

When I was a young medical student, an interesting discussion was going on about how antibody diversity was encoded; was the antibody molecule encoded in many genes, one for each specificity, or were they general adaptor proteins that were formed on an antigen template. I understood the arguments, which were laid out by my laboratory instructor, Hugh McDevitt. He had just discovered immune response genes in the mouse and was soon to map them to the major histocompatibility complex, which was at that time only known to encode the antigens that provoked rapid graft rejection. I worked the summer after my second year at Harvard Medical School in Hugh's lab, after which I went to the National Institute for Medical Research to work with John Humphrey. In Hugh's lab, I was given the project of trying to make antibodies to L- and D-enantiomers of the same basic amino acids, called L- and D-TGA (for tyrosine, glutamic acid, and alanine). I spent 2 years in Humphrey's laboratory, spent much time in the library reading papers, and published three or four of my own. This was a period when little was known about immunology, as the idea of cutting and splicing genes lay ahead in the future. Antibodies were the only known specific products of lymphocytes, so we measured them in detail. But everyone I talked to in those years was full of ideas about how antibody diversity was generated and how, once generated, it remained the same or very similar throughout the course of a specific immune response.
What I will describe in this essay is the growth of three ideas. The first is that the immune system is actually made up of two arms, one specific for infectious non-self, and the other specific for all non-self-antigens. These two arms are called innate immunity and adaptive immunity, the latter of which makes the success of vaccines possible, but which is also responsible for all autoimmune diseases. The second concept I will describe is how the adaptive immune system is in essence self-referential, in that it is selected on self-molecules, sustained by self-molecules, and activated in the presence of self-molecules. These ideas apply to both the T cells with which I have worked for most of my career and to the B cells that were my initial source of fascination and have returned to give me a big surprise in recent years. Finally, I will describe experiments that have been published recently by others, and replicated in my laboratory, that suggest that autoimmune disease is caused by expansions of T cells and B cells with clonally distributed receptors that are susceptible to regulation by other conventional T cells. These T cells, variously called T suppressor cells (Ts) or regulatory T cells (Treg), appear to have both the ability to respond to autoantigens and the ability to produce immunoregulatory cytokines such as IL-10 and TGF-β. I will argue that such cells can be immunized by the epicutaneous route and that they could be used to vaccinate against autoimmunity. I hope to live long enough to see the first vaccines against the two autoimmune syndromes I work on, type I or insulin-dependent diabetes mellitus and a mouse model for multiple sclerosis, called experimental allergic encephalomyelitis.
Innate Immunity Discriminates Between Infectious Non-Self and Non-Infectious Self
The innate immune system is based on non-clonally distributed receptors that recognize certain molecular patterns, found in microbes but not in self-tissues (Table 1). These patterns come in many flavors, as they can be found in bacterial lipopolysaccharide in the cell walls of Gram-negative bacteria, as well as in fungi, viruses, Gram-positive bacteria, and in other forms of Gram-negative bacteria. We refer to such molecules as pathogen-associated molecular patterns. The receptors that recognize these pathogen-associated molecular patterns are known as pattern recognition receptors. One key feature of pattern recognition receptors is that they do not recognize any self-structure. This is because they are selected over evolutionary time, and any receptor that bound to a self-ligand could lead to death of the organism that expressed such a receptor. This prevents autoimmunity from happening when the only available recognition system is the innate immune system.
Table 1.
Innate and adaptive immunity
| Property | Innate immune system | Adaptive immune system |
|---|---|---|
| Receptors | Fixed in genome Rearrangement not necessary | Encoded in gene segments Rearrangement necessary |
| Distribution | Nonclonal All cells of a class identical | Clonal All cells of a class distinct |
| Recognition | Conserved molecular patterns (LPS, LTA, mannans, glycans) | Details of molecular structure (proteins, peptides, carbohydrates) |
| Self–non-self-discrimination | Perfect: selected over evolutionary time | Imperfect: selected in individual somatic cells |
| Action time | Immediate activation of effectors | Delayed activation of effectors |
| Response | Costimulatory molecules Cytokines (IL-1β, IL-6) Chemokines (IL-8) | Clonal expansion or anergy IL-2 Effector cytokines: (IL-4, IFNγ) |
One of the most important events triggered by the activation of the innate immune system is the surface expression of the costimulatory molecules B7.1 and B7.2, or CD80 and CD86 as they are now called. In one of our early studies, we demonstrated that costimulatory molecules had to be presented by the same cell as the ligand for the T cell receptor (1). Recently, we cloned a gene for a human toll-like receptor (later renamed TLR4) that was the first mammalian member of a family of genes now numbering ten (2). The mutation giving rise to nonresponsiveness to lipopolysaccharide was mapped precisely to the TLR4 gene, meaning that TLR4 was the lps gene. These lps mutant strains were hypersusceptible to the Gram-negative organism Salmonella typhimurim, which provided a confirmation for us that we were studying genes that played a central role in this early phase of the induction of adaptive immunity (3, 4). Therefore, one apparent role of the innate immune system is to regulate the expression of the dominant costimulatory molecules, CD80 and CD86, as I had earlier predicted (5).
Thus, the innate immune system appears to be important in the induction of adaptive immunity (Fig. 1). But it also plays many other roles, and it is much more ancient than the adaptive immune system, as elements of this system can be detected in all multicellular organisms, including vertebrates, invertebrates, and plants. These roles include, but are not limited to, resisting infection in the first place by preventing entry across the surface epithelia that protect our bodies, the skin, and the linings of our internal organs, which collectively are the sites of entry of all infections. The innate immune system of macrophages and polymorphonuclear neutrophilic leukocytes mediate inflammation and the early phases of the innate immune response to those infectious agents that can cross one or other barriers to cause an infection. Many cells found at the surface epithelia of the body secrete antimicrobial peptides, such as the cryptidins made by the Paneth cells that lie at the base of intestinal crypts.
Figure 1.

Various pathogen-associated molecular patterns recognized by cognate pattern recognition receptors expressed on APCs induce the expression of B7 molecules, which signal the presence of pathogens and allow activation of lymphocytes specific for antigens derived from the pathogens. Shown are lipopolysaccharide recognition by TLR-4, proteoglycan recognition by TLR-2, and the recently reported role of TLR-9 in the recognition of CpG DNA.
One could go on describing the innate immune system for a long time, but an even greater mystery is contained in the adaptive immune system, which appeared only recently, at the time of the differentiation of vertebrates between the hagfish and lampreys, which are the only known survivors in the vertebrate line that have no adaptive immunity, and the appearance of jawed fish and all species descended from them. These have a full set of immunoglobulins and T cells, as well as MHC molecules, and so are equipped to make specific antibody and specific T cell responses. Thus, the adaptive immune system arose relatively recently in evolution (6, 7).
Therefore, although much valuable work has been done in the area of self-non-self discrimination, I believe that virtually always this distinction is made by the innate immune system, which primes the adaptive immune system when a pathogen is present, but fails to prime such responses in the absence of infection. By infection, I mean either a real infection by a pathogen or a dead pathogen that is a component of virtually all effective adjuvants. Adjuvants are substances that increase adaptive immune responses to proteins with which they are mixed. A third way to stimulate adaptive immunity is by vaccines, which effectively mimic the microbes from which they were derived and therefore induce protective, specific immunity. This is crucial for the next part of this essay, in which I lay out the functioning of the adaptive immune system as self-referential, that is, as a system that functions on internal cues to prepare it to see the vast range of external cues that it encounters over a lifetime.
The Adaptive Immune Response
The self-referential nature of adaptive immunity, which is based on antigen-specific responses of T and B lymphocytes, is now well established for T cells, and, based on recent experiments showing positive selection of immature B cells in a receptor-specific fashion, it appears to be true of B lymphocytes as well. I will lay out these arguments as follows. First, I will present evidence, published by my laboratory and many others, that T cell development occurs on self-peptide:self-MHC ligands. I will also lay out preliminary evidence from studies performed in my laboratory that B cells similarly depend on self-ligands and that the most likely self-ligand for the B cell receptor is secreted Ig itself. Next, I plan to address how such naïve T cells persist in the periphery, again by recognizing self-peptide:self-MHC ligands with which they interact continuously during their lives. B lymphocytes may also be under the constraint of making meaningful interactions with self-immunoglobulins, although the evidence for this is slim and cries out for reinforcement. Then, I will propose, and present one example of, the role of self-peptide:self-MHC ligands in signaling for activation of T cells. Lastly, I will discuss evidence for a new (or old) class of T cells called variously T suppressor cells (Ts) or regulatory T cells (Treg) that are absolutely needed to hold the autoimmune response in check and, when eliminated in mice-bearing transgenic T cell antigen receptor (TCR) for self-ligands, allow flagrant autoimmunity.
The Source of Adaptive Immunity: Invasion of a Retroposon
Adaptive immunity only became possible after the acquisition and dissociation of a retroposon that invaded the genome of an unknown organism many millions of years ago. This organism had to have been a vertebrate, as only vertebrates have both of the elements of the retroposon: the two genes that encode a site-specific recombinase, known as RAG-1 and RAG-2, and the two sites that apparently were used by the retroposon to invade a member of the primordial Ig gene family, the recognition signal sequences. These are short DNA sequences that are found adjacent to all Ig and T cell receptor gene segments. One of these is made up of a heptamer–12-bp nonamer, and the other is made up of a heptamer–23-bp nonamer. These recognition signal sequences and the DNA that lies between them must be removed by the RAG-1:RAG-2 heterodimer to form two joints, one of which is re-ligated to form a coding joint that encodes the variable exon of all immunoglobulins and T cell receptors. The invasion of a primordial Ig gene by a retroposon has only recently been described, but the evidence for it is so strong that it almost has to be correct.
The site-directed recombinase, RAG-1:RAG-2, acts on germ-line gene segments to produce all antibody molecules and T cell receptors of the adaptive immune system, as proven the total inability of RAG-1 and/or RAG-2 knockout mice to rearrange their receptor gene segments. These receptors, once formed and delivered to the cell surface, have to survive two types of selective processes. One selects for utility by asking: Does the receptor recognize anything in its environment, be this the thymus or the bone marrow? When the former happens, the result is conventionally described as positive selection. The second asks: Does this recognition event trigger a response? When this happens in immature lymphocytes, the result is almost always immediate cell death by apoptosis. When nothing is recognized, at least in T cells, the result is called “death by neglect.” This happens to most developing T cells because they need to bind something in their immediate environment and because their ligands consist of self-peptides bound to self-MHC molecules. Because of the extensive polymorphism of MHC molecules and their bound self-peptides, most T cells die early in development as nothing can be recognized.
Positive selection is very specific, as it has to be provided at a subtly defined binding of the receptors by ligands that don't lead to full signaling. Conventionally, for T cells, such ligands are called weak agonists or antagonist ligands. They must originate from a self-protein, generating self-peptides that, in turn, bind to self-MHC molecules. Such self-peptide:self-MHC ligands have to drive the process of thymocyte positive selection, as self-peptides are the only peptides available, and the mature MHC molecules require a peptide to stabilize them at the surface of the cell. Therefore, the T cell receptor repertoire is positively selected on self-peptide:self-MHC complexes.
The next test for developing T cells is whether they recognize self too strongly. This drives a process known as negative selection, in which cells that can be activated by aggregation (or at least dimerization) of their receptors are deleted. The sum total of these selective processes is a T cell receptor repertoire that is weakly self-reactive but is unable to be fully activated by self-ligands.
This weak reactivity to self, seen as partial activation and a low rate of ligand-driven turnover, is true for all T cells that survive the gauntlet of intrathymic selection. The T cells that emigrate from the thymus and join the peripheral pool divide slowly and die off slowly, maintaining a homeostatic number of T cells. As a result of these two processes, peripheral T cells are maintained at a constant number, and the ratio of CD4 MHC class II-restricted T cells to CD8 MHC class I-restricted T cells is basically fixed in the absence of an adaptive immune response. Only when an infectious agent enters the system and induces an innate immune response leading to expression of the B7 costimulatory molecules does an adaptive immune response occur that activates T cells and upsets this homeostasis. Some viruses can even stimulate a response in which up to 80% of peripheral blood CD8 T cells can bind a single viral peptide:self-MHC class I ligand (8). After this tremendous response, the numbers of antigen:MHC ligand binding cells returns to nearly its original number. The remaining peptide:MHC binding cells become memory T cells, which have a lifespan that essentially coincides with the life of the host. Although it is known that such cells continue their homeostatic division in the absence of stimulation by self-ligands, it is not clear whether such memory T and B cells, which mediate accelerated responses to foreign or non-self-molecules, are regulated by some other stimulus; various cytokines have been proposed (9, 10).
The case for the positive selection of B lymphocytes is less well-explored, but we have recently derived data that are consistent with positive selection mediated by soluble antibody. The data that led us to ask this question appeared recently in Proceedings (11), and, at that time, we had no clue as to what the selecting ligand would be. However, when we compared positive selection of B cells in mice whose heavy chain transgene could be secreted, with the same process in mice bearing the identical heavy chain that could not be secreted, we obtained a different and, quite frankly, unexpected result. The previously observed positive B cell selection in mice whose B cells could secrete antibody did not occur with B cells from mice whose only known difference was the inability to secrete their surface Ig as antibody molecules. As this selection occurred in mature, naïve B cells, we infer from this experiment that secreted Ig can select the B cell repertoire in a receptor-specific manner (11). Thus, we can conclude, tentatively, that the postulate of a network of interacting idiotypes by Niels Jerne, which won him a Nobel Prize, actually regulates the naïve, mature B cell receptor repertoire as well (12).
Thus, both the mature, naïve T cell receptor repertoire and the mature, naïve B cell receptor repertoire are generated by interaction with self-ligands rather than non-self-ligands. These self-ligands can signal the B and T lymphocytes to mature and to survive, but they deliver only a partial rather than a fully activating signal.
The Adaptive Immune System Interacts with the Innate Immune System to Generate Adaptive Immunity
The adaptive immune system is absolutely essential for normal, healthy life. I learned this at an early age from my father, one of the first clinical immunologists, although he was first and foremost the finest clinician I have ever observed. My father was the first to identify a series of infants that failed to make any class of antibody or immunoglobulins. In fact, it was from the study of his patients that it was realized that the adaptive humoral immune response could be broken down into the major classes of Ig (13). He accumulated 17 patients with no circulating antibody or Ig, all of whom had recurrent infection with pyogenic (pus-forming) bacteria. This discovery only became possible with the introduction of the “wonder drug” penicillin, as up to that time many normal children also died of such infections. This disease, the first defect in an adaptive immune response, has led to many other discoveries over the last half-century, including the identification of the kinase that was necessary to drive B cell development and that was missing in such cases, called Btk (for Bruton's tyrosine kinase) (14). Bruton reported a single case, for which my father generously ran a serum immunoelectrophoresis. My father went on to explore intramuscular injection of pooled Ig and, eventually, to be the first human given i.v. immunoglobulin, which led to nearly fatal complications because of the presence of many aggregates of Ig in the preparation. The discovery of this immunodeficiency disease in 1953, and its effective therapy with Ig pooled from many donors (15), opened the door to the founding, over the last 48 years, of the discipline of clinical immunology and the establishment of literally hundreds of clinical immunology divisions and laboratories around the world. I think it is a great tribute to my always modest father that from a humble start, so many wonderful discoveries have come.
The second piece of evidence that the adaptive immune response is absolutely critical to human health is the global spread of the AIDS, which is caused by infection with HIV-1. This disease is a particularly nasty infection, for which there is currently no cure. AIDS kills a growing number of people each year, especially in sub-Saharan Africa. But it also impacts on individuals around the world. Well over 33 million people are infected at the present time (16), and more infections are happening worldwide at an ever-increasing rate. The recent burst of optimism brought on by the invention of powerful anti-viral agents has been sobered by the realization that the virus can rapidly produce mutants in a single infected individual that can circumvent the most potent of these drugs. Infection with HIV-1 leads in almost all people to the development of AIDS. This immunodeficiency disease leads to death from pathogens that are normally controlled by the adaptive immune system, such as Mycobacterium avium and Candida albicans. This provides dramatic proof that both the innate and the adaptive immune systems are needed, and neither one on its own can provide effective protection against infectious disease.
The reason that I devoted so much space to my father's seminal discovery is not because he is my father, but rather that immunodeficiency virtually always affects the adaptive rather than the innate immune system. This could reflect an argument I made in 1989 in the introduction to the 54th Cold Spring Harbor Symposium in Quantitative Biology (5), that the innate immune system was based on germ line-encoded receptors, like the TOLL receptor that I briefly described in the first part of this essay. Such receptors are essential to a healthy life, while various parts of adaptive immunity, such as the ability to secrete antibodies, can be lost, as long as this loss is compensated with antibiotics during infection as well as passive intramuscular injection of immunoglobulins, or, more recently, with i.v. immunoglobulin. The fact of the matter is that defects in innate immunity are rare by comparison with defects in adaptive immunity. From this I infer that, of the two immune systems, the innate immune system is more important than the adaptive immune system, in which numerous defects have been described. However, before antibiotics were available, one could die from defects in either system. Thus, it is clear that to live a long and healthy life, one needs both an effective innate immune system and an effective adaptive immune system, and defects in either one can lead to disease, difficulty living a normal, healthy life, and early death from infection.
Evidence That T and B Lymphocytes Are Fundamentally Self-Referential
We now come to the crux of my argument, which is that the adaptive immune system is made up of T and B lymphocytes that are fundamentally self-referential. For T lymphocytes, I think that this is obvious, as they are selected on self-peptide:self-MHC molecules in the thymus, leading to positive selection of all thymocytes whose receptor can recognize such ligands. The majority of thymocytes do not recognize any self-peptide:self-MHC complexes because of the great polymorphism of MHC molecules, and these are lost by a process known as “death by neglect.” The surviving T cell receptor positive thymocytes then face screening for self-reactivity, in which there is a window of affinities that is greater than can be tolerated in the periphery, and at this juncture they are deleted by a process called negative selection. Negative selection also happens on self-peptide:self-MHC complexes, again as these are the only complexes available in the central lymphoid organ where T cells develop, the thymus. Thereafter, the naive, peripheral repertoire of CD4 and CD8 T cells is able to survive by contact with the same or a similar self-peptide:self-MHC complex on which they were positively selected, said by some to be carried by the most important antigen-presenting cell, the dendritic cell (17).
B Lymphocytes Also Appear to Depend on the Self-Ligands in Their Environment for Their Positive Selection and Onward Survival
Recently, we have been studying positive selection of B cells passing from the immature B cells that emerge from the bone marrow to the mature B cells that make up the majority of the long-lived peripheral B cell pool. These mature B cells loose 70–90% of their numbers in this transition, and we wondered why. Was it just a stochastic loss of B cells, or was there a selective event about which we did not know? And what was the selecting agent? We set out to study this by using a similar system to that which we used in our previous analysis of the T cell receptor repertoire, in which we used a TCRβ chain transgene to infer what was influencing the choice of TCRα chain sequences (18).
It was previously published that the peripheral B cells needed some signal to survive, as amputating their receptors had a marked effect on B cell survival in heavy chain transgenic mice. That is, the receptor-less mature B lymphocytes rapidly disappeared from the circulation (19). But this did not say whether this disappearance was simply a function of B cell dynamics or whether there was a receptor-specific element in this loss. To ask this question, we placed an Ig H chain transgene into the germ line of mice that were homozygous for a deletion in the necessary JH gene segments. We then asked: What light chains can these transgenic mice express? This meant sorting many B cells into immature, heat-stable antigen high cells and mature, heat-stable antigen low cells. Then we isolated genomic DNA, cloned it, and sequenced many κ light chains from the immature and mature, naïve B cell populations. What we found in this analysis surprised us. There was indeed apparent selection for particular light chain sequences, and the set of light chains selected was dependent on the heavy chain transgene chosen (11). Thus, it was apparent that the particular transition from peripheral immature to peripheral mature B cells was dependent on some sort of selective signal that was specific to the heavy:light chain pairs expressed on the surfaces of these cells. This could have been some sort of foreign antigen, but as we were not looking at activated B cells but rather at naive, immature and naïve, mature B cells, this seemed highly unlikely. We saw no evidence for activation of the mature B cells that would be expected if they were stimulated with antigen.
We then went on to explore this phenomenon further. We ruled out stimulation by the normal flora by using gnotobiotic mice, provided to us by Edward Balish of the University of Wisconsin. We ruled out a role for unexpected TdT expression, by using mice provided to us by John Kearny of the University of Alabama. But when we asked whether soluble Ig affected the mature B cell receptor repertoire, we found that indeed it could, and, moreover, that it did. This experiment needs to be described in detail so that it can be understood.
Ig heavy chain transgenic mice carrying identical VH genes, and whose endogenous JH genes were deleted, were engineered to secrete their Ig or, by manipulating their intracytoplasmic regions, were engineered to have only cell-surface Ig (20). We then asked the same question that we had asked earlier, but now by a colony hybridization method, which meant that we could assess far more cells for positive selection than by the previous sequencing method. Once again, we got a surprise. As Niels Jerne had predicted over 30 years ago, but had never confirmed, soluble Ig could regulate the structure and selection of the receptors on the surfaces of B cells. We demonstrated this point by the loss of the positive selection that we had observed earlier with B cells carrying the same heavy chain transgene that could secrete their Ig. This positive selection was absent in B cells that could not secrete their surface Ig, which pointed very strongly to the idiotypic network (21). We still have to confirm this result by resupplying the secreted Ig and restoring positive selection, but we are confident that we can perform this final proof. Thus, Jerne's idiotypic network, which was originally proposed to account for immune regulation after immunization, actually can act on the naive B cell receptor repertoire to positively select certain heavy:light Ig chain pairs over others. Secreted Ig can play a role in the Jernian idiotypic network, although this role is quite different from the role for which the Jernian idiotypic network was originally proposed. Our experiments are not able to tell us whether this network also plays a regulatory role as originally proposed by Niels Jerne (12).
Ongoing Survival of T Cells Depends on Peripheral Expression of Self-Peptide:Self-MHC Ligands
My laboratory, and those of many others, have observed that mature, naïve T cells can go through the cell cycle, as determined by labeling them with the fluorescent dye carboxy fluorescein succinimide ester (CFSE). Most of these studies were carried out in mice that were irradiated, which raised the possibility of a radiation-induced artifact, or in immunodeficient mice where this cycling was said to “fill up the spaces” (22–24). Therefore, we repeated these studies in unirradiated, normal mice. We found that the cycling was less vigorous than that seen previously in irradiated or lymphopenic mice, but it happened nonetheless (C. Viret and C.A.J., unpublished results). Thus, we infer from the ability of CFSE-labeled T cells to cycle that the cycling we observe is important in peripheral selection and may also play a role in the homeostasis of the T cell receptor repertoire.
We also collected T cells raised in H2-DM−/− mice. These T cells are reactive to a variety of self-peptides, as they have lost expression of the peptide exchange factor, H2-DM. We took such cells, labeled them with CFSE, and transferred them to normal or H2-DM−/− recipients. T cells from H2-DM−/− mice respond strongly to the self-MHC class II molecule I-Ab, leading to rapid expansion in the strain C57BL6, which expresses this MHC allele. T cells raised in an H2-DM−/− thymus show homeostatic cycling in H2-DM−/− recipients. The lesson from all of these experiments, and many others performed by other groups, was that survival depended on seeing the same or similar self-peptide:self-MHC complexes on which they were raised.
Survival of B Cells Depends on the B Cell Receptor
Given what we had found for CD4 T cells, that they needed to be exposed to the same or a similar self-peptide:self-MHC complex as that upon which they were positively selected to survive in the periphery, we can cite two studies which showed that a functional B cell receptor is also necessary for long-term survival of naïve B cells. In the first of these, the transgenic B cell receptor heavy chains were flanked by lox-p sites, and mice were made transgenic for these “floxed” genes, as well as for the cre recombinase, which acts specifically on genes flanked by lox-p sites. The cre recombinase was under the control of an IFN-inducible promoter, and when IFN was given to such mice, all of their receptors were deleted. All of the B cells that had lost their receptors then died, as they could not interact with their selecting ligand (19). A second line of evidence came from Syk−/− mice, which again could not survive in the periphery, which told us that the B cell receptor must be able to signal to provide for effective survival (25).
However, neither of these studies said anything about the specificity of the B cell receptors nor of their ligands. Thus, these studies prove that the expression of a B cell receptor is needed for long-term survival of B cells, but they tell us nothing about what is providing the signal for survival, or even if there is a specific ligand. The advantage of our system, while laborious, was that it told us that we needed a specific ligand and then allowed us to identify it as secreted Ig, presumably acting through the idiotypic network.
T Cell Activation Requires Expression of Self-Peptide:Self-MHC Complexes as Well as Agonist Peptide:Self-MHC Complexes
I will now outline how we demonstrated that agonist peptides can only trigger T cell activation in the presence of self-peptide:self-MHC ligands. This demonstration rests on the breeding of two different transgenic mice, one carrying a T cell receptor that recognizes a peptide expressed on B10.A(5R) cells, which is also recognized by the monoclonal antibody called Y-Ae (26). This monoclonal antibody, produced in my laboratory at Yale and characterized by my former colleague there, Dr. Donal Murphy, recognizes a 17-mer peptide derived from the Eα chain (Eα 52–68), which is bound to the MHC class II molecule I-Ab. This monoclonal antibody reacts with the B10.A(5R) strain, as both genes (I-Ab and I-Eα) are found in these mice, but it does not recognize the cells of C57BL/6 mice, which lack the gene for Eα. We then immunized C57BL/6 mice with the Eα52–68 peptide and derived several T cell hybrids, some of which recognized stimulator cells from the strain B10.A(5R). One of these, called 1H3.1, was selected for the formation of transgenic mice. The genes for the TCRα and the TCRβ chains were isolated and cloned into expression vectors.
When TCR transgenic mice were prepared from such genes and bred to C57BL/6 mice, which lacked the ability to stimulate the parental hybrid, we observed positive selection of the transgenic T cells. Despite the apparent absence of the Y-Ae epitope in such mice, we could inhibit positive selection of cells by giving the Y-Ae antibody, proving that some Y-Ae-like epitope was expressed there (27). When the same TCR was bred to B10.A(5R) mice, we saw deletion of all of the double-positive T cells, indicating that the TCR transgene worked as predicted from many previous studies in other labs.
We then began to cross the 1H3.1 transgenic mice to mice that were engineered in the laboratory of my colleague, Phillippa Marrack, to express only the I-Ab-Eα peptide on their cell surfaces (so called AbEp mice). This was achieved by covalent linkage of the Eα peptide to the N terminus of the A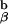 chain. To retain the Eα peptide in the peptide binding groove of the I-Ab molecule, Kappler, Marrack, and Ignatowitz had to breed in two gene knock-outs, the β chain of the I-Ab molecule and the MHC class II invariant chain, Ii (27). The further breeding to the 1H3.1 T cell receptor transgenic mice was an onerous task, as we had to repeat the breeding done by the Marrack lab, and at the same time be sure that the 1H3.1 TCR was carried along with it. As the breeding progressed, we saw mainly intense negative selection in mice that were positive for the 1H3.1 transgene and the AbEp transgene. It was only when the mice were transgenic for the 1H3.1 TCR, the AbEp transgene, and deleted for both the invariant chain (Ii−/−) and the A
chain. To retain the Eα peptide in the peptide binding groove of the I-Ab molecule, Kappler, Marrack, and Ignatowitz had to breed in two gene knock-outs, the β chain of the I-Ab molecule and the MHC class II invariant chain, Ii (27). The further breeding to the 1H3.1 T cell receptor transgenic mice was an onerous task, as we had to repeat the breeding done by the Marrack lab, and at the same time be sure that the 1H3.1 TCR was carried along with it. As the breeding progressed, we saw mainly intense negative selection in mice that were positive for the 1H3.1 transgene and the AbEp transgene. It was only when the mice were transgenic for the 1H3.1 TCR, the AbEp transgene, and deleted for both the invariant chain (Ii−/−) and the A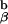 molecule (A
molecule (A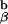 −/−) that we got a big surprise. These mice positively selected and exported to the periphery fully active 1H3.1 TCR positive cells (C. Viret and C.A.J., unpublished results). My preferred explanation for this result is shown in Figs. 2–4.
−/−) that we got a big surprise. These mice positively selected and exported to the periphery fully active 1H3.1 TCR positive cells (C. Viret and C.A.J., unpublished results). My preferred explanation for this result is shown in Figs. 2–4.
Figure 2.

Demonstration that the binding of the TCR to agonist peptides induces dimerization of TCRs, leading to two TCRs binding stably to one peptide: MHC class I molecule, whereas antagonist peptides that induce positive selection lead to binding but not TCR dimerization. The results with agonist peptide can be seen only at 37°C and not at 25°C. Data from Alam et al. (29).
Figure 4.

Data derived from in vivo positive and negative selection as interpreted by the experiments shown in Figs. 2 and 3. Shown are T cell receptors binding to agonist peptides and to antagonist peptides. The agonist peptides used to bind to the TCR induce dimer formation, but only in the presence of self-peptides, leading to activation followed by deletion in the thymus (panel 1). Y-Ae analogue peptide, which can positively select thymocytes, causes binding but not deletion. These self-peptides do not induce dimer formation, although they bind to the TCR. As a result of binding, these peptides induce positive selection in the thymus and sustain T cells in the periphery (27). Panel 3 shows what happens when the TCR does not bind to any peptide, leading to death by neglect. Our favored interpretation of the paradoxical positive selection seen with covalently linked agonist peptide in the absence of invariant chain and the endogenous I-A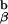 (panel 4) shows that all of the TCRs on a thymocyte adopt the same conformation and thus cannot aggregate, leading to positive selection. See text for further details.
(panel 4) shows that all of the TCRs on a thymocyte adopt the same conformation and thus cannot aggregate, leading to positive selection. See text for further details.
Fig. 2 shows the effects of a specific TCR binding to agonist or antagonist peptide:MHC complexes at 25°C or at 37°C in an instrument called a BIACor, which measures binding as surface plasmon resonance. This shows clearly that agonist peptides can induce temperature-dependent conformational changes in the T cell receptor, which leads to the formation of TCR dimers. This only occurs when the MHC binds to an agonist peptide and the MHC:agonist peptide complex is bound to the sensor chip (29). It should be noted that this result was observed with several related TCRs tested at 37°C, but not at 25°C (room temperature).
Fig. 3 shows a model to explain why the TCR dimerizes upon binding to a complex of MHC and agonist peptides. The model in Fig. 3 shows that the interaction requires a TCR that, on binding the agonist peptide:MHC, alters its conformation, and a second TCR is recruited in its native conformation. This is presumed to bind to an adjacent MHC molecule, which almost certainly will be bound to a self-peptide:self-MHC complex that can bind to the TCR but cannot alter TCR conformation. The argument that it be a self-peptide:self-MHC complex is based on the observation that fewer than 100 agonist peptides can trigger a T cell, whereas the APC surface has ≈105 MHC molecules.
Figure 3.

My personal interpretation of the results reported by Alam et al. (28), showing the dimerization to the TCR when one is bound to an agonist peptide and the other is bound to a self-peptide in its native conformation.
Fig. 4 shows how we interpret the paradoxical positive selection of this particular TCR. As one can only obtain positive selection in the absence of TCR aggregation, this may occur in two different instances. A positively selecting peptide that does not alter TCR conformation is found on its own, surrounded by a sea of self-peptide:self-MHC molecules, as presumably occurs in C57BL/6J mice. The other way one can obtain such binding without multimerization is in our unique experimental system in which all of the TCRs are conformationally altered upon binding to the agonist peptide, which leads to all 1H3.1 TCRs having the bound conformation and so being unable to aggregate. This can also presumably signal for positive intrathymic selection. Whenever we had Ii+/− or A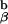 +/− molecules in the mice, we saw massive deletion of the 1H3.1 transgene-bearing T cells.
+/− molecules in the mice, we saw massive deletion of the 1H3.1 transgene-bearing T cells.
The Self-Referential Nature of the Adaptive Immune System Lays the Groundwork for Autoimmunity and Its Regulation
Having said that all lymphocytes are selected on self-ligands, sustained on self-ligands, and stimulated by foreign antigen in the presence of self-ligands (at least in the case of T cells), what might one expect of autoimmune disease? My personal opinion is that autoimmunity would be nearly inevitable were it not for the action of regulatory or suppressor T cells. Such T cells have been suspected since Richard Gershon proposed their existence and importance (30), but support for this notion fell apart when various genes or proteins were found to be missing by molecular techniques, in particular the I–J locus and secreted antigen-binding TCRα chains. All of that is changed now that regulatory T cells have been described. The clearest examples for such cells are those in which a TCR transgene directed at an autoantigen is placed in the germ line of mice and causes no problem for these mice, although the vast majority of the T cells can make autoreactive receptors. When the same receptors are bred to RAG−/− mice, which are unable to rearrange their endogenous receptors, autoimmunity develops, as shown by Juan Lafaille et al. (31), Jeff Bluestone et al. (32), and several others. This means that the reason that no autoimmunity happens in normal autoreactive TCR transgenic mice is due to the inhibitory actions of regulatory or suppressor T cells. Thus, if one has no other T cell receptors to participate in this process, then the inevitable result of having made the TCR self-reactive is autoimmunity.
Therefore, the reason for the general absence of autoimmunity is the presence and, I will argue, the activation of such suppressor T cells. Such cells recognize antigen just as do all other T cells, but they are distinguished from them by secreting various immunoregulatory cytokines such as IL-10 and TGF-β that prevent the attack on the cells or the tissue in question. We have seen hints of such cells, but the best data come from the work of other investigators, so I describe that here. However, the major lesson I have learned from this is to never accept what looks like a plausible explanation until you have explored all possible alternative explanations. So suppressor cells are back, under the banner of regulatory T cells, and I for one am happy with this result. My only sadness is that they are called Treg rather than Ts, but I guess that I can understand that in view of the many layers of Ts cells that used to exist in the minds of many of us.
I would like to think that we can learn how to reliably induce such cells, and some investigators are trying various ways to do this, as I know we are. One promising route is to administer antigen across epithelial barriers: skin, gut, respiratory, urogenital, and other routes. In organ-specific autoimmunity, like insulin-dependent diabetes mellitus, there are specific targets to aim at. One other advantage of inducing regulatory or suppressor T cells is that they seem to home to the organ producing their ligand at the highest level. There, they secrete immunoregulatory cytokines that can protect not only against attack directed at the specific antigen used to induce such regulatory T cells but other antigens as well, due to bystander regulatory effects. If we could learn how to manipulate the induction of such cells reliably, we can envision a future in which, along with vaccinations against measles and chicken pox, one could receive immunization against diabetes mellitus and other autoimmune diseases in childhood, before they can do irreparable damage to the host.
Conclusions
I began this essay with four important points: the innate immune response is very important as a discriminator between self and non-self, and it does so by regulating the expression of costimulatory molecules on the surfaces of antigen-presenting cells. I then pointed out that the adaptive immune system of T and B lymphocytes is referential to self-ligands but does not respond to them under normal conditions. Third, I reviewed the evidence that foreign antigen can only be recognized in comparison to self-antigen, at least in the one system we have been able to analyze to date. Finally, I would like to welcome back suppressor/regulatory T cells from their 15–20-year sojourn in the wilderness. They may be our best hope for preventing autoimmune disease.
Acknowledgments
First, I acknowledge the coworkers in my own laboratory, without whose dedication these studies could never have been done: Ruslan Medzhitov, F. Susan Wong, Christophe Viret, Derek Sant'Angelo, and Matthew Levine. I thank the many friends and colleagues that I have met over the last 35 years for teaching me about the immune system; these are too numerous to list. I also thank the members of the National Academy of Sciences for electing me to membership, giving me the highest honor available to a scientist in this country. I thank my secretary, Jennifer Boucher-Reid, for typing many drafts of this manuscript. I thank the many people who have trained in my lab and who have enriched my experiences by performing experiments that I asked them to, and also experiments I did not. I express special thanks to the many wonderful students to whom I have had the privilege of teaching the always fascinating science of immunology over the past 23 years at the Yale University School of Medicine. Were it not for their probing questions, I probably would never have made the few contributions outlined in this essay. It has been a wonderful life for me.
Abbreviation
- TCR
T cell antigen receptor
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on May 2, 2000.
References
- 1.Liu Y, Janeway C A., Jr Proc Natl Acad Sci USA. 1992;89:3845–3849. doi: 10.1073/pnas.89.9.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medzhitov R, Preston-Hurlburt P, Janeway C A., Jr Nature (London) 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 3.Poltorak A, He X, Smirnova I, Liu M Y, Huffel C V, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 4.Qureshi S T, Lariviere L, Leveque G, Clermont S, Moore K J, Gros P, Malo D. J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janeway C A., Jr Cold Spring Harbor Symp Quant Biol. 1989;54:1–13. [Google Scholar]
- 6.Agrawal A, Eastman Q M, Schatz D G. Nature (London) 1998;394:744–751. doi: 10.1038/29457. [DOI] [PubMed] [Google Scholar]
- 7.Hiom K, Melek M, Gellert M. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- 8.Murali-Krishna K, Altman J D, Suresh M, Sourdive D J, Zajac A J, Miller J D, Slansky J, Ahmed R. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 9.Swain S L, Hu H, Huston G. Science. 1999;286:1381–1383. doi: 10.1126/science.286.5443.1381. [DOI] [PubMed] [Google Scholar]
- 10.Murali-Krishna K, Lau L L, Sambhara S, Lemonnier F, Altman J, Ahmed R. Science. 1999;286:1377–1381. doi: 10.1126/science.286.5443.1377. [DOI] [PubMed] [Google Scholar]
- 11.Levine M H, Haberman A M, Sant'Angelo D B, Hannum L G, Cancro M P, Janeway C A, Jr, Shlomchik M J. Proc Natl Acad Sci USA. 2000;97:2743–2748. doi: 10.1073/pnas.050552997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jerne N K. Ann Immunol. 1974;125:373–389. [PubMed] [Google Scholar]
- 13.Gitlin D, Hitzig W H, Janeway C A. J Clin Invest. 1956;35:1199–1204. doi: 10.1172/JCI103374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerner J D, Appleby M W, Mohr R N, Chien S, Rawlings D J, Maliszewski C R, Witte O N, Perlmutter R M. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 15.Janeway C A, Rosen F S, Merler E, Alper C A. The Gamma Globulins. Brown, Boston: Little; 1967. [Google Scholar]
- 16.Cohen J. Science. 2000;288:2150–2153. doi: 10.1126/science.288.5474.2150. [DOI] [PubMed] [Google Scholar]
- 17.Brocker T. J Exp Med. 1997;186:1223–1232. doi: 10.1084/jem.186.8.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sant'Angelo D B, Waterbury P G, Cohen B E, Martin W D, Van Kaer L, Hayday A C, Janeway C A., Jr Immunity. 1997;7:517–524. doi: 10.1016/s1074-7613(00)80373-8. [DOI] [PubMed] [Google Scholar]
- 19.Lam K P, Kuhn R, Rajewsky K. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 20.Cheng A M, Rowley B, Pao W, Hayday A, Bolen J B, Pawson T. Nature (London) 1995;378:303–306. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 21.Levine M H. Ph.D. thesis. New Haven, CT: Yale Univ.; 2000. [Google Scholar]
- 22.Hannum L G, Haberman A M, Anderson S M, Shlomchik M J. J Exp Med. 2000;192:931–942. doi: 10.1084/jem.192.7.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanchot C, Lemonnier F A, Perarnau B, Freitas A A, Rocha B. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 24.Viret C, Wong F S, Janeway C A., Jr Immunity. 1999;10:559–568. doi: 10.1016/s1074-7613(00)80055-2. [DOI] [PubMed] [Google Scholar]
- 25.Goldrath A W, Bevan M J. Immunity. 1999;11:183–190. doi: 10.1016/s1074-7613(00)80093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janeway C A, Jr, Dianzani U, Portoles P, Rath S, Reich E P, Rojo J, Yagi J, Murphy D B. Cold Spring Harbor Symp Quant Biol. 1989;54:657–666. doi: 10.1101/sqb.1989.054.01.077. [DOI] [PubMed] [Google Scholar]
- 27.Ignatowicz L, Kappler J, Marrack P. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]
- 28.Viret C, He X, Janeway C A., Jr J Immunol. 2000;165:6183–6192. doi: 10.4049/jimmunol.165.11.6183. [DOI] [PubMed] [Google Scholar]
- 29.Alam S M, Davies G M, Lin C M, Zal T, Nasholds W, Jameson S C, Hogquist K A, Gascoigne N R, Travers P J. Immunity. 1999;10:227–237. doi: 10.1016/s1074-7613(00)80023-0. [DOI] [PubMed] [Google Scholar]
- 30.Gershon R K, Kondo K. Immunology. 1971;21:903–914. [PMC free article] [PubMed] [Google Scholar]
- 31.Olivares-Villagomez D, Wang Y, Lafaille J J. J Exp Med. 1998;188:1883–1894. doi: 10.1084/jem.188.10.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salomon B, Lenschow D J, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone J A. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]