

Abstract
We have previously shown that chronic administration of the selective A3 receptor agonist N6-(3-iodobenzyl)-5′-N-methyl-carboxoamidoadenosine (IB-MECA) leads to a significant improvement of postocclusive cerebral blood flow, and protects against neuronal damage and mortality induced by severe forebrain ischemia in gerbils. Using immunocytochemical methods we now show that chronic with IB-MECA results in a significant preservation of ischemia-sensitive microtubule associated protein 2 (MAP-2), enhancement of the expression of glial fibrillary acidic protein (GFAP), and a very intense depression of nitric oxide synthase in the brain of postischemic gerbils. These changes demonstrate that the cerebroprotective actions of chronically administered IB-MECA involve both neurons and glial cells, and indicate the possibility of distinct mechanisms that are affected in the course of chronic administration of the drug.
Keywords: Ischemia, Adenosine A3 receptor, GFAP (glial fibrillary acidic protein), MAP-2 (microtubule associated protein), Nitric oxide (NO), Synthase, (Gerbil)
1. Introduction
Among the three major adenosine receptor types (i.e., A1, A2, and A3) the function and the biological significance of the A3 receptor are the least known. Although all adenosine receptors belong to the G protein-coupled super-family they are associated with different second messenger systems. A3 receptors are associated with adenylate cyclase and phospholipase C. Upon stimulation, adenosine A3 receptor (in similarity to the A1 type) depresses cAMP levels (Zhou et al., 1992; Kim et al., 1994), and (contrary to A1 type) activates phospholipase C (Ramkumar et al., 1993; Abbracchio et al., 1995a). In vitro studies also revealed that at least in the human leukemia cell line (HL-60), intense activation of adenosine A3 receptors results in enhanced influx of Ca2+, release of intracellular calcium stores, and apoptosis (Kohno et al., 1996). Ceruti et al. (1996) and Abbracchio et al. (in preparation) have also shown that astrocytes are sensitive to stimulation with adenosine A3 receptor agonists. Astrocytic response to these drugs is curious in that at low concentrations (i.e., 10–100 nM) 2-chloro-N6-(3-iodobenzyl)-5′-N-methyl-carboxamido-adenosine (Cl-IB-MECA) induces transformation of astrocytes from the protoplasmic to the fibrous state, while exposure to micromolar amounts (> 30 μM) of Cl-IB-MECA leads to DNA fragmentation (Ceruti et al., 1996; Abbracchio et al., in preparation). Finally, we have shown that chronic administration of IB-MECA in gerbils results in a significant improvement of postischemic cerebral blood perfusion following 10 min forebrain ischemia in gerbils. The same treatment regimen also results in a striking reduction of postischemic mortality and neuronal damage in the hippocampal CA1 sector (Von Lubitz et al., 1994). Finally, there are indications that, in gerbils, chronic exposure to IB-MECA results in a very significant preservation of postischemic spatial memory and learning ability both of which can be demonstrated as late as 60 days after 10 min forebrain ischemia (Von Lubitz, 1997).
The association of the adenosine A3 receptor with at least two different effector mechanisms, and the broad range of effects elicited by its stimulation indicate the possibility that several distinct neuronal and glial systems may be affected. Nonetheless, there are no studies indicating the possible nature of these systems. Hence, the mechanisms involved in the cerebroprotective impact of the chronic treatment with adenosine A3 receptor agonists, while unquestionably complex, are equally uncertain. We have therefore studied the effect of a very long preischemic exposure to the selective adenosine A3 receptor agonist IB-MECA on immunocytochemical responses of neurons and astrocytes following severe brain ischemia in gerbils.
2. Materials and methods
2.1. Animals
Female gerbils (70–80 g, Tumblebrook Farms, Tumble Brook, MA) were used in the study. Prior to the experiments all animals were tested for susceptibility to spontaneous seizures following the method of Lee et al. (1984). Convulsing animals were rejected.
2.2. Drug and its administration
N6-(3-iodobenzyl)-5′-N-methylcarboxoamidoadenosine (IB-MECA) was obtained from Research Biochemicals International (RBI, Natick, MA). The drug was dissolved in a 20:80 mixture of Alkamuls EL-620 (Rhône-Poulenc, Cranbury, NJ) and phosphate-buffered saline (pH 7.4). The drug was administered i.p. in a 0.15 cc volume at 100 μg/kg. Treated animals (N = 20) received a single daily injection for 60 days prior to ischemia followed by one drug-free day immediately preceding occlusion of the carotid arteries. Prior studies have shown that prolonged treatment with IB-MECA does not affect any of the studied parameters. Hence, controls (N = 20) were injected only with the vehicle. The schedule was identical to that of the drug. No injections were given during the recovery.
2.3. Ischemia
The details of induction of forebrain ischemia were described in our previous publications (Von Lubitz et al., 1994, 1996). During the present experiments both carotid arteries were occluded for 10 min using Prolene 7.0 sutures with the temperature of the animals monitored and kept at the immediately preischemic level as described previously (Von Lubitz et al., 1994). Five non-injected animals were sham operated and served as overall controls for the histo- and immunocytochemical studies.
2.4. Histo- and immunocytochemistry
Seven days after ischemia, the surviving gerbils (8 controls and 16 IB-MECA treated) were anesthetized with Nembutal (50 mg/kg i.p.) and perfused with saline-buffered (pH 7.4) solution of 3.4% paraformaldehyde (Lin et al., 1990). The brains were removed and, after 24 h immersion in fresh fixative, were transferred for the following 24 h to a 20% (v/v) solution of sucrose in phosphate-saline buffer (PBS). Serial frozen sections were cut at 40 μm.
2.4.1. NADPH-diaphorase staining
NADPH-diaphorase (nitric oxide synthase (NO synthase) histochemical staining was performed using according to the method of Hope et al. (1991). The sections were incubated at 37°C for 2–8 h in 0.1 M PBS containing 0.3% Triton X-100, 0.01% NADPH and 0.02% Nitro Blue Tetrazolium (all from Sigma, St. Louis, MO). Following a brief rinse in 0.1 M PBS, the sections were mounted on gelatin glass slides, air-dried, and mounted using DPX medium (BDH Labs., Poole, UK).
2.4.2. MAP-2 and GFAP staining
Immunocytochemical and immunofluorescent staining methods (Lin et al., 1995) were used for visualization of microtubule associated protein (MAP-2) and glial fibrillary acidic protein (GFAP). To avoid possible differences in the intensity of staining caused by random factors, all appropriate sections (i.e., GFAP or MAP-2) were grouped and processed simultaneously under exactly the same conditions.
The sections were incubated at 4°C overnight using either the primary polyclonal GFAP antibody (Incstar, Stillwater, MN) diluted at 1:50–200), or the primary monoclonal MAP-2 antibody (Sigma, St. Louis, MO) diluted at 1:100–1000. The sections were rinsed three times with 0.1 M PBS and then incubated for 1 h at room temperature with the appropriate secondary antibodies of fluorescein isothiocyanate (FITC), rhodamine isothiocyanate (RITC), or Texas Red-conjugated IgG (dilution 1:100–200), or ABC kit (Vector, Burlingame, CA). The immunofluorescent labelling patterns were studied with an epifluorescent microscope (Labophot) equipped with appropriate filters (Nikon B2 and G1B).
2.4.3. Quantitative analysis of GFAP and MAP-2
GFAP and MAP-2 stained sections (altogether 15 sections/stain) were randomly selected from all 5 brains of the sham operated animals (N = 5) and from 5 randomly selected postischemic brains of both vehicle and IB-MECA-injected gerbils. All sections were taken from the region approximately 1.5–1.9 mm posterior to the bregma (Loskota et al., 1974).
In order to establish volume density (Vv) of astrocytes expressing GFAP, a micrograph containing the medial segment of CA1 was made from each section (see also Von Lubitz et al., 1989). The micrographs (total magnification × 1100) were overlaid with a rectangular grid (54 intersection points) placed with its upper edge against the lower border of stratum pyramidale and with the intersection points distributed over the underlying stratum radiatum. The number of grid intersection points falling over GFAP-expressing astrocyte profiles was counted on each micrograph, followed by the analysis of Vv performed by means of standard stereological techniques (Elias and Hyde, 1980) described extensively in an earlier paper (Von Lubitz and Diemer, 1982).
As in our previous study (Von Lubitz et al., 1996, in press), the density of MAP-2 staining dendrites was determined using a computer image analyzing system (Biographics, Winston-Salem, NC). The density was measured within 5 randomly chosen areas of CA1 stratum radiatum in each of the previously selected MAP-2 stained sections (see above).
2.5. Statistical analysis of immunocytochemical data
Since the analysis (ANOVA) of absolute values of either GFAP-expressing astrocytes or MAP-2 density revealed no statistically significant differences among individual sections of the same brain, the values were pooled and expressed as meanrbrain. The statistical significance of group (i.e., sham, vehicle- and IB-MECA-treated) differences was analyzed using ANOVA followed by Dunnett’s test. Sham animals served as the controls, and P≤0.05 indicated statistical significance. The data were then expressed as % change compared to the control group.
3. Results
The intensity of histochemical staining for nitric oxide synthase (NOS) and immunocytochemical reaction for GFAP and MAP-2 in non-ischemic, sham operated animals is shown in Figs. 1A, 2A and 3A.
Fig. 1.

(A) NO synthase staining in the hippocampus of a sham-operated (normal) animal. Weak reaction product is evenly distributed along pyramidal and molecular layers of the entire hippocampal formation and the dentate gyrus. (B) Postischemic NO synthase staining in the vehicle injected control animal. Note the very intense staining in the pyramidal layer of CA1, 2, and 3 sectors and its absence in the dentate. (C) Postischemic NO synthase staining in the animal injected chronically with IB-MECA. There is a marked absence of the reaction product within the entire pyramidal layer of the hippocampus and within the dentate. Weak staining, similar to that seen in the sham-operated animals is present in the molecular layer of CA1 and CA2.
Fig. 2.

(A) Sham-operated gerbil. There is a pronounced paucity of astrocytes expressing GFAP. (B) Postischemic gerbil injected chronically with the vehicle, and showing a pronounced increase in the number of GFAP expressing astrocytes present within the hippocampal stratum radiatum. (C) Postischemic animal injected with IB-MECA. The number of activated, GFAP-expressing astrocytes is perceptibly higher than in the vehicle injected control (compare with 2B).
Fig. 3.

(A) MAP-2 staining in the stratum radiatum of a sham-operated gerbil. (B) Postischemic control (chronic vehicle injections). There is a very substantial loss of MAP-2 reaction product. The few fibers showing its presence have a distinctly ‘beaded’ appearance indicating their possible impairment. (C) Postischemic animal injected chronically with IB-MECA. While the expression of MAP-2 immunoreactivity is lower than in the sham-operated gerbils, MAP2 staining resembles that observed in the sham-operated animals.
3.1. Postischemic changes
Neuronal preservation in the control group and in the animals treated with IB-MECA was comparable to that reported by us in our previous studies (Von Lubitz et al., 1994; Von Lubitz, 1996, 1997.
3.1.1. NADPH-diaphorase
Seven days after ischemia, the intensity of NADPH-diaphorase staining intensity was strikingly elevated in the pyramidal layer along the entire length of the hippocampal formation in all animals injected with the vehicle (Fig. 1B). The dentate, on the other hand, were remarkably free of the NADPH-diaphorase reaction product (Fig. 1A,B).
In the animals treated with IB-MECA, postischemic staining of NADP-diaphorase was virtually absent within the pyramidal layer of all hippocampal sectors (Fig. 1C). Faint staining within the molecular layer of CA1rCA2 sectors was comparable to that seen in the sham operated group (Fig. 1A,C).
3.1.2. GFAP
Compared to sham operated animals, the volume density (Vv) of postischemic astrocytes expressing GFAP increased significantly (P < 0.01) in the animals receiving chronic injections of the vehicle (Figs. 2A, B and 4. In the animals treated with IB-MECA a statistically significant increase of GFAP-marked astrocytes was present (Figs. 2A, B, C and 4) when compared to both sham operated gerbils and the animals injected with the vehicle (P < 0.01 in both cases).
Fig. 4.
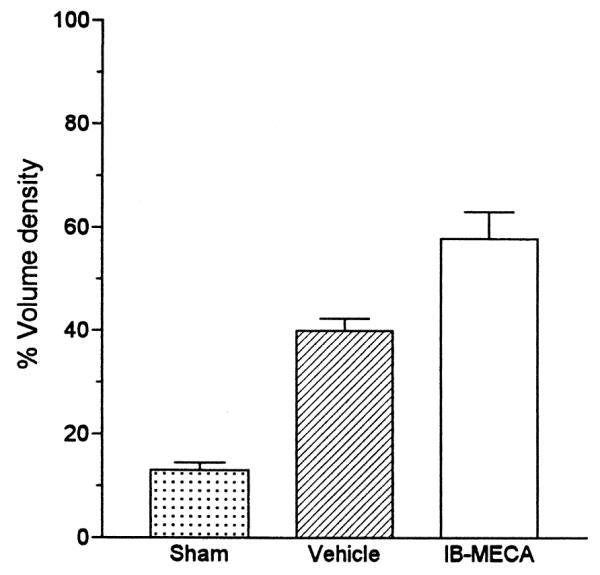
Quantitative analysis of the effect of 10 min ischemia on the number of GFAP expressing astrocytes in animals injected chronically either with the vehicle or with IB-MECA. Abbreviations: (a) P < 0.05 vs. sham-operated gerbils; (b) P < 0.05 vs. IB-MECA.
3.1.3. MAP-2
Compared to sham operated animals, the density of MAP-2 expressing dendrites decreased significantly in the ischemic animals injected with the vehicle (Figs. 3A, B and 5). The postischemic density of MAP-2 containing dendrites in the IB-MECA treated group was significantly lower than in the sham-operated gerbils (78% of sham, p < 0.05). However, the extent of MAP-2 preservation in the IB-MECA group was very significantly higher (p < 0.001) than in the vehicle injected animals (Figs. 3B, C and 5).
Fig. 5.
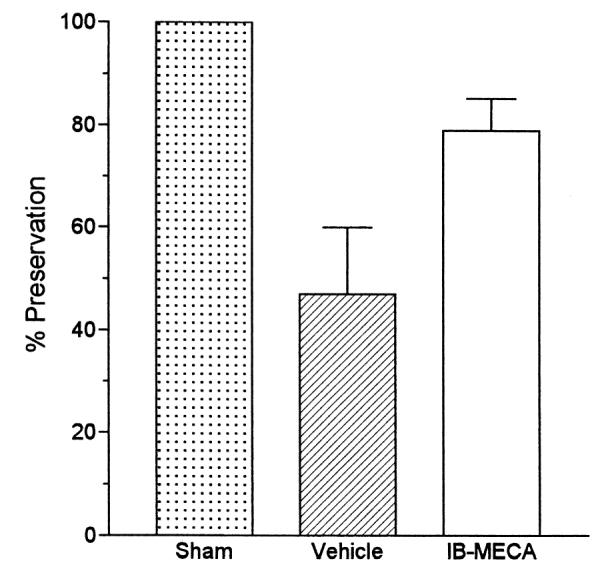
Quantitative analysis of MAP-2 expression in sham-operated gerbils (100%) vs. vehicle- and IB-MECA injected postischemic groups. Abbreviations as in Fig. 4.
4. Discussion
The most striking effect of chronic treatment with IB-MECA is its effect on postischemic expression of NO synthase. Although NADP-diaphorase staining is now considered as a standard marker for NO synthase (Wallace, 1996; Li et al., 1997), the histochemical methods used in this study are not specific enough to define the nature of the suppressed NO synthase (Šestan and Kostovi, 1994), i.e., whether its constitutive or inducible form is affected. Neurons, astrocytes, perivascular nerves, and the endothelium of cerebral vasculature have been shown to form nitric oxide (NO) during cerebral ischemia (Endoh et al., 1994; Kato et al., 1994; Loesch et al., 1994; Nakashima et al., 1995; Sparrow, 1995). However, while the activity of the constitutive NO synthase may be increased during ischemia (Dawson et al., 1991; Nagafuji et al., 1995), several studies have shown that its maximal stimulation apparently takes place at intracellular concentrations of Ca2+ that are well below those encountered during cerebral ischemia (Knowles et al., 1989; Silver and Erecinska, 1990; see also the review by Aoki et al., 1995). Moreover, expression of the calciumrcalmodulin-dependent constitutive form of NO synthase appears to depend largely on small transients of intracellular Ca2+ (Aoki et al., 1995). The inducible isoform of NO synthase, on the other hand, is independent of Ca2+ transients (Dalkara et al., 1994; Sparrow, 1995) and, furthermore, late and sustained release of nitric oxide may be the result of enhanced expression of inducible NO synthase. Very recently, Iadecola et al. (1995) showed enhanced activity of inducible NO synthase in hypertensive rats subjected to focal ischemia with the peak occurring on the 2nd day after the insult. Seven days after ischemia, NO synthase level returned to the preischemic baseline. Although in our study the controls showed very intense NO synthase staining as late as seven days after ischemia, it is likely that the effect is also related to the enhanced postischemic expression of the inducible NO synthase. Hence, it may be expected that chronic treatment with IB-MECA affects the same NO synthase isoform as well.
Whether the demonstrated reduction of NO synthase activity constitutes the primary mechanism resulting in postischemic protection of hippocampal neurons observed in this (data not shown) and our previous study of chronic IB-MECA administration is unclear. Studies in which specific NO synthase inhibitors have been used in either focal (Dalkara et al., 1994) or global ischemia (Shapira et al., 1994) indicate the dual role of nitric oxide, wherein its lower concentrations appear to be protective, while excessive release results in neuronal demise. The neuroprotective effect of NO synthase appears to be related to its hemodynamic effects (reviewed by Iadecola et al., 1994). Interestingly, our preliminary observations indicate that the depression of NO synthase expression in the postischemic brain following chronic treatment with IB-MECA is much more severe in the cerebral parenchyme than either in the blood vessels or in the immediately surrounding tissue. Furthermore, the same treatment also causes a significant improvement of the postischemic blood flow (Von Lubitzet al., 1994). Hence, it is conceivable that rapid normalization of the postocclusive blood perfusion is the result of vasodilatory actions of NO released both by the endothelium and by the perivascular end-feet of the activated astrocytes. Such conclusion is also supported by the very significantly enhanced presence of GFAP-expressing (i.e., activated) astrocytes in the IB-MECA treated group.
Rapid postischemic increase in the number of astrocytes exhibiting GFAP reaction is a well known phenomenon (Petito and Babiak, 1982; Petito et al., 1990). This increase appears to have two distinct forms (Petito et al., 1990): either a rapid and reversible postischemic proliferation of astrocytes indicating the presence of reconstitutive processes (Abbracchio et al., 1995b), or persistent reactive astrocytosis characterized by hyperplasia and hypertrophy that are typical of the damaged regions (Petito et al., 1990). Our preliminary data indicate that in the hippocampus of animals treated chronically with IB-MECA, the number of GFAP-staining astrocytes returns to the baseline in less than 4 weeks after ischemia, i.e., the time period which corresponds well to the duration of the reversible phase.
Activation of adenosine A3 receptors results in stimulation of phospholipase C and protein kinase C (Ramkumar et al., 1993; Abbracchio et al., 1995a) resulting in the enhancement of extracellular Ca2+ influx and liberation of intracellular calcium stores (Kohno et al., 1996). It is, however, unknown whether adenosine A3 receptor-mediated elevation of intracellular Ca2+ is sufficient to activate proteolytic enzymes and Ca2+-activated kinases, e.g., calpain or calmodulin (Goodman and Zagon, 1986) whose activation has been suggested to play a major role in the development of ischemic damage. We have shown in our previous study that preischemically administered IB-MECA enhances both mortality and neuronal destruction in gerbils (Von Lubitz et al., 1994). Chronic treatment with IB-MECA improved the outcome in all studied measures (Von Lubitz et al., 1994; Von Lubitz, 1996, 1997). Although the extensive preservation of MAP-2 reported in this study does not provide the definitive proof of phospholipase C attenuation induced by chronic treatment with IB-MECA, it is consistent with the hypothesis of diminished intraneuronal levels of Ca2+, and the consequent depression of calpain activity (Matesic and Lin, 1994).
Elevation of intracellular Ca2+ is necessary in order to activate Ca2+/calmodulin-dependent protein kinase II and subsequent phosphorylation of GFAP (Yano et al., 1994). Hence, the postulate that chronically administered IB-MECA depresses intracellular calcium levels may not be true for astrocytes since chronic exposure to the drug results in the increased number of GFAP-expressing astrocytes. The latter observation, may, however, indicate that either the interactions of astrocyte A3 receptors with other adenosine/non-adenosine receptor types or receptor–second messenger systems are different from those observed in neurons.
As mentioned previously, adenosine A3 receptors are associated with at least two second messenger mechanisms, i.e., adenylate cyclase (inhibition) and phospholipase C (activation). Recently, it has been also shown that acute exposure of cultured astrocytes to very low concentrations (nanomolar) of selective adenosine A3 agonists induces their differentiation and reinforcement of their cytoskeleton (i.e., enhanced expression of GFAP), while higher levels of the agonist (micromolar) induce their apoptotic death (Ceruti et al., 1996). These observations suggest the possibility that both submaximal acute- and chronic stimulation of astrocyte A3 receptors may attenuate phospholipase C activity, and consequently (and in similarity to adenosine A1 receptors) promote neuroprotective effects associated with the reduction of cAMP. While the notion of such shift requires experimental verification, there is no doubt that the present results confirm the fact that adenosine A3 receptors may play an important role in the development of ischemic brain damage. The wide range of the effects induced by their chronic stimulation also indicate that adenosine A3 receptors may be involved in other cerebral pathologies as well.
References
- Abbracchio MP, Brambilla R, Cerutti S, Kim HO, Von Lubitz DKJE, Jacobson KA, Cattabeni F. G Protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol. Pharmacol. 1995a;48:1038–1045. [PubMed] [Google Scholar]
- Abbracchio MP, Ceruti S, Burnstock G, Cattabeni F. Purinoceptors on glial cells of the central nervous system: functional and pathologic implications. In: Belardinelli L, Pelleg A, editors. Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Kluwer Academic Publishers; Boston: 1995b. pp. 271–280. [Google Scholar]
- Aoki E, Takeuchi IK, Shoji R. Nitric oxide: an attractive signalling molecule. Acta Histochem. Cytochem. 1995;2:97–100. [Google Scholar]
- Ceruti S, Barbieri D, Franceschi C, Giammaroli AM, Rainaldi G, Malorni W, Kim HO, Von Lubitz DKJE, Jacobson KA, Cattabeni F, Abbracchio MP. Effects of adenosine A3 receptor agonists on astrocytes: induction of cell protection at low and cell death at high concentrations. Drug. Dev. Res. 1996;3:177. [Google Scholar]
- Dalkara T, Yoshida T, Irikura K, Moskowitz MA. Dual role of nitric oxide in focal cerebral ischemia. Neuropharmacology. 1994;33:1447–1452. doi: 10.1016/0028-3908(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cell culture. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias H, Hyde DM. An elementary introduction to stereology. Am. J. Anat. 1980;159:412–446. doi: 10.1002/aja.1001590407. [DOI] [PubMed] [Google Scholar]
- Endoh, Maiese K, Wagner J. Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Res. 1994;651:92–100. doi: 10.1016/0006-8993(94)90683-1. [DOI] [PubMed] [Google Scholar]
- Goodman SR, Zagon IS. The neural cell spectrin skeleton: a review. Am. J. Physiol. 1986;250:347–360. doi: 10.1152/ajpcell.1986.250.3.C347. [DOI] [PubMed] [Google Scholar]
- Hope BT, Mitchell GJ, Knigg KM, Vincent SR. Neuronal NADP diaphorase is a nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2811–2820. doi: 10.1073/pnas.88.7.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Pellegrino DA, Moskowitz MA, Lassen N. Nitric oxide synthase inhibition and cerebrovascular regulation. J. Cereb. Blood Flow Metab. 1994;14:175–192. doi: 10.1038/jcbfm.1994.25. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Xu X, Zhang F, el-Ferkany EE, Ross ME. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1995;1:52–59. doi: 10.1038/jcbfm.1995.6. [DOI] [PubMed] [Google Scholar]
- Kato H, Kogure K, Liu Y, Araki T, Itoyama Y. Induction of NADPH-diaphorase activity in the hippocampus in a rat model of cerebral ischemia and ischemic tolerance. Brain Res. 1994;652:71–75. doi: 10.1016/0006-8993(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Kim HO, Ji X.-d., Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 1994;21:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles RG, Palacios M, Palmer RMJ, Moncada S. Formation of nitric oxide from L-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble gunanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5159–5162. doi: 10.1073/pnas.86.13.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno Y, Sei Y, Koshiba M, Kim HO, Jacobson KA. Induction of apoptosis in HL-60 human promyelocytic leukemia cells by adenosine A3 receptor agonists. Biochem. Biophys. Res. Commun. 1996;219:904, 910. doi: 10.1006/bbrc.1996.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RJ, Bajorek JG, Lomax P. Similar anticonvulsive, but unique behavioural effects of opioid agonists in the seizure-sensitive Mongolian gerbil. Neuropharmacology. 1984;23:517–524. doi: 10.1016/0028-3908(84)90024-8. [DOI] [PubMed] [Google Scholar]
- Lin RCS, Matesic DF, Marvin M, McKay RDG, Brüstle O. Re-expression of the intermediate filament nestin in reactive astrocytes. Neurobiol. Dis. 1995;2:79–85. doi: 10.1006/nbdi.1995.0008. [DOI] [PubMed] [Google Scholar]
- Li J, Chen S, Lin RCS, Smith SS. Cerebellar nitric oxide synthase is expressed within granule cell patches innervated by specific mossy fiber terminals: a development profile. Dev. Neurosci. 1997;19:274–282. doi: 10.1159/000111216. [DOI] [PubMed] [Google Scholar]
- Lin C-S, Polsky K, Nadler JV, Crain B. Selective neocortical and thalamic cell death in the gerbil after transient ischemia. Neuroscience. 1990;2:289–299. doi: 10.1016/0306-4522(90)90083-g. [DOI] [PubMed] [Google Scholar]
- Loesch A, Belai A, Burnstock G. An ultrastructural study of NADPH-diaphorase and nitric oxide synthase in the perivascular nerves and vascular endothelium of the rat basilar artery. J. Neurocytol. 1994;23:49–59. doi: 10.1007/BF01189816. [DOI] [PubMed] [Google Scholar]
- Loskota WJ, Lomax P, Verity MA. Ann Arbor Sci. Publ.; Ann Arbor, MI: 1974. A Stereotaxic Atlas of the Mongolian Gerbil Brain. [Google Scholar]
- Matesic DF, Lin RCS. Microtubule-associated protein 2 as an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain. J. Neurochem. 1994;63:1012–1020. doi: 10.1046/j.1471-4159.1994.63031012.x. [DOI] [PubMed] [Google Scholar]
- Nagafuji T, Sugiyama M, Matsui T, Muto A, Naito S. Nitric oxide synthase in cerebral ischemia: possible contribution of nitric oxide synthase activation in brain microvessels to cerebral ischemic injury. Mol. Chem. Neuropathol. 1995;26:107–157. doi: 10.1007/BF02815009. [DOI] [PubMed] [Google Scholar]
- Nakashima MN, Yamashita K, Kataoka Y, Yamashita YS. Time course of nitric oxide synthase activity in neuronal, glial, and endothelial cells of rat striatum following focal cerebral ischemia. Cell Mol. Neurobiol. 1995;15:341–349. doi: 10.1007/BF02089944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petito CK, Babiak T. Early proliferative changes in astrocytes in postischemic noninfarcted rat brain. Ann. Neurol. 1982;11:510–518. doi: 10.1002/ana.410110511. [DOI] [PubMed] [Google Scholar]
- Petito CK, Morgello S, Felix JC, Lesser ML. The two patterns of reactive astrocytosis in postischemic rat brain. J. Cereb. Blood Flow Metab. 1990;10:850–859. doi: 10.1038/jcbfm.1990.141. [DOI] [PubMed] [Google Scholar]
- Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3 AR is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem. 1993;268:16887–16890. [PubMed] [Google Scholar]
- Šestan N, Kostovi I. Histochemical localization of nitric oxide synthase in the CNS. Trends Pharmacol. Sci. 1994;3:106. doi: 10.1016/0166-2236(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Shapira S, Kadar T, Weissman BA. Dose-dependent effect of nitric oxide synthase inhibition following transient forebrain ischemia in gerbils. Brain Res. 1994;668:80–84. doi: 10.1016/0006-8993(94)90513-4. [DOI] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Intracellular and extracellular (Ca2+ ) in hypoxia and ischemia in rat brain in vivo. J. Gen. Physiol. 1990;95:837–866. doi: 10.1085/jgp.95.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR. Inducible nitric oxide synthase in the central nervous system. J. Mol. Neurosci. 1995;5:219–229. doi: 10.1007/BF02736723. [DOI] [PubMed] [Google Scholar]
- Von Lubitz DKJE. Trembly B, Slikker W, editors. Adenosine A3 receptor and brain: a culprit, a hero, or merely yet another receptor. Neuroprotective Agents: Clinical and Experimental Aspects II. Ann. New York Acad. Sci. 1996;825:49–67. doi: 10.1111/j.1749-6632.1997.tb48413.x. [DOI] [PubMed] [Google Scholar]
- Von Lubitz DKJE. Adenosine A3 receptor and brain: a culprit, a hero, or merely yet another receptor. Proc. N.Y. Acad. Sci. 1997;825:49–67. doi: 10.1111/j.1749-6632.1997.tb48413.x. [DOI] [PubMed] [Google Scholar]
- Von Lubitz DKJE, Diemer NH. Complete cerebral ischemia in the rat: an ultrastructural and stereological analysis of the distal stratum radiatum in the hippocampal CA-1 region. Neuropathol. Appl. Neurobiol. 1982;8:197–215. doi: 10.1111/j.1365-2990.1982.tb00275.x. [DOI] [PubMed] [Google Scholar]
- Von Lubitz DKJE, Dambrosia JM, Redmond DJ. Protective effect of cyclohexyl adenosine in treatment of cerebral ischemia in gerbils. Neuroscience. 1989;2:451–462. doi: 10.1016/0306-4522(89)90265-0. [DOI] [PubMed] [Google Scholar]
- Von Lubitz DKJE, Lin RC-S, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur. J. Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz DKJE, Lin RC-S, Paul IA, Beenhakker M, Boyd M, Bischofberger N, Jacobson KA. Postischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur. J. Pharmacol. 1996:171–179. doi: 10.1016/s0014-2999(96)00667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MN. NADP-diaphorase activity in activated astrocytes representing inducible nitric oxide synthase. Methods Enzymol. 1996;268:497–503. doi: 10.1016/s0076-6879(96)68051-3. [DOI] [PubMed] [Google Scholar]
- Yano S, Fukunaga K, Ushio Y, Miyamoto M. Activation of Ca2+/calmodulin-dependent protein kinase II and phosphorylation of intermediate filament proteins by stimulation of glutamate receptors in cultured cortical astrocytes. J. Biol. Chem. 1994;261:5428–5439. [PubMed] [Google Scholar]
- Zhou QY, Li CY, Olah ME, Johnson RA, Stiles GL, Civelli O. Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]