastus-Corallium clade. Within this clade, Paragorgia cf. coralloides also shares gene order D with C. konojoi (Thoma J, Brugler, MR, France SC, personal communication). Successful attempts to PCR-amplify across novel gene junctions (e.g., cob-mtMutS or nad4-nad4l) suggest that another species in this clade, Notodysiferus dhondtae, may share gene order E with P. aldersladei, although that result has not yet been confirmed by sequencing (S. Brockman, unpublished data). Sequencing across the nad4l–mtMutS and cox1–cox2 gene junctions indicates, however, that Eleutherobia flammicerebra, several species in the genus Anthomastus, and an undescribed genus of Alcyoniidae included in McFadden et al. (2006) (Alcyoniidae n. gen. WAM Z13105) do not share gene orders C, D, or E with other members of the clade, but most likely have retained gene order A (fig. 4).
Fig. 4.—

Cladogram of Calcaxonia-Pennatulacea and Anthomastus-Corallium clades of Octocorallia showing inferred ancestral states of clades (circled letters) and points at which genome rearrangements (colored bars) have occurred. Letters A–E refer to the five gene arrangements shown in figure 2, colored bars correspond to conserved gene blocks 1−4 (heavy line below numbers indicates gene blocks encoded on light strand). Circled letters next to taxon names indicate the gene order as known from complete mt genome sequences (shaded circles) or inferred from gene junction screening (open circles).
Discussion
Phylogenetic Distribution of Gene Order Changes within Octocorallia
Comparisons of the five mitochondrial gene orders now known from Octocorallia suggest that inversions of large blocks of protein-coding genes underlie all of the gene order changes known to have occurred within this sub-class of cnidarians (fig. 2). Among the five different genomes, four large blocks of genes have remained conserved and have been inverted or translocated as a unit: block 1: cox1–rns–nad1–cob; block 2: nad6–nad3–nad4l; block 3: mtMutS–rnl–nad2–nad5–nad4; and block 4: trnM–cox3–atp6–atp8–cox2. The documented changes in gene order all involve the inversion of one or more of these four blocks, such that half to two-thirds of the genes in the mt genome are always encoded on the heavy strand in one contiguous stretch.
Modifications of gene order are considered to be rare genetic changes in animals (Boore and Brown 1998; Boore 1999). The broad phylogenetic distribution of gene order A suggests, therefore, that it represents the ancestral gene order within Octocorallia; each of the four alternative gene orders B-E is restricted to a single, small clade and appears to have evolved only once (figs. 3 and 4). Gene orders B, C, and D can all be generated from A by a series of local inversions, each requiring double-stranded breakage of the genome on either side of a conserved block of genes (fig. 2a). For example, within the Calcaxonia clade, the inversion of block 3 gave rise to gene order B in the lineage leading to sub-family Keratoisidinae. Gene junction screening suggests that gene order B is shared by all genera in that sub-family, whereas ancestral gene order A is retained in other sub-families of Isididae and in families Primnoidae and Chrysogorgiidae (Brugler and France 2008) (fig. 4).
Within the Anthomastus-Corallium clade, results from gene junction screening suggest that both Anthomastus and E. flammicerebra retain ancestral gene order A. The phylogeny supports a sister relationship between Anthomastus and the (Paracorallium-Corallium-Paragorgia) clade (fig. 3). Although the relationships among Paracorallium, Corallium, and Paragorgia are not well resolved (Herrera et al. 2010), we found moderate support for a sister relationship between Corallium and Paragorgia. That relationship is consistent with the hypothesis that an inversion of blocks 2−3−4 occurred following the divergence of the (Paracorallium-Corallium-Paragorgia) lineage from Anthomastus, generating gene order C observed in P. japonicum (figs. 2a and 4). Subsequently, a second inversion of blocks 3 and 4 occurred in the lineage leading to C. konojoi and Paragorgia, giving rise to gene order D. Uda et al. (2011) presented two alternative models for the evolution of gene orders C and D from ancestral gene order A. Their models are complex, requiring gene rearrangement to have occurred either by gene duplication followed by inversion and random loss, or by double-stranded breakage, blunting of overhangs, and re-integration of blunt-ended fragments into the genome in inverted orientation. In addition, they assumed that rearrangement of the C. konojoi genome (D) preceded that of P. coralloides (C), an evolutionary progression that is not supported by our phylogenetic reconstruction (figs. 3 and 4).
The phylogeny supports P. aldersladei as the earliest-diverging genus in the Anthomastus-Corallium clade (fig. 3). That relationship suggests that gene order E evolved directly from ancestral gene order A, a change that would have required the inversion and long-distance translocation of gene block 2 (fig. 2b). In contrast to the local inversions hypothesized to generate gene orders B, C, and D (fig. 2a), translocation would have required three rather than two double-stranded breaks in the DNA. Inversions of gene blocks accompanied by translocation (so-called long-range inversions) have, however, been observed in other metazoan taxa, particularly those in which tRNA genes are frequently rearranged (Grande et al. 2008; Dowton et al. 2009; Rawlings et al. 2010).
Mechanisms of Gene Rearrangement in Octocorallia
Among metazoans, mitochondrial genome rearrangements resulting from inversions of large blocks of protein-coding genes are relatively rare, and many of the “extensive” changes in gene order that have been reported and widely studied involve the movement of tRNA genes only (e.g., Xu et al. 2006; Gissi et al. 2008; Dowton et al. 2009). Some notable exceptions include the inversion of a block of six protein-coding genes and four tRNAs that has occurred in several genera of chalcidoid wasps (Dowton et al. 2009; Xiao et al. 2011); a block of six protein-coding genes, rns, rnl, and 8−10 tRNAs that has been inverted between the ancestral arthropod gene order and Priapulida (Rota-Stabelli et al. 2010), and independently in Caenogastropoda (Grande et al. 2008; Rawlings et al. 2010); the inversion of a block that includes two protein-coding genes, rnl, and a large cluster of tRNA genes in two classes of echinoderms (Smith et al. 1989); and a clade of placozoans and two species of Demospongiae that have each had half to two-thirds of the genome inverted (Signorovitch et al. 2007; Wang and Lavrov 2008). In all but the latter case, however, inversion of a large block of protein-coding genes apparently has occurred only once in the history of a major lineage. We are aware of no other cases in which multiple genome rearrangements within a lineage have occurred by successive inversions of large blocks of protein-coding genes.
The duplication/random loss model that is frequently invoked as the primary mechanism underlying changes in mitochondrial gene order in metazoans (Boore 2000) cannot account for gene inversions. Mechanisms that generate inversions require the double-stranded breakage of DNA on either side of the inverted region followed by subsequent re-integration of the excised fragment back into the circular genome in opposite orientation (Dowton and Campbell 2001). Re-integration can occur either at the original breakage point to generate a local inversion, or elsewhere in the genome in the case of long-range inversions. Most of the rearrangements in Octocorallia appear to have been local inversions, with the exception of gene order E, which requires a long-range inversion to have evolved directly from gene order A.
The evolution of gene rearrangements by the frequent inversion of large, conserved blocks of genes suggests (a) that there may be selective advantages to some genes remaining together; and (b) that the junctions between blocks may represent hotspots that promote the double-stranded breakage necessary to explain gene inversion (Dowton and Campbell 2001). There is little evidence from other metazoan taxa, however, that selection favors particular mitochondrial gene orders (Dowton et al. 2009); within Cnidaria that conclusion is supported by the highly variable gene orders observed within subclass Hexacorallia, in which almost no gene boundaries are shared with Octocorallia or among hexacorallian orders (fig. 5).
Fig. 5.—

Mitochondrial gene arrangements compared among Demospongiae (group G1, Wang and Lavrov 2008), Medusozoa (AMGO = ancestral medusozoan gene order, Kayal et al. 2012), Octocorallia (presumed ancestral gene order A), and five orders of Hexacorallia. Gene blocks conserved among Demospongiae, Medusozoa, and Octocorallia are indicated using the same colors as figure 2 (blue: block 2; yellow: block 3; and green: block 4). Blocks of genes that are conserved among Hexacorallian orders are indicated using other colors. Heavy line below gene names indicates genes encoded on light strand.
Hotspots that promote gene rearrangements have, however, been identified in other taxa. Sequence motifs associated with such hotspots include stable stem-loop structures such as tRNAs (Xu et al. 2006) and origins of replication (Boore 1999, 2000); inverted repeats (Dowton et al. 2003); and short direct repeats (Kajander et al. 2000; Dowton and Campbell 2001). In octocorals, the locations of oriH, oriL and trnM all coincide with the junctions between frequently rearranged blocks of genes. oriH is associated with the 5'-end of block 1, oriL always appears to be located downstream of nad4 and thus represents the 3'-end of block 3, and block 4 is bounded on its 3'-end by trnM. The presence of at least one stable stem-loop structure is a characteristic of each of these locations (Brugler and France 2008).
To date, the only octocoral genomes in which inverted repeats have been found to be associated with gene rearrangements are C. konojoi and P. coralloides, both of which have inverted repeats of 44- and 47-bp flanking gene blocks that have been inverted (Uda et al. 2011). Although Uda et al. attempted to explain these inverted repeats as products of gene rearrangement, the a priori existence of inverted repeats is known to promote local inversion by site-specific recombination (Nash 1996; Hallet and Sherratt 1997; Dowton et al. 2003). According to our scenario (fig. 2a), site-specific recombination between inverted repeats a would have caused a local inversion of blocks 2−3−4 to generate gene order C. Subsequent site-specific recombination between sequence b repeats would generate a local inversion of blocks 3−4 (gene order D). Inverted repeats have not, however, been found in the mt genomes of Keratoisidinae (Brugler and France 2008) or in P. aldersladei, suggesting either that the gene inversions observed in those species have occurred by a mechanism other than site-specific recombination, or, alternatively, that the telltale inverted repeats have been lost subsequent to the evolution of a novel gene order.
Short direct repeats are also known to promote intramolecular recombination in mitochondrial genomes (Lunt and Hyman 1997; Kajander et al. 2000; Dowton and Campbell 2001). In contrast to the inversions that result from recombination among inverted repeats, site-specific recombination among direct repeats results in the excision of a circular “sublimon” from the genome (Nash 1996; Hallet and Sharrett 1997; Kajander et al. 2000). Re-integration of excised genes by intermolecular recombination can then occur either in the original or inverted orientation (Dowton and Campbell 2001); re-integration at a location distant from the original point of excision can generate a long-range inversion. The short length of direct repeats capable of promoting site-specific recombination—e.g., human mt genomes have been shown to undergo recombination at direct repeats of less then 12 bp (Kajander et al. 2000)—makes these motifs difficult to detect because they may be easily generated and subsequently lost in the course of evolution.
Mitochondrial Gene Order and Phylogenetic Relationships among Cnidarian Classes
Based on evidence that includes morphological and life cycle characters, mitogenomic structure (i.e., linear vs. circular mt genome), and molecular phylogenetic analyses, phylum Cnidaria has long been assumed to consist of two reciprocally monophyletic sister clades, Anthozoa and Medusozoa (Daly et al. 2007). The latter clade includes four classes—Cubozoa, Hydrozoa, Scyphozoa and Staurozoa—whose members all have linear mt genomes (Kayal et al. 2012; Smith et al. 2012). Class Anthozoa includes two large sub-classes—Hexacorallia and Octocorallia—whose members all have circular mt genomes (with the possible exception of Ceriantharia, Brugler M, unpublished MSc thesis). In contrast to all other lines of evidence, however, several recent phylogenetic reconstructions of Cnidaria based on concatenated mitochondrial protein-coding sequences have recovered a paraphyletic Anthozoa, with sub-class Octocorallia sister to a monophyletic Medusozoa (Kayal and Lavrov 2008; Park et al. 2012). This unorthodox result has generally been dismissed as an artifact either of incomplete taxon sampling (Kayal and Lavrov 2008) or of the known disparities in substitution rates among cnidarian lineages (Park et al. 2012).
Comparisons of mitochondrial gene orders among classes of cnidarians reveal additional similarities between Octocorallia and Medusozoa (fig. 5). Several of the same gene blocks that have been conserved and successively inverted within the octocoral mt genome are also conserved, in full or partially, in the linear mt genomes of Medusozoa (Kayal et al. 2012). These include block 2 (nad6–nad3–nad4l), block 4 (trnM–cox3–atp6–atp8–cox2, with the addition of trnW), and part of block 3 (nad2–nad5) (fig. 5). These same blocks are also found in the circular mt genomes of several sponge species (Vaceletia sp., Hippospongia lachnea) belonging to the G1 group of Demospongiae (Wang and Lavrov 2008) (fig. 5). Among Demospongiae, whose mt genomes vary greatly in gene content and order (Wang and Lavrov 2008), the G1 group shares with Cnidaria the loss of all tRNA genes other than trnM and trnW. Although trnW also falls within gene block 2 in Demospongiae group G1, it is in a different position within that block relative to its position in Medusozoa (fig. 5).
In extreme contrast to the conservation of gene blocks observed among Medusozoa and Octocorallia, the mitochondrial gene orders in subclass Hexacorallia show virtually no homology to other cnidarians. In Hexacorallia, all genes in the mt genome are encoded on the same strand. Moreover, the five orders of Hexacorallia for which complete mt genomes have been sequenced differ radically from one another in gene order (Medina et al. 2006; Uda et al. 2011) (fig. 5). Orders Actiniaria and Antipatharia differ by the long-range translocation of a block of three protein-coding genes (cox2–nad4–nad6); this block and several other blocks of 2−4 genes each have been conserved between these two orders and Zoanthidea (fig. 5). Only two gene boundaries (nad4–nad6, trnM–rnl) are conserved, however, between those three orders and Corallimorpharia, and only trnM–rnl is conserved across all five hexacorallian orders (fig. 5). Octocorallia shares only a single gene boundary (atp8–atp6) with Actiniaria, Antipatharia, and Zoanthidea, and has no gene boundaries in common with Corallimorpharia or Scleractinia.
The conservation of large gene blocks between Octocorallia and Medusozoa to the exclusion of Hexacorallia could be interpreted as further support for a sister relationship between those two clades (Kayal and Lavrov 2008; Park et al. 2012). An alternative, perhaps more plausible, explanation is that the gene blocks shared between Octocorallia and Medusozoa are plesiomorphic character states in Cnidaria. Although the presence of the same conserved gene blocks in group G1 sponges would seem to offer further support for this interpretation, the G1 mt genome does not appear to represent the ancestral state in Demospongiae (Wang and Lavrov 2008). To the contrary, the loss of almost all tRNA genes in group G1 suggests that they have a highly derived mt genome relative to other groups of sponges that can have up to 27 mt-encoded tRNAs (Wang and Lavrov 2008). Based on the phylogenetic position of group G1 within Demospongiae it appears, therefore, that the gene blocks it shares with Octocorallia and Medusozoa reflect convergence rather than shared ancestry.
The assumption that Octocorallia and Medusozoa retain shared ancestral character states implies that mt gene orders have remained relatively static in both groups since their divergence. It has been suggested that linearization has stabilized the genome and reduced the rate of genome rearrangement (other than fragmentation, e.g., Smith et al. 2012) in Medusozoa (Kayal et al. 2012). In comparison, the mt genomes of Hexacorallia have undergone extensive rearrangements since they last shared a common ancestor with other cnidarians. The extreme shuffling of genes among orders and the lack of gene inversions in Hexacorallia suggest that their mt genomes have evolved by mechanisms very different from those underlying the changes observed in Octocorallia or Medusozoa.
A Role for mtMutS in Genome Rearrangement?
It is tempting to speculate that the unique presence of mtMutS in the octocoral genome is responsible not only for its slow rate of nucleotide substitution (Bilewitch and Degnan 2011), but also for the unique patterns and mechanisms of genome rearrangement observed within the sub-class. A functional DNA mismatch repair system in octocoral mt genomes could reduce the incidence of slipped-strand mispairing, one of the mechanisms inferred to lead to the gene duplications proposed to account for the types of gene shuffling observed in Hexacorallia (Boore 2000). In addition to functioning in DNA mismatch repair, MutS genes with endonuclease domains similar to that of octocoral mtMutS (Abdelnoor et al. 2006) have also been demonstrated to mediate homologous recombination through double-stranded break repair in plant mitochondrial genomes (Davila et al. 2010). The double-stranded break repair activity of plant MSH1 has been suggested to account for both the low nucleotide substitution rate and high rates of genome rearrangement observed in plant mt genomes (Davila et al. 2010). If mtMutS is similarly capable of double-stranded break repair, its activity could promote the recombinational processes that underlie the evolution of mt gene order in Octocorallia. Exploration of the functional role of mtMutS is an exciting area for future research that could shed additional light on the unusual pattern of mitochondrial gene arrangements observed to date in Octocorallia.
Conclusion
The phylogenetic distribution of mitochondrial gene arrangements in Octocorallia supports the most commonly encountered arrangement A (fig. 2) as the ancestral gene order in this cnidarian subclass. All four of the alternative gene orders that are currently known have been derived from A by the local or long-range inversion of one or more conserved blocks of 3−5 protein-coding genes. Although inversions of large blocks of protein-coding genes have been observed in some other metazoan mt genomes, their occurrence is rare, and in no other taxa are multiple successive inversions known to have occurred. The frequency of inversions and the absence of any other types of mt genome rearrangements suggest a unique mode of mt gene order evolution in Octocorallia, especially when compared to its presumed sister clade, Hexacorallia. Among the mt gene orders found in Hexacorallia there have been no gene inversions, but there has instead been extreme shuffling of genes, changes that are typically explained by a model of gene duplication followed by random loss. In contrast, the prevalence of inversions implies that the primary mechanism of rearrangement in octocoral mt genomes has been intramolecular recombination, which requires double-stranded breakage and subsequent repair of the DNA molecule. We speculate that mtMutS, a mismatch repair protein that is uniquely found in the octocoral mt genome, may play a role in mt genome evolution by simultaneously reducing the incidence of slipped-strand mispairing while promoting recombination by double-stranded break repair. Further work on the functional properties of mtMutS is necessary to elucidate its contributions, if any, to the evolution of the octocoral mitochondrial genome.
Supplementary Material
Supplementary figures S1–S3 and tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank S. Abdalla for laboratory assistance, S.C. France and J. Thoma for discussion and for sharing unpublished data, and the Coral Reef Research Foundation in Palau (P. Colin and L. Colin) for collection support. The manuscript was improved by the helpful comments of three anonymous reviewers. This work was supported by the National Science Foundation grants EF-0531570 to C.S.M. and EF-0531779 to P. Cartwright and by the Arnold and Mabel Beckman Foundation (Beckman Scholars Program Award to Harvey Mudd College and S.A.B.).
Literature Cited
- Abdelnoor RV, et al. Mitochondrial genome dynamics in plants and animals: convergent gene fusions of a MutS homolog. J Mol Evol. 2006;63:165–173. doi: 10.1007/s00239-005-0226-9. [DOI] [PubMed] [Google Scholar]
- Aziz RK, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagley CT, et al. Mitochondrial genomes of Anthozoa (Cnidaria) In: Palmieri F, editor. Progress in cell research. Vol. 5. Amsterdam: Elsevier Science; 1995. pp. 149–153. [Google Scholar]
- Beagley CT, Okada NA, Wolstenholme DR. Two mitochondrial group I introns in a metazoan, the sea anemone Metridium senile: one intron contains genes for subunits 1 and 3 of NADH dehydrogenase. Proc Natl Acad Sci U S A. 1996;93:5619–5623. doi: 10.1073/pnas.93.11.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagley CT, Okimoto R, Wolstenholme DR. The mitochondrial genome of the sea anemone Metridium senile (Cnidaria): introns, a paucity of tRNA genes, and a nearstandard genetic code. Genetics. 1998;148:1091–1108. doi: 10.1093/genetics/148.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaton MJ, Roger AJ, Cavalier-Smith T. Sequence analysis of the mitochondrial genome of Sarcophyton glaucum: conserved gene order among octocorals. J Mol Evol. 1998;47:697–708. doi: 10.1007/pl00006429. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntson EA, Bayer FM, McArthur AG, France SC. Phylogenetic relationships within the Octocorallia (Cnidaria: Anthozoa) based on nuclear 18S rRNA sequences. Marine Biol. 2001;138:235–246. [Google Scholar]
- Bilewitch JP, Degnan SM. A unique horizontal gene transfer event has provided the octocoral mitochondrial genome with an active mismatch repair gene that has potential for an unusual self-contained function. BMC Evol Biol. 2011;11:228. doi: 10.1186/1471-2148-11-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In: Sankoff D, Nadeau JH, editors. Comparative genomics. Dordrecht, the Netherlands: Kluwer Academic Publishers; 2000. pp. 133–147. [Google Scholar]
- Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8:668–674. doi: 10.1016/s0959-437x(98)80035-x. [DOI] [PubMed] [Google Scholar]
- Bridge D, Cunningham CW, Schierwater B, DeSalle R, Buss LW. Class-level relationships in the phylum Cnidaria: evidence from mitochondrial genome structure. Proc Natl Acad Sci U S A. 1992;89:8750–8753. doi: 10.1073/pnas.89.18.8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugler MR, France SC. The mitochondrial genome of a deep-sea bamboo coral (Cnidaria, Anthozoa, Octocorallia, Isididae): genome structure and putative origins of replication are not conserved among octocorals. J Mol Evol. 2008;67:125–136. doi: 10.1007/s00239-008-9116-2. [DOI] [PubMed] [Google Scholar]
- Burger G, Lavrov DV, Forget L, Lang FB. Sequencing complete mitochondrial and plastid genomes. Nat Protoc. 2007;2:603–614. doi: 10.1038/nprot.2007.59. [DOI] [PubMed] [Google Scholar]
- Burger G, Yan Y, Javadi P, Lang FB. Group I-intron trans-splicing and mRNA editing in the mitochondria of placozoan animals. Trends Genet. 2009;25:381–386. doi: 10.1016/j.tig.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Chen C, Chiou C-Y, Dai C-F, Chen CA. Unique mitogenomic features in the scleractinian family Pocilloporidae (Scleractinia: Astrocoeniina) Marine Biotechnol. 2008;10:538–553. doi: 10.1007/s10126-008-9093-x. [DOI] [PubMed] [Google Scholar]
- Chen I, -P, et al. Comparative analyses of coding and non-coding DNA regions indicate that Acropora (Anthozoa: Scleractinia) possesses a similar evolutionary tempo of nuclear vs. mitochondrial genomes as in plants. Marine Biotechnol. 2009;11:141–152. doi: 10.1007/s10126-008-9129-2. [DOI] [PubMed] [Google Scholar]
- Daly M, et al. The phylum Cnidaria: A review of phylogenetic patterns and diversity 300 years after Linnaeus. In: Zhang Z-Q, Shear WA, editors. Linnaeus tercentenary: progress in invertebrate taxonomy. 2007. 1668:1–766. [Google Scholar]
- Davila JI, et al. Double-strand break repair processes drive evolution of the mitochondrial genome in Arabidopsis. BMC Biol. 2010;9:64. doi: 10.1186/1741-7007-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellaporta SL, et al. Mitochondrial genome of Trichoplax adhaerans supports Placozoa as the basal lower metazoan phylum. Proc Natl Acad Sci U S A. 2006;103:8751–8756. doi: 10.1073/pnas.0602076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton M, Cameron SL, Dowavic JI, Austin AD, Whiting MF. Characterization of 67 mitochondrial tRNA gene rearrangements in the Hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol Biol Evol. 2009;26:1607–1617. doi: 10.1093/molbev/msp072. [DOI] [PubMed] [Google Scholar]
- Dowton M, Campbell NJH. Intramitochondrial recombination—is it why some mitochondrial genes sleep around? Trends Ecol Evol. 16. 2001:269–271. doi: 10.1016/s0169-5347(01)02182-6. [DOI] [PubMed] [Google Scholar]
- Dowton M, Castro LR, Campbell SL, Bargon SD, Austin AD. Frequent mitochondrial gene rearrangements at the hymenopteran nad3–nad5 junction. J Mol Evol. 2003;56:517–526. doi: 10.1007/s00239-002-2420-3. [DOI] [PubMed] [Google Scholar]
- Gelfand Y, Rodriguez A, Benson G. TRDB—the tandem repeats database. Nucleic Acids Res. 2006;35(Database issue):D80–D87. doi: 10.1093/nar/gkl1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- Grande C, Templado J, Zardoya R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol Biol. 2008;8:61. doi: 10.1186/1471-2148-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haen KB, Lang BF, Pomponi SA, Lavrov DV. Glass sponges and bilaterian animals share derived mitochondrial genomic features: a common ancestry or parallel evolution? Mol Biol Evol. 24. 2007:1518–1527. doi: 10.1093/molbev/msm070. [DOI] [PubMed] [Google Scholar]
- Hallet B, Sheratt DJ. Transposition and site-specific recombination: adapting DNA cut-and-paste mechanisms to a variety of genetic rearrangements. FEMS Microbiol Rev. 1997;21:157–178. doi: 10.1111/j.1574-6976.1997.tb00349.x. [DOI] [PubMed] [Google Scholar]
- Hellberg ME. No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol. 2006;6:24. doi: 10.1186/1471-2148-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera S, Baco A, Sánchez JA. Molecular systematics of the bubblegum coral genera (Paragorgiidae, Octocorallia) and description of a new deep-sea species. Mol Phylogen Evol. 2010;55:123–135. doi: 10.1016/j.ympev.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Huang D, Meier R, Todd PA, Chou LM. Slow mitochondrial COI sequence evolution at the base of the metazoan tree and its implications for DNA barcoding. J Mol Evol. 2008;66:167–174. doi: 10.1007/s00239-008-9069-5. [DOI] [PubMed] [Google Scholar]
- Kajander OA, et al. Human mtDNA sublimons resemble rearranged mitochondrial genomes found in pathological states. Hum Mol Gen. 2000;9:2821–2835. doi: 10.1093/hmg/9.19.2821. [DOI] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–513. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayal E, Lavrov DV. The mitochondrial genome of Hydra oligactis (Cnidaria, Hydrozoa) sheds new light on animal mtDNA evolution and cnidarian phylogeny. Gene. 2008;410:177–186. doi: 10.1016/j.gene.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Kayal E, et al. Evolution of linear mitochondrial genomes in medusozoan cnidarians. Genome Biol Evol. 2012;4:1–12. doi: 10.1093/gbe/evr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrov DV. Key transitions in animal evolution: a mitochondrial DNA perspective. Integr Comp Biol. 2007;47:734–743. doi: 10.1093/icb/icm045. [DOI] [PubMed] [Google Scholar]
- Lavrov DV, Forget L, Kelly M, Lang BF. Mitochondrial genomes of two demosponges provide insights into an early stage of animal evolution. Mol Biol Evol. 2005;22:1231–1239. doi: 10.1093/molbev/msi108. [DOI] [PubMed] [Google Scholar]
- Lobry J. A simple vectorial representation of DNA sequences for the detection of replication origins in bacteria. Biochemie. 1996;78:323–326. doi: 10.1016/0300-9084(96)84764-x. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt DH, Hyman BC. Animal mitochondrial DNA recombination. Nature. 1997;387:247. doi: 10.1038/387247a0. [DOI] [PubMed] [Google Scholar]
- McFadden CS, France SC, Sánchez JA, Alderslade P. A molecular phylogenetic analysis of the Octocorallia (Coelenterata: Anthozoa) based on mitochondrial protein-coding sequences. Mol Phylogen Evol. 2006;41:513–527. doi: 10.1016/j.ympev.2006.06.010. [DOI] [PubMed] [Google Scholar]
- McFadden CS, Sánchez JA, France SC. Molecular phylogenetic insights into the evolution of Octocorallia: a review. Integr Comp Biol. 2010;50:389–410. doi: 10.1093/icb/icq056. [DOI] [PubMed] [Google Scholar]
- McFadden CS, Tullis ID, Hutchinson MB, Winner K, Sohm JA. Variation in coding (NADH dehydrogenase subunits 2, 3, and 6) and noncoding intergenic spacer regions of the mitochondrial genome in Octocorallia (Cnidaria: Anthozoa) Mar Biotechnol. 2004;6:516–526. doi: 10.1007/s10126-002-0102-1. [DOI] [PubMed] [Google Scholar]
- McFadden CS, et al. Limitations of mitochondrial gene barcoding in the cnidarian sub-class Octocorallia. Mol Ecol Res. 2011;11:19–31. doi: 10.1111/j.1755-0998.2010.02875.x. [DOI] [PubMed] [Google Scholar]
- Medina M, Collins AG, Takaoka TL, Kuehl JV, Boore JL. Naked corals: skeleton loss in Scleractinia. Proc Natl Acad Sci U S A. 2006;103:9096–9100. doi: 10.1073/pnas.0602444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrazek J, Karlin S. Strand compositional asymmetry in bacterial and large viral genomes. Proc Natl Acad Sci U S A. 1998;95:3720–3725. doi: 10.1073/pnas.95.7.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash HA. Site-specific recombination: integration, excision, resolution, and inversion of defined DNA segments. In: Neidhardt FC, et al., editors. Escherichia coli and Salmonella typhimurium: cellular and molecular biology. 2nd ed. Vol. 2. Washington, DC: American Society for Microbiology; 1996. pp. 2363–2376. [Google Scholar]
- Park E, Kim B, Won Y-J. The complete mitochondrial genome of Dendronephthya gigantea (Anthozoa: Octocorallia: Nephtheidae) Korean J Syst Zool. 2010;26:197–201. [Google Scholar]
- Park E, Song J-I, Won Y-J. The complete mitochondrial genome of Calicogorgia granulosa (Anthozoa: Octocorallia): potential gene novelty in unidentified ORFs formed by repeat expansion and segmental duplication. Gene. 2011;486:81–87. doi: 10.1016/j.gene.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Park E, et al. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Mol Phylogen Evol. 2012;62:329–345. doi: 10.1016/j.ympev.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Pont-Kingdon G, et al. A coral mitochondrial MutS gene. Nature. 1995;375:109–11. doi: 10.1038/375109b0. [DOI] [PubMed] [Google Scholar]
- Pont-Kingdon G, et al. Mitochondrial DNA of the coral Sarcophyton glaucum contains a gene for a homologue of bacterial MutS: a possible case of gene transfer from the nucleus to the mitochondrion. J Mol Evol. 1998;46:419–431. doi: 10.1007/pl00006321. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Rawlings TA, MacInnis MJ, Bieler R, Boore JL, Collins TM. Sessile snails, dynamic genomes: gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genomics. 2010;11:440. doi: 10.1186/1471-2164-11-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot C, Goldfarb I, Ilan M, Huchon D. Putative cross-kingdom horizontal gene transfer in sponge (Porifera) mitochondria. BMC Evol Biol. 2006;6:71. doi: 10.1186/1471-2148-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota-Stabelli O, et al. Ecdysozoan mitogenomics: evidence for a common origin of the legged invertebrates, the Panarthropoda. Genome Biol Evol. 2010;2:425–440. doi: 10.1093/gbe/evq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Graf S, Chaga OY, Lavrov DV. Mitochondrial genome of the moon jelly Aurelia aurita (Cnidaria, Scyphozoa): a linear DNA molecule encoding a putative DNA-dependent DNA polymerase. Gene. 2006;381:92–101. doi: 10.1016/j.gene.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Shearer TL, van Oppen MJH, Romano SL, Wörheide G. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria) Mol Ecol. 2002;11:2475–2487. doi: 10.1046/j.1365-294x.2002.01652.x. [DOI] [PubMed] [Google Scholar]
- Signorovitch AY, Buss LW, Dellaporta SL. Comparative genomics of large mitochondria in placozoans. PLoS Genet. 2007;3:e13. doi: 10.1371/journal.pgen.0030013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Banfield DK, Doteval K, Gorski S, Kowbel DJ. Gene arrangement in sea star mitochondrial DNA demonstrates a major inversion event during echinoderm evolution. Gene. 1989;76:181–185. doi: 10.1016/0378-1119(89)90022-x. [DOI] [PubMed] [Google Scholar]
- Smith DR, et al. First complete mitochondrial genome sequence from a box jellyfish reveals a highly fragmented linear architecture and insights into telomere evolution. Genome Biol Evol. 2012;4:52–58. doi: 10.1093/gbe/evr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JM, Horspool D, Brown G, Tcherepanov V, Upton C. GraphDNA: a Java program for graphical display of DNA composition analyses. BMC Bioinform. 2007;8:21. doi: 10.1186/1471-2105-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda K, et al. Complete mitochondrial genomes of two Japanese precious corals, Paracorallium japonicum and Corallium konojoi (Cnidaria, Octocorallia, Coralliidae): notable differences in gene arrangement. Gene. 2011;476:27–37. doi: 10.1016/j.gene.2011.01.019. [DOI] [PubMed] [Google Scholar]
- van der Hamm JL, Brugler MR, France SC. Exploring the utility of an indel-rich intergenic region as a molecular barcode for bamboo corals (Octocorallia: Isididae) Mar Genomics. 2009;2:183–192. doi: 10.1016/j.margen.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Van Oppen MJH, et al. The mitochondrial genome of Acropora tenuis (Cnidaria: Scleractinia) contains a large group I intron and a candidate control region. J Mol Evol. 2000;55:1–13. doi: 10.1007/s00239-001-0075-0. [DOI] [PubMed] [Google Scholar]
- Voigt O, Erpenbeck D, Wörheide G. A fragmented metazoan organellar genome: the two mitochondrial chromosomes of Hydra magnipapillata. BMC Genomics. 2008;9:350. doi: 10.1186/1471-2164-9-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lavrov DV. Mitochondrial genome of the homoscleromorph Oscarella carmela (Porifera, Demospongiae) reveals unexpected complexity in the common ancestor of sponges and other animals. Mol Biol Evol. 2007;24:363–373. doi: 10.1093/molbev/msl167. [DOI] [PubMed] [Google Scholar]
- Wang X, Lavrov DV. Seventeen new complete mtDNA sequences reveal extensive mitochondrial genome evolution within the Demospongiae. PLoS One. 2008;3(7):e2723. doi: 10.1371/journal.pone.0002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J-H, Jia J-G, Murphy RW, Huang D-W. Rapid evolution of the mitochondrial genome in chalcidoid wasps (Hymenoptera: Chalcidoidea) driven by parasitic lifestyles. PLoS One. 2011;6(11):e26645. doi: 10.1371/journal.pone.0026645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Jameson D, Tang B, Higgs PG. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol. 2006;63:375–392. doi: 10.1007/s00239-005-0246-5. [DOI] [PubMed] [Google Scholar]
- Zucker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]