

Abstract

Macrocyclic Hedgehog (Hh) pathway inhibitors have been discovered with improved potency and maximal inhibition relative to the previously reported macrocycle robotnikinin. Analogues were prepared using a modular and efficient build-couple-pair (BCP) approach, with a ring-closing metathesis step to form the macrocyclic ring. Varying the position of the macrocycle nitrogen and oxygen atoms provided inhibitors with improved activity in cellular assays; the most potent analogue was 29 (BRD-6851), with an IC50 of 0.4 μM against C3H10T1/2 cells undergoing Hh-induced activation, as measured by Gli1 transcription and alkaline phosphatase induction. Studies with Patched knockout (Ptch–/–) cells and competition studies with the Smoothened (Smo) agonists SAG and purmorphamine demonstrate that in contrast to robotnikinin, select analogues are Smo antagonists.
Keywords: macrocycle, diversity-oriented synthesis (DOS), Sonic Hedgehog pathway, Smoothened antagonist, C3H10T1/2
The Hedgehog (Hh) signaling pathway regulates cell growth and migration during embryonic development. It is normally dormant in adult cells, but elevated activity of this pathway is associated with cancers such as medulloblastoma and basal cell carcinoma.1 In recent years, antagonism of different members of this pathway has been investigated as a novel mode of cancer chemotherapy, as described in several reviews.2−6 Inhibition of Smoothened (Smo), a seven-pass transmembrane receptor with similar topology to G-protein-coupled receptors (GPCRs), shows promise for the treatment of cancers driven by activating mutations to the Hh pathway and also for a subset of epithelial cancers that may require Hh for growth via a paracrine mechanism.7−10 Several Smo antagonists have proceeded to advanced clinical trials,11−13 including GDC-0449 (vismodegib),14 which was recently approved by the Food and Drug Administration (FDA) for treatment of advanced basal cell carcinoma.
In 2009, a macrocyclic compound derived from a diversity-oriented synthesis (DOS) library [robotnikinin (1)] was described that inhibits the Hh signaling pathway.15,16 Hit compounds were identified by their interaction with Sonic Hedgehog (Shh) in a small-molecule microarray screen. We aimed to build upon this work by developing analogues with improved activity in cellular assays. We ultimately used an assay based on the differentiation of murine mesenchymal C3H10T1/2 cells, induced by the Shh protein.17 The differentiation of these cells results in the expression of alkaline phosphatase, which is readily quantified with fluorescent substrates; Gli1 expression levels (mRNA) provide another readout of Hh pathway activation. Our primary objective was thus to find analogues with improved potency and maximal inhibition in these assays.18
The preparation of macrocyclic analogues of robotnikinin used a build/couple/pair19 strategy related to previous reports15,16,20 that affords rapid access to diverse analogues. Our general strategy is illustrated in Scheme 1, with only one of numerous accessible stereoisomers depicted. Amino alcohols and diamines were coupled with successive alkenoic acid building blocks, and the resulting dienes were paired in a ring-closing metathesis (RCM) step. Many compounds underwent further elaborations at the functional handles included with the carboxylic acid building blocks.
Scheme 1. General Strategy for Macrocycle Synthesis.

One focus of our medicinal chemistry studies was the determination of the optimal linker joining the alkenoic acids. To this end, a variety of amino alcohols and diamines were obtained or prepared, and these building blocks were incorporated into different macrocyclic products. A selection of these compounds is depicted in Chart 1, along with their half-maximal inhibitory concentrations in the Shh-induced21 C3H10T1/2 alkaline phosphatase assay, and their maximal activity relative to the prototypical Shh pathway inhibitor cyclopamine.
Chart 1. Analogues with Alternative Amino Alcohol Linkers.

Cyclopamine produced an half-maximal inhibitory concentration (IC50) of 0.6 μM and reduced the alkaline phosphatase activity to levels measured in the cells without Shh treatment. As previously reported, robotnikinin (1) proved to be only weakly active in this assay.18 Removal of the 2-phenyl substituent from the macrocycle of 1 obviated all activity (compound 2). Norephedrine-based compound 3 and norpseudoephedrine-based 4 had improved maximal activity over 1 and slightly improved potency in the C3H10T1/2 assay, as did the prolinol derivative 5. A significant improvement was observed with compound 6 (IC50 = 5 μM), where the positions of the macrocyclic oxygen and nitrogen are reversed. Indane 7 was also prepared, but its potency and maximal activity were poor.
We systematically explored several other structure–activity relationships (SARs) (Charts 2 and 3 and Table 1). Methylation of the macrocyclic nitrogen (compound 8) gave a slight improvement versus 3, but inversion of stereochemistry at the 2-position of 6 (compound 9) decreased potency. Substitution at the 11-position was well-tolerated; methyl (10) and benzyl-substituted (11) analogues of 6 maintained potency with good to excellent maximal activity. The 11-isopropylamino-substituted analogue 12 was weakly active. Certain modifications of the olefin were also tolerated. For example, compound 13, possessing a Z-olefin derived from the minor product of a metathesis reaction, was more potent than the analogous E-olefin 10. A number of hydrogenated analogues showed activity comparable to the parent olefin series; for example, ephedrine derivative 14 showed moderate potency but low maximal activity. Noncyclic dienes such as 15 were inactive.
Chart 2. Analogues Probing Key SARs.

Chart 3. Analogues with Alternative Side Chains at the 6-Position.

Table 1. Activity of 2-Substituted Macrocycles in Shh-Induced C3H10T1/2 Cell Differentiation.

Relative to cyclopamine (100%).
Measured by alkaline phosphatase activity.
Measured by Gli1 levels. See the Supporting Information for details.
We next modified the substituent at position 6 (Chart 3) of the various scaffold variants. Truncated analogues such as 16 and 17 were inactive in the cell assay or were only partial inhibitors. Amides such as 18 possessing solubilizing groups had poor activity, suggesting that a lipophilic chain is necessary at position 6. Compound 19, possessing a trifluorobutyl group in place of the 4-chlorobenzylamide, showed a dose–response in this assay, but with poor maximal inhibition. Compounds 20 (IC50 = 7 μM) and 21 (IC50 = 8 μM) demonstrate that the amide moiety is not critical for activity. Interestingly, movement of the aromatic chloride of 1 from the para to the meta position (22) gave improved potency in this assay relative to 1 (IC50 = 8 μM), although the moderate maximal inhibition was not improved and reached only 50%. The macrocyclic carbamate 23 was prepared to remove the chiral center at the 6-position and because it would be expected to have improved plasma stability. Unfortunately, it showed poor activity and decreased maximal inhibition relative to 6.
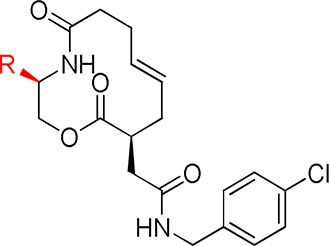
Using 6 as a lead compound, we reexamined the SAR at the 2-position of the scaffold (Table 1). The potency was maintained when the arene was replaced with a cyclohexyl (24) or benzyl group (25); however, replacement with a n-butyl group gave weak, difficult-to-quantify activity (26), and the isopropyl analogue 27 had significantly weaker activity than 6. Potency and maximal inhibition were unaffected by introduction of a fluorine to the para position of the arene of 6 (compound 28). A log gain in activity [IC50 = 0.6 μM (alkaline phosphatase readout); 0.4 μM (Gli1 expression)] was observed with the introduction of a 4-chloro substituent (29), and this compound also attained the maximal inhibition of cyclopamine. The synthesis of 29 is depicted in the Supporting Information. Heteroarene 30 had lower activity, pointing to the importance of a hydrophobic aromatic ring at the 2-position of the scaffold.
To confirm specificity of the new macrocyclic inhibitors for the Shh pathway, a SAG rescue test was performed, in which inhibition of Shh-induced Gli1 expression in C3H10T1/2 cells was measured in the presence of the Smo agonist SAG22,23 for two of the most potent compounds, 25 and 29. We used SAG at 20 nM concentration as it was the minimal concentration that produced a nearly maximal effect in this assay (see the Supporting Information). Gli1 mRNA transcript levels were measured using real-time polymerase chain reaction (PCR) (Figure 1). Similar to cyclopamine (Figure 1A), both macrocycles showed a significant rightward shift of the dose–response curve in the presence of 20 nM SAG, resulting in 18- and 32-fold increases in the IC50 values for 25 and 29, respectively (Figure 1B,C). These findings suggest that both compounds act at or above Smo in the Shh-signaling pathway.
Figure 1.

Use of Smo agonists and antagonists demonstrates Shh pathway specificity of macrocyclic inhibitors. Shh-induced activation of C3H10T1/2 cells is inhibited by cyclopamine (A), 25 (B), and 29 (C). Pathway inhibition is rescued by the Smo agonist SAG (20 nM). Gli1 mRNA levels were measured by qPCR using Actb as an internal control.
To narrow the range of possible targets of 25 and 29, we used constitutively active Ptch–/– mouse embryonic fibroblast (MEF) cells carrying Gli-responsive β-galactosidase (β-gal) reporter.17 Because Patched functions upstream of Smo and acts as its repressor, inhibition of the Shh pathway at or upstream from Patched is prevented in this cell line. Measuring both β-gal activity and Gli1 transcription levels, we found that both 25 [IC50 = 8.8 μM (Gli1 readout); 8.1 μM (β-gal readout)] and 29 [IC50 = 3.5 μM (Gli1 readout); 2.0 μM (β-gal readout)] behave similar to cyclopamine [IC50 = 1.5 μM (Gli1 readout); 1.9 μM (β-gal readout)] and fully inhibit Shh pathway activity. This indicates that in contrast to robotnikinin, part or all of their activities are derived from interactions downstream from Patched. As in the studies with the wild-type (Ptch+/+) cell line, these activities are abrogated by the addition of 20 nM SAG. A representative dose–response curve for 25 is shown in Figure 2. Additional evidence of interaction with Smo was provided by BODIPY-cyclopamine competitive binding assays15,23 (see the Supporting Information).
Figure 2.

Dose–response curves of 25 in Ptch–/– MEF cells carrying β-gal reporter gene. The activity in this assay is evidence for action of the inhibitor downstream of Patched. Pathway inhibition is rescued by the Smo agonist SAG (20 nM). β-Gal activity levels in untreated cells (dashed line) and in cells treated with 100 nM KAAD-cyclopamine (complete inhibition, dotted line) are indicated.
To obtain further mechanistic insight, a Schild type analysis24 was undertaken using double titration experiments with the Smo agonist SAG (Figure 3). We measured downstream Gli1 expression induced by different concentrations of SAG in C3H10T1/2 cells in the presence of different concentrations of the inhibitors 1 (robotnikinin), 25 and 29, as well as the Smo antagonists cyclopamine and GDC-0449 (vismodegib). Cyclopamine (Figure 3A) and GDC-0449 (Figure 3B) both induced rightward shifts in the dose–response curves without affecting the maximal induction by SAG, consistent with their actions as competitive inhibitors of SAG at Smo. In contrast, robotnikinin (1) showed only marginal effects on the induction of Gli1 transcription by SAG (see the Supporting Information). Interestingly, the related macrocycles 25 (see the Supporting Information) and 29 (Figure 3C) behaved differently by strongly decreasing maximal activity of SAG and only weakly (25) or moderately (29) increasing its EC50 value. This is consistent with a mechanism that involves allosteric inhibition of SAG at Smo.25
Figure 3.

Double-titration experiment with the Smo agonists SAG and purmorphamine (PUR) in the presence of cyclopamine (A, D), GDC-0449 (vismodegib) (B, E), and 29 (C, F). Gli1 mRNA levels were measured by qPCR using Actb as an internal control.
These double-titration studies were repeated with the alternative Smo agonist purmorphamine.26,27 In contrast to its effects with SAG, cyclopamine had a very weak effect on purmorphamine-induced Gli1 expression (Figure 3D), and GDC-0449 affected Gli1 expression strongly but in a manner consistent with allosteric inhibition25 (Figure 3E). Robotnikinin showed little effect on purmorphamine-induced Gli-expression (see the Supporting Information), consistent with its weak inhibition in the original C3H10T1/2 screen (Chart 1). Interestingly, the responses to increasing concentrations of 25 (see the Supporting Information) and in particular to 29 (Figure 3F) were more pronounced. This resulted in the reduction of purmorphamine EC50 without a strong effect on its maximal induction, thus supporting a competitive interaction between 25 and 29 and purmorphamine. It should be emphasized that although the Gli mRNA levels may be modulated by binding events at the Smo receptor, it is impossible to rule out additional interactions with downstream pathway components, so interpretations should be made cautiously.
The studies described in Figure 3 support a two-site binding model at Smo, whereby SAG and purmorphamine bind to unique sites on the receptor. Related competition studies with alternative Smo antagonists that also provide evidence for allosteric binding modes have been reported by Rominger24 and Tao.28 Such novel allosteric inhibitors of Smo could show important utility for the treatment of cancers with mutated forms of Smo, such as the D473H mutation characterized after clinical treatment with GDC-0449 (vismodegib).29
In summary, several novel macrocyclic compounds are reported that appear to block the Shh pathway by inhibiting the membrane protein Smo. These compounds were assembled with a modular build/couple/pair synthetic strategy using different olefin-containing carboxylic acid and amino alcohol building blocks. SAR studies determined that a lipophilic side chain at the 6-position is required for activity. Several amino alcohol linkers provided decent activity; interestingly, improved potency was observed when the macrocycle nitrogen and oxygen of 1 were reversed. With this scaffold, maximal activity was observed with an aromatic moiety at the 2-position with (R) stereochemistry. The most potent compound was the 4-chlorophenyl analogue 29 (IC50 = 0.4 μM). Competition studies with the Smo agonists SAG and purmorphamine, as well as activity in a Ptch–/– cell line, suggest that 25 (BRD-0607) and 29 (BRD-6851), in contrast to robotnikinin, act predominantly as Smo antagonists. It is interesting to note that the subtle structural changes between robotnikinin (a direct binder of Shh) and compounds such as 29 (characterized here as a Smo antagonist) apparently lead to an additional mode of Shh pathway inhibition. These studies also provided evidence for a two-site binding model at Smo. Compound 29 is particularly interesting for its activity as an allosteric inhibitor of SAG but a competitive inhibitor of purmorphamine-induced Gli1 expression. Its activity in cell lines carrying Smo mutations is presently under investigation.
Acknowledgments
We thank James Chen (Stanford University) for Light-2, Ptch–/– cells, and Smo expression constructs; Stephen Johnston, Chris Johnson, and Mike Lewandowski for analytical chemistry support; Giannina Schafer and Tom Hasaka for help with Smo binding assays; Lili Wang, Yan-Ling Zhang, Katie Doud, LaTese Briggs, and Angela Koehler for additional studies not described here; and Robert Gould, Lee Peng, and Aly Shamji for helpful comments.
Glossary
Abbreviations
- β-gal
β-galactosidase
- DOS
diversity-oriented synthesis
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FDA
Food and Drug Administration
- GPCR
G-protein-coupled receptor
- Hh
Hedgehog
- IC50
half-maximal inhibitory concentration
- MEF
mouse embryonic fibroblasts
- NPG
nitrogen with protecting group
- PCR
polymerase chain reaction
- RCM
ring-closing metathesis
- SAR
structure–activity relationship
- Shh
Sonic Hedgehog
- Smo
Smoothened
Supporting Information Available
Assay protocols, structures of tool compounds, double-titration experimental data with 25, BODIPY-cyclopamine competitive binding assay data, and protocols for the preparation of 25 and 29. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding for this work was provided in part by the Broad Institute Gift (M.M.N., J.R.P., and C.S.) and NIH Genomics Based Drug Discovery Grants RL1GM084437 and UL1RR024924, administratively linked to NIH Grants RL1HG004671 and RL1CA133834. This work was supported in part by GM38627 (awarded to S.L.S.).
The authors declare no competing financial interest.
Author Contributions
§ These authors contributed equally to this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- See, for example,Ng J. M. Y.; Curran T. The Hedgehog's tale: Developing strategies for targeting cancer. Nat. Rev. Cancer 2011, 11, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestone A. J.; Chen J. K.. Small-molecule inhibitors of the Hedgehog pathway. In Hedgehog Signaling Activation in Human Cancer and Its Clinical Applications; Xie J., Ed.; Springer: New York, NY, 2011; pp 163–186. [Google Scholar]
- Peukert S.; Miller-Moslin K. Small-molecule inhibitors of the Hedgehog signaling pathway as cancer therapeutics. ChemMedChem 2010, 5, 500–512. [DOI] [PubMed] [Google Scholar]
- Tremblay M. R.; McGovern K.; Read M. A.; Castro A. C. New developments in the discovery of small molecule Hedgehog pathway antagonists. Curr. Opin. Chem. Biol. 2010, 14, 428–435. [DOI] [PubMed] [Google Scholar]
- Stanton B. Z.; Peng L. F. Small-molecule modulators of the Sonic Hedgehog signaling pathway. Mol. BioSyst. 2010, 6, 44–54. [DOI] [PubMed] [Google Scholar]
- Mahindroo N.; Punchihewa C.; Fujii N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 3829–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch R. L.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [DOI] [PubMed] [Google Scholar]
- Nolan-Stevaux O.; et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H.; et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. 2009, 106, 4254–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen J.-W.; de Sauvage F. J. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009, 69, 6007–6010. [DOI] [PubMed] [Google Scholar]
- Tremblay M. R.; et al. Discovery of a potent and orally active Hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [DOI] [PubMed] [Google Scholar]
- Pan S.; et al. Discovery of NVP-LDE225, a potent and selective Smoothened antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munchhof M.; Li Q.; Shavnya A.; Borzillo G. V.; Boyden T. L.; Jones C. S.; LaGreca S. D.; Martinez-Alsina L.; Patel N.; Pelletier K.; Reiter L. A.; Robbins M. D.; Tkalcevic G. T. Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med. Chem. Lett. 2012, 3, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin C. M.; Hann C. L.; Laterra J.; Yauch R. L.; Callahan C. A.; Fu L.; Holcomb T.; Stinson J.; Gould S. E.; Coleman B.; LoRusso P. M.; Von Hoff D. D.; de Sauvage F. J.; Low J. A. Treatment of medulloblastoma with Hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton B. Z.; Peng L. F.; Maloof N.; Nakai K.; Wang X.; Duffner J. L.; Taveras K. M.; Hyman J. M.; Lee S. W.; Koehler A. N.; Chen J. K.; Fox J. L.; Mandinova A.; Schreiber S. L. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 2009, 5, 154–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L. F.; Stanton B. Z.; Maloof N.; Wang X.; Schreiber S. L. Syntheses of aminoalcohol-derived macrocycles leading to a small-molecule binder to and inhibitor of Sonic Hedgehog. Bioorg. Med. Chem. Lett. 2009, 19, 6319–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J.; Chen J. K.; Cooper M. K.; Wang B.; Mann R. K.; Milenkovic L.; Scott M. P.; Beachy P. A. The effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [DOI] [PubMed] [Google Scholar]
- The maximal inhibition of C3H10T1/2 differentiation with 1 (measured via alkaline phosphatase activity) was only ∼47% at 31.3 μM concentration. See the Supporting Information of ref (15).
- Nielsen T. E.; Schreiber S. L. Towards the optimal screening collection: A synthesis strategy. Angew. Chem., Int. Ed. 2007, 47, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.; Sello J. K.; Schreiber S. L. A strategy for macrocyclic ring closure and functionalization aimed toward split-pool syntheses. J. Am. Chem. Soc. 1999, 121, 10648–10649. [Google Scholar]
- A conditioned medium containing native Shh was used at a concentration giving maximal induction of alkaline phosphatase. See the Supporting Information for further details.
- Frank-Kamenetsky; Zhang X. M.; Bottega S.; Guicherit O.; Wichterle H.; Dudek H.; Bumcrot D.; Wang F. Y.; Jones S.; Shulok J.; Rubin L. L.; Porter J. A. Small-molecule modulators of Hedgehog signaling: Identification and characterization of Smoothened agonists and antagonists. J. Biol. 2002, 1, 10.1–10.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. K.; Taipale J.; Young K. E.; Maiti T.; Beachy P. A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14071–14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rominger C. M.; et al. Evidence for allosteric interactions of antagonist binding to the Smoothened receptor. J. Pharmacol. Exp. Ther. 2009, 329, 995–1005. [DOI] [PubMed] [Google Scholar]
- Taylor P.; Insel P. A.. Quantitation of pharmacologic antagonism. In Principles of Drug Action: The Basis of Pharmacology; Pratt W. P., Taylor P., Eds.; Churchill Livingstone: New York, 1990; pp 61–66. [Google Scholar]
- Wu X.; Ding S.; Ding Q.; Gray N. S.; Schultz P. G. A small molecule with osteogenesis-inducing activity in multipotent mesenchymal progenitor cells. J. Am. Chem. Soc. 2002, 124, 14520–14521. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Chen J. K. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nature Chem. Biol. 2002, 2, 29–30. [DOI] [PubMed] [Google Scholar]
- Tao H.; Jin Q.; Koo D.-I.; Liao X.; Englund N. P.; Wang Y.; Ramamurthy A.; Schultz P. G.; Dorsch M.; Kelleher J.; Xu W. Small molecule antagonists in distinct binding modes inhibit drug-resistant mutant of Smoothened. Chem. Biol. 2011, 18, 432–437. [DOI] [PubMed] [Google Scholar]
- Dijkgraaf G. J. P.; Alicke B.; Weinmann L.; Januario T.; West K.; Modrusan Z.; Burdick D.; Goldsmith R.; Robarge K.; Sutherlin D.; Scales S. J.; Gould S. E.; Yauch R. L.; de Sauvage F. J. Small molecule inhibition of GDC-0449 refractory Smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.