

Abstract
The M1-selective muscarinic receptor antagonist pirenzepine (5,11-dihydro-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b] [1,4]benzodiazepin-6-one) was derivatized to explore points of attachment of functionalized side chains for the synthesis of receptor probes and ligands for affinity chromatography. The analogues prepared were evaluated in competitive binding assays versus [3H]-N-methylscopolamine at four muscarinic receptor subtypes (m1AChR-m4AChR) in membranes from rat heart tissue and transfected A9L cells. 9-(Hydroxymethyl)pirenzepine, 8-(methylthio)pirenzepine, and a series of 8-aminosulfonyl derivatives were synthesized. Several 5-substituted analogues of pirenzepine also were prepared. An alternate series of analogues substituted on the 4-position of the piperazine ring was prepared by reaction of 4-desmethylpirenzepine with various electrophiles. An N-chloroethyl analogue of pirenzepine was shown to form a reactive aziridine species in aqueous buffer yet failed to affinity label muscarinic receptors. Within a series of aminoalkyl analogues, the affinity increased as the length of the alkyl chain increased. Shorter chain analogues were generally much less potent than pirenzepine, and longer analogues (7–10 carbons) were roughly as potent as pirenzepine at m1 receptors, but were nonselective. Depending on the methylene chain length, acylation or alkyl substitution of the terminal amine also influenced the affinity at muscarinic receptors.
Muscarinic cholinergic receptors (mAChRs) mediate the actions of the neurotransmitter acetylcholine in the central and peripheral nervous systems,1 gastrointestinal system,2 heart,3 endocrine glands,4 lungs,5 and other tissues. At least five distinct gene products6–8 have been identified that code for five distinct mAChRs, termed m1 through m5. The m1, m2, and m3 receptors correlate pharmacologically to the M1 M2, and M3 (M2 glandular) receptors, respectively.9 The m1 and m3 receptors have been shown to be coupled preferentially to the stimulation of phosphoinositide metabolism,7 and m2 and m4 receptors have been shown to be coupled preferentially to the inhibition of adenylate cyclase.7,10 Other effector systems such as voltage-dependent11 and calcium-dependent12 potassium channels are coupled to muscarinic receptors.
In the brain, five genetic subtypes have been localized through hybridization with oligonucleotide probes.6a The working hypothesis that administration of a centrally active, selective muscarinic agonist would relieve the memory loss associated with Alzheimer’s disease was deduced from the observed degeneration of cholinergic cells in the nucleus basalis region of these patients.13 Subsequent research prompted by this observation has led to the identification of a number of classes of muscarinic agonists.14
In addition to the development of new agonists, there is need to develop more highly selective muscarinic antagonists, both as pharmacological tools and as potential therapeutic agents. For example, the first known m1-selective antagonist, pirenzepine,15 is useful clinically in the inhibition of gastric acid production.2 An m3-selective antagonist would be useful in treating airway disease5 and atonic conditions of the gut and bladder.16 An m3-selective antagonist would be useful in the control of hyperreactivity of smooth muscle.16
Our previous studies of structure–activity relationships in analogues of the muscarinic agonist oxotremorine utilized a functionalized congener approach,17 i.e., chemically functionalized chains were incorporated at sites on the pharmacophore that were insensitive to this modification in receptor binding. Functionalized congeners in other drug classes have been useful in affinity chromatography to purify receptors, in the synthesis of selective affinity labels, and in prodrug design.18 We now extend the functionalized congener approach to analogues of the selective muscarinic antagonist pirenzepine 1.
Results and Discussion
Chemistry
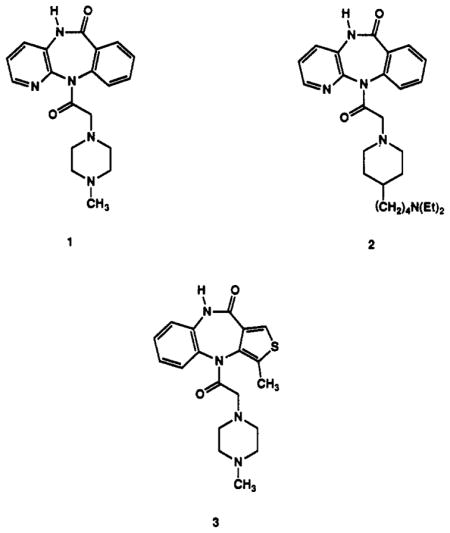
The tricyclic skeleton and the piperazine ring of the pirenzepine molecule were explored as possible sites for the attachment of derivatized chains in making functionalized congeners of pirenzepine. These two regions contain a hydrogen-bond acceptor atom (usually a carbonyl oxygen) and a basic nitrogen atom, two key structural elements involved in binding to muscarinic receptors. Since a number of classes of muscarinic antagonists have major structural differences at the “carbonyl-end” of the ligand,14 we surmised that there would be sufficient steric freedom for chain attachment on one of the aromatic rings of pirenzepine. In addition, a number of muscarinic ligands have been reported in which the basic nitrogen side chain is varied.19–21 It appears that structural modifications at the “amine-end” of muscarinic ligands frequently cause major changes in the selectivity profile of the compound. For example, AQ-RA 741 (2), which contains the same tricyclic ring system as pirenzepine, is selective for M2 receptors.20 On the other hand, certain modifications of pirenzepine in the tricyclic moiety such as in the more potent antagonist telenzepine (3) preserve selectivity for M1 receptors.22
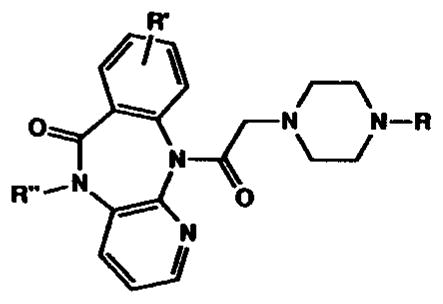
Pirenzepine analogues containing sites for functionalization and chain extension were synthesized and fully characterized (Table I). Analogues containing substituents located on the benzene ring of pirenzepine in the 8- and 9-positions (Schemes I–III) were included. Small (11a and 16), extended chain (11c–e), or sterically large substituents (15) were prepared. The position of aromatic substitution was verified by a comparative proton NMR study, which is summarized in Table II. The C-8 aromatic proton (para to the amino group) appears at approximately 6.9 ppm from tetramethylsilane in the tricyclic intermediates (6 and 14) and at approximately 7.4 ppm when the amine is acylated, as in 1 and 15. This signal is absent in compounds 8, 9, and 11, which are substituted at the C-8 position. The tricyclic intermediate 6 was synthesized by published procedures,21 starting with 3-amino-2-chloro-pyridine (4) and methyl anthranilate (5) (Scheme I). Chlorosulfonation of 6 resulted in the formation of the 8-chlorosulfonyl intermediate 7 (not isolated), which was then reduced with triphenylphosphine.23 The resulting thiol was alkylated to give the corresponding thioether 8. Prior efforts to substitute pirenzepine (1) directly by a variety of electrophilic reagents (e.g. by chlorosulfonation, nitration, chloromethylation) resulted in recovery of unreached pirenzepine. The extreme inertness of pirenzepine in comparison to 6 can be rationalized partially by the planarity of 6, which enables delocalization of the N-11 electron pair within the benzene ring. Such activation of the benzene ring is not possible in the nonplanar, “butterfly” shape of pirenzepine.20 The acylation of the exocyclic amine may also contribute to the relative un-reactivity of the tricyclic moiety of pirenzepine.
Table I.
Synthesis and Binding Affinity of Novel Pirenzepine Analogues. All Compounds Were Analyzed for C, H, and N within ±0.4%, Except Where Noted. Ki Values (±sem) for Inhibition of Binding of [3H]-N-Methylscopolamine Are Given for m1–m4 Muscarinic Receptors, for One to Three Determinations, Each Performed in Triplicate. R″ = H, Except for Compound 17 As Indicated. Binding Was Carried Out for 90 min at 37 °C, Using Membranes from Rat Heart Tissue (m2 Receptors) and Transfected A9L Cells (m1 and m3 Receptors) and in NG108-15 Cells (m4 Receptors) in the Presence of 0.5 nM [3H]-NMS. Ki Values Are Expressed in Units of Micromolar
| ||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R = | R′ = | mp, °C | Anal. | m1 | m2 | m3 | m4 |
| 1 | CH3 | H | 0.029 | 0.61 ± 0.05 | 0.24 | 0.33 | ||
| 3 | 0.0018 | 0.0503 | 0.0069 | 0.0174 | ||||
| 11a | CH3 | 8-SCH3 | >205dec | C20H23N5O2S·1HCl·1.5H2O | 4.66 ± 1.02 | 11.0 | 3.62 ± 0.14 | n.d.l |
| 11b | CH3 | 8-SO2NH2 | C19H22N6O4S·0.5H2Oa | >30 | >30 | >30 | n.d. | |
| 11c | CH3 | 8-SO2NH(CH2)2NHBoc | 210–213 dec | C26H35N7O6S·3oxalate·0.25H2O | >30 | n.d. | >30 | n.d. |
| 11d | CH3 | 8-SO2NH(CH2)2NH2 | 155–158 | C21H27N7O4S·1.5oxalate·0.5H2O | >30 | n.d. | >30 | n.d. |
| 11e | CH3 | 8-SO2NH(CH2)2NHAc | 190–193 | C23H29N7O5S·2oxalate·0.5H2O | >30 | n.d. | >30 | n.d. |
| 15 | CH3 | 9-CH2OSi(CH3)2-t-Bu | 245–248 | C26H37N5O3Si·0.25H2O | >30 | 4.4 ± 0.3 | >30 | n.d. |
| 16 | CH3 | 9-CH2OH | C20H23N5O3·1.5H2O | 4.4 | n.d. | 4.4 | n.d. | |
| 17aj | CH3 | H | 108–111 | C21H25N5O2·1HCl·1.75H2O | 7.7 ± 2.1 | 8.00 | n.d. | n.d. |
| 17bk | CH3 | H | oil | C26H32N6O2·0.5H2O | 8.9 ± 4.7 | n.d. | n.d. | n.d. |
| 18 | H | H | >230 | 0.37 | 3.34 | 1.31 | 1.67 | |
| 19a | COCH3 | H | >290 | C20H21N5O3·0.25H2O | >100 | >100 | >100 | >100 |
| 19b | COC6H5 | H | 202–205 | C25H23N5O3·HCl·0.75H2O | >100 | >100 | >100 | >100 |
| 20 | SO2CH3 | H | 180–183 | C19H21N5O4S·HCl | >100 | >100 | >100 | >100 |
| 21 | CSNHC6H5 | H | 204–206 | C25H24N6O2S·0.75H2O | >100 | >100 | >100 | >100 |
| 22 | CH2≡CH | H | 249–251 | C21H21N5O2 | 1.67 ± 0.06 | 1.37 ± 0.06 | n.d. | n.d. |
| 23 | CH2C6H5 | H | 145–148 | C25H25N5O2·oxalate·H2Ob | 0.33 | 0.24 ± 0.10 | 0.24 | 0.22 |
| 24a | CH2C6H4-p-CONHCH3 | H | 191–193 | C27H28N6O3·oxalate·0.5H2O | 2.84 | 2.65 | 4.02 | 2.94 |
| 24b | CH2C6H4-p-CONH(CH2)2NHBoc | H | 155–158 | C33H39N7O5·oxalate·H2Oc | n.d. | 2.50 | n.d. | n.d. |
| 24c | CH2C6H4-p-CONH(CH2)2NH2 | H | 208–212 | C26H31N7O3·oxalate·5H2Od | 1.39 | 0.96 | 3.65 | 1.04 |
| 24d | CH2C6H4-p-CONH(CH2)4NHBoc | H | 173–175 | C35H43N7O5·oxalate·0.5H2O | 1.68 | 1.09 | 3.47 | n.d. |
| 25 | (CH2)2OH | H | 197–200 | C20H23N5O3 | 5.8 ± 1.0 | 3.2 | 9.5 | n.d. |
| 26 | (CH2)2Cl | H | 188–190 | C20H22N5O2Cl·2HCl·1.5H2O | 0.052 | 0.21 | 0.078 | 0.13 |
| 27 | (CH2)2NH2 | H | 190–193 | C20H24N6O2·1H2O | 1.19 | 3.04 | 1.97 | 2.84 |
| 28 | (CH2)2NHAc | H | 93–95 | C22H26N6O3·0.75H2O | 0.67 | 2.52 | 0.97 | 2.57 |
| 29a | (CH2)2NHCO(CH2)2NHBoc | H | 112–115 | C28H37N7O5·1CHCl3·0.5H2O | 13.3 ± 5.95 | 6.06 | 6.19 ± 0.28 | n.d. |
| 29b | (CH2)2NHCO(CH2)3NHBoc | H | 130–133 | C29H39N7O5e | 4.80 ± 1.36 | 3.34 | 4.10 ± 0.52 | n.d. |
| 30a | (CH2)2NHCO(CH2)2NH2 | H | 85–88 | C23H29N7O3·2.67TFA·1.55H2O | 4.34 ± 0.26 | 5.84 | 6.65 ± 0.43 | n.d. |
| 30b | (CH2)2NHCO(CH2)3NH2 | H | C24H31N7O3·3TFA | 9.53 ± 2.46 | 6.34 | 11.5 ± 1.98 | n.d. | |
| 31a | (CH2)2NHCO(CH2)2NHAc | H | 180–183 | C25H31N7O4·0.75H2O | 39.4 ± 16.6 | 34.5 | >100 | n.d. |
| 31b | (CH2)2NHCO(CH2)3NHAc | H | 162–165 | C26H33N7O4·2.75H2Of | 9.65 ± 2.98 | 8.82 | 10.8 ± 0.45 | n.d. |
| 32d | (CH2)6NHBoc | H | C29H40N6O4·oxalate·H2Og | 0.0371 ± 0.0017 | 0.0214 ± 0.0039 | 0.0460 ± 0.0155 | 0.0232 ± 0.0053 | |
| 33a | (CH2)3NPth | H | 202–204 | C29H28N6O4·0.75H2O | 1.81 ± 0.40 | 0.84 ± 0.35 | n.d. | n.d. |
| 33b | (CH2)4NPth | H | 195–196 | C30H30N6O4·oxalate·0.5H2O | 0.17 ± 0.006 | 0.089 ± 0.005 | 0.12 | 0.21 |
| 33c | (CH2)5NPth | H | 120–122 | C30H32N6O4 | 0.060 ± 0.001 | 0.020 ± 0.001 | n.d. | n.d. |
| 33d | (CH2)6NPth | H | 157–158 | C32H34N6O4·1DMF | 0.018 ± 0.008 | 0.018 ± 0.003 | n.d. | n.d. |
| 34a | (CH2)3NH2 | H | 111–113 | C21H26N6O2·1.5H2O | 3.87 ± 0.12 | 5.12 ± 0.93 | n.d. | n.d. |
| 34b | (CH2)4NH2 | H | 121–123 | C22H28N6O2·1H2O | 3.08 ± 0.22 | 1.74 ± 0.03 | n.d. | n.d. |
| 34c | (CH2)5NH2 | H | 200–202 | C23H30N6O2·1H2O | 1.17 ± 0.01 | 5.89 ± 0.13 | n.d. | n.d. |
| 34d | (CH2)6NH2 | H | 138–140 | C24H32N6O2·0.5CHCl3·0.67H2Oh | 1.31 ± 0.02 | 1.34 ± 0.12 | n.d. | n.d. |
| 34e | (CH2)7NH2 | H | 72–75 | C25H34N6O2·1.5-H2O | 0.066 | 0.057 | n.d. | n.d. |
| 34f | (CH2)8NH2 | H | 65–68 | C26H36N6O2·H2O | 0.020 ± 0.001 | 0.026 ± 0.0005 | n.d. | n.d. |
| 34g | (CH2)9NH2 | H | 140–143 | C27H38N6O2 | 0.024 ± 0.001 | 0.024 ± 0.007 | n.d. | n.d. |
| 34h | (CH2)10NH2 | H | 95–97 | C28H40N6O2·1.5H2O | 0.0158 ± 0.0009 | 0.0117 ± 0.0007 | n.d. | n.d. |
| 35g | (CH2)9NHAc | H | 170–173 | C29H40N6O3·2oxalate | 0.052 ± 0.001 | 0.050 ± 0.018 | n.d. | n.d. |
| 35h | (CH2)10NHAc | H | 87–90 | C30H48N6O3·0.5H2O | 0.075 ± 0.013 | 0.043 ± 0.025 | n.d. | n.d. |
| 36 | (CH2)10NEt2 | H | 168–171 | C32H48N6O2·4oxalate·3H2O | 0.0030 ± 0.0004 | 0.0135 ± 0.0009 | n.d. | n.d. |
| 37 | (CH2)10NHCH2-(o-MeO)Ph | H | 153–156 | C36H48N6O3· 4oxalate·4H2Oi | 0.0033 ± 0.0006 | 0.022 ± 0.0012 | n.d. | n.d. |
Calcd 5.28 H, found 5.87.
Calcd 13.08 N, found 11.93.
Calcd 13.58 N, found 12.58.
Calcd 6.25 H, found 5.45.
Calcd 17.33 N, found 16.73.
Calcd 6.97 H, found 6.17.
Calcd 13.04 N, found 12.26.
Calcd 16.53 N, found 18.01.
Calcd 8.04 N, found 6.84.
R″ = Et.
R″ = (CH2)6CN.
n.d. = not determined.
Scheme Ia.

aBoc = CO2C(CH3)3.
Scheme III.

Table II.
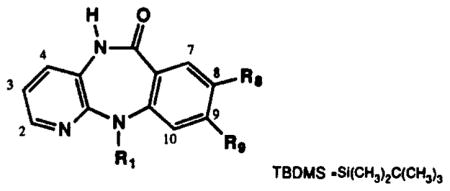
NMR Resonances for Aromatic Protons of Pirenzepine and Analogues. Chemical shift of Each Resonance in ppm from Tetramethylsilane, Multiplicity, Coupling Constant J (in Hz) Are Given
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| no. | solvent | R8 | R9 | R1 | H2 | H3 | H4 | H7 | H8 | H9 | Hl0 |
| 1 | CDCl3 | H | H |
|
8.29 br s | 7.32 dd J = 7.9, 7.8 |
7.58 dd J = 7.9, 1.4 |
7.98 d J = 7.6 |
7.44 m | 7.6 m | 7.64 m |
| 6 | DMSO-d6 | H | H | H | 7.9 dd J = 4.7, 1.6 |
7.0 m | 7.3 dd J = 8.6, 1.6 |
7.94 dd J = 8.0, 1.6 |
6.95 m | 7.36 J = 8.3, 1.6 |
7.13 d J =7.4 |
| 8 | CDCl3 | SMe | H | H | 7.91 d J = 4.5 |
6.9 dd J = 7.7, 4.4 |
7.06 d J = 8.2 |
7.8 d J =2.3 |
7.24 dd J = 8.3, 2.3 |
6.66 d J =8.3 |
|
| 9 | DMSO-d6 | SO2NH2 | H | H | 7.94 d J =4.6 |
7.0 dd J = 7.7, 4.6 |
7.23 d J = 7.6 |
8.23 d J = 2 |
7.73 dd J = 8.5, 2 |
7.25 d J = 8.5 |
|
| 11a | CDCl3 | SMe | H |
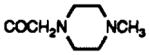
|
8.3 br s | 7.34 dd J = 7.9, 4.4 |
7.6 dd J = 7.9, 1.2 |
7.78 d J = 2.1 |
7.47 dd J = 8.6, 2.1 |
7.53 d J = 8.6 |
|
| 11b | CD3OD | SO2NH2 | H |
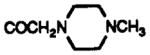
|
8.38 br s | 7.44 dd J = 8.0, 4.5 |
7.7 m | 8.28 br s | 8.11 dd J = 8, 2.2 |
7.65 m | |
| 14 | CDCl3 | H | CH2OTBDMS | H | 7.92 m | 6.9 m | 7.1 d J =7.7 |
7.92 m | 6.88 m | 6.75 s | |
| 15 | CDCl3 | H | CH2OTBDMS |
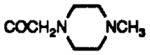
|
8.28 br s | 7.3 dd J = 7.9, 3.15 |
7.6 dd J = 7.9, 1.3 |
7.90 J =8.1 |
7.41 d J = 8.1 |
7.54 s | |
Aminosulfonyl compounds 9 and 10 were also prepared from intermediate 6 according to Scheme I. Reaction of intermediates 8,9, and 10 with chloroacetyl chloride and then with N-methylpiperazine gave the 8-substituted pirenzepine analogues 11a, 11b, and 11c, respectively (Scheme II). Compound 11c was used to prepare analogues 11d and 11e by deprotection with TFA and reaction with acetyl chloride. The 9-substituted analogues 15 and 16 were prepared from aminoterephthalate 12 according to Scheme III. The carboxymethyl group of compound 12 meta to the amine was selectively reduced with Super-Hydride24 (Aldrich) followed by protection of the hydroxyl groups as the tert-butyldimethylsilyl ether25 to give 13. Intermediate 13 was subsequently condensed with 4 and then cyclized to 14, using a carefully controlled quantity of p-toluenesulfonic acid (<0.1 equiv, pH ≈ 7.0) in order to prevent hydrolysis of the silyl ether. Addition of the (4-methyl-1-piperazinyl)acetyl side chain to form 15 used the same one-pot procedure as shown in Scheme II. Removal of the silyl ether with tetrabutylammonium fluoride25 afforded the final 9-hydroxymethyl analogue of pirenzepine, 16.
Scheme II.

The 5-position of pirenzepine was derivatized through alkylation of the amide NH, resulting in the N-ethyl and N-6-cyanohexyl derivatives 17a and 17b, respectively (Table I). This reaction was carried out at the stage of the tricyclic intermediate26 6, followed by elaboration of the N-methylpiperazine side chain, as in Scheme II. Although analogues bearing small alkyl substituents at the position were reported in the patent literature27 to be active, no details concerning selectivity were reported.
Much variation in the substitution of the piperazine ring of pirenzepine is tolerated at the muscarinic antagonist binding sites. The desmethyl analogue of pirenzepine28 (18) was prepared from compound 1, by treatment with α-chloroethyl chloroformate and tertiary base,29 or from 6, by acetylation with chloroacetyl chloride followed by reaction with piperazine. Compound 18 served as an intermediate for model compounds derivatized at the 4-position of the piperazine ring (Scheme IV) through acylation (compounds 19–21) or alkylation with propargyl (compound 22), benzyl (compound 23), and 4-substituted benzyl (compounds 24a–d) groups. Compounds 24a–d offered the possibility of chain extension, dependent on the toleration of an aromatic group close to the piperazine ring.
Scheme IV.

Straight-chain N-alkyl substitution was examined as an alternative to chain extension through a benzyl substituent. A functionalized ethyl group was introduced at the pirenzepine 4-position through alkylation of 18 with 2-iodoethanol, as shown in Scheme V. The subsequent conversion of the 2-hydroxyethyl derivative 25 to a chloroethyl intermediate, 26, allowed the synthesis of ethyl-amino congener 27 and various extended amide derivatives (28–31). Compound 26 was also of interest as a potential alkylating agent for irreversible inhibition through formation of the aziridinium species.30–32
Scheme V.

Homologues of the aminoethyl functionalized congener 27 containing alkyl chains up to 10 methylenes in length were prepared. An ω-amino side chain was appended to 18 in one of three ways according to Scheme VI: (1) reductive amination using a Boc-protected ω-amino aldehyde to give compound series 32, (2) alkylation with ω-phthaloyl bromides to give series 33, and (3) alkylation with alkyl dibromides and subsequent amination with ammonium hydroxide to give the primary amine analogues 34. The route utilizing alkyl dibromides was preferred and gave the highest yields. Intermediates 32 and 33 could easily be converted to 34, using TFA or hydrazine, respectively. The primary amines were subsequently acetylated with acetic anhydride to give the N-acetyl derivatives 35. These homologous series were designed to probe the effects of varying chain length and the nature of the distal terminal group on the affinity of the analogue in binding to muscarinic receptors.
Scheme VI.

Pharmacology
The analogues were screened in binding assays using membranes from cells or tissues containing primarily single subtypes of m1 through m4 muscarinic receptors. Transfected A9L cells33 were used as a source of homogeneous populations of m1 and m3 receptors, and NG108-15 cells were the source of m4 receptors. Rat heart tissue was the source of m2 muscarinic receptors. The ability of the analogue to displace [3H]-N-methylscopolamine ([3H]NMS) was measured, and the results are summarized in Table I.
Substitution of the 8- and 9-positions dramatically decreased affinity at all of the muscarinic receptor subtypes. Only the 8-methylthio and 9-hydroxymethyl analogues 11a and 16, respectively, caused any detectable displacement of [3H]NMS from the receptor. The inactivity of the aryl-modified analogues was surprising in light of the report of activity of the 8-methyl and 8-ethyl derivatives of pirenzepine.21 In addition, compounds 19–21, in which the most distal nitrogen of the piperazine ring exists in a neutral, acylated form, were totally inactive as muscarinic antagonists.
Alkylation of the endocyclic amide, to yield compounds 17a and 17b, resulted in analogues of low affinity. The only promising strategy for chain functionalization appears to be alkylation of the 4-amino position of the pirenzepine ring. Among the latter group of derivatives, the shorter chain substituents, e.g. the N-propargyl (22) and 2-substituted ethyl derivatives (25, 27, and 28) were weak inhibitors of [3H]NMS binding with Ki values in the vicinity of 10−6 M. Curiously, these rather modest modifications of the methyl group of pirenzepine abolish selectivity. In this series, only the chloromethyl derivative 26 retained selectivity for m1 receptors. The selectivity ratio for 26 was 4.4-fold in favor of m1 versus m2 receptors, compared to a ratio of 21 for pirenzepine (1). Telenzepine (3) has a ratio of 25 in favor of m1 versus m2 receptors. The N-benzyl derivative 23 was nonselective for m1 versus m2 receptors. Although larger sterically than the propargyl derivative 22, compound 23 had greater affinity at m1 and m2 muscarinic receptors. This indicated that there may be a region on the receptor accessory to the pharmacophore binding site that favors a hydrophobic group. This observation prompted the synthesis of compounds 24a–d, in which functionalized chains were extended via a carboxylic group at the para position of 23. However, a decrease in binding affinity was observed in this series.
Amino acid derivatives (29–31, a and b) of the ethyl-amino congener indicated a loss in potency as the chain was extended by amide bond formation. There was no apparent correlation in receptor affinity among charged (amino group, compound 30), small neutral (acetylamino, compound 31), and neutral, hydrophobic ((tert-butyloxy-carbonyl)amino, compound 29) terminal substituents.
The chloroethyl mustard derivative 26 was examined as a potential irreversible inhibitor of muscarinic receptors. A similar mustard derivative of the dopamine receptor antagonist fluphenazine has been reported to be an irreversible inhibitor of D1 and D2 dopamine receptors and of calmodulin.32 At muscarinic receptors, propylbenzylcholine mustard apparently binds covalently to an aspartyl residue at the ligand binding site.30 A chloroethyl mustard derivative of the muscarinic agent McN-A-343 has also been reported.31 The active alkylating agent is thought to be the aziridinium species, which forms spontaneously in neutral or slightly basic aqueous medium. To ascertain that this intermediate would indeed form under the conditions selected for an incubation prior to the receptor binding assay, we carried out an NMR study of the stability and chemical transformations of 26. The cyclization experiment was performed in 100 mM phosphate buffered D2O (pD – 7.4) at 37 °C. The hydrochloric acid salt of 26 was dissolved in deuterated phosphate buffer to liberate the free base. Spectra (Figure 1) were then recorded at various times during a 3-h period. An aziridinium species formed rapidly, followed by the appearance of the hydroxyethyl analogue 25 and its O-phosphate ester. The four hydrogens of the aziridinium ring appeared as a singlet at δ 3.21. The CH2Cl, CH2OH, and CH2OPO32− protons appeared as multiplets at δ 3.88, 4.0, and 4.18, respectively. The intensity of the signal related to the aziridinium ion increased initially and then decreased as the reaction progressed. Figure 2 shows the time course of the spontaneous transformation of 26 to the aziridinium species, leading to 25 and the O-phosphate ester. During a 45-min incubation a major fraction of the material exists in the reactive aziridinium form.
Figure 1.

Superimposed 500-MHz 1H NMR spectra of 26 at various times following dissolution in aqueous medium. The time after addition of pD 7.4 potassium phosphate buffer (0.1 M) is indicated on the left side. Major resonance peaks have been assigned as (δ in ppm from tetramethylsilane): 3.22 and 3.63 (aziridinium ion); 3.42, 3.62, and 4.16 (O-phosphate ester); 3.89 (26, CH2 α to Cl); 4.01 (25, CH2 α to OH).
Figure 2.

Kinetics of chemical transformation of the chloroethyl derivative (26) to form the aziridinium species, which subsequent leads to the hydroxyethyl (25) and O-phosphate species. Values were obtained through a nonlinear curve analysis using a computer program and the data obtained from the NMR study (Figure 1).
Rat brain membranes were exposed to compound 26 in phosphate buffered saline, under conditions either similar to (pH 8.0, at 24 °C for 60 min) or nearly equivalent (pH 7.0, at 37 °C for 45 min) to those of the NMR kinetic experiment, to assay for irreversible inhibition of muscarinic receptors. In both cases, even a very high concentration of 26 failed to effect irreversible inhibition of radioligand binding. Scatchard analyses before and after treatment of the membranes with 26 were compared. Kd and Bmax values at binding sites for [3H]NMS were found to be 357 pM and 0.74 pmol/mg protein, respectively. Preincubation of membranes at pH 8.0 with a concentration of 1.0 mM compound 26 produced no change in the Kd and Bmax values. Thus, 26 is not an irreversible inhibitor of m1 receptors. The lack of irreversible inhibition in spite of high binding affinity of 26 suggests that there is no nucleophilic group at the binding site in close proximity to the aziridine ring.
Among straight-chain terminally functionalized N-alkyl derivatives, a marked enhancement of affinity at muscarinic receptors was observed as the number of methylene groups was increased from 2 to 10. With seven or more methylene units, the Ki values for the free amines (34e–h) were below 100 nM. Moreover, for the intermediate chain lengths (3–6 methylenes), analogues having large, hydrophobic groups at the terminal position, e.g., phthaloyl amino (33) and (tert-butyloxycarbonyl)amino (32d), were more potent than the corresponding free amines (34). In the series having longer chain lengths of nine and ten methylene units, derivatives bearing a free amino group (34g and 34h) had higher affinities than the corresponding derivatives with the small neutral acetylamino terminal group (35g and 35h). Aminodecyl derivative 34h had Kd values of less than 20 nM at both m1 and m2 receptors. The free amino derivative with five methylenes 34c displayed a 5-fold selectivity for m1 versus m2 receptors. The other chain-extended primary amino derivatives were nonselective, as were the small methyl-modified pirenzepine analogues such as 22.
Compounds 36 and 37 were further analogues of compound 34h, designed with amine substituents similar to the terminal groups of the m2-selective antagonists AQ-RA-74120 and methoctramine,3 respectively, and however displayed m1-selectivity.
Discussion
A functionalized congener approach to drug design has been shown to be useful for enhancing potency and selectivity of ligands that bind to extracellular receptor sites, in affinity chromatography38 or affinity labeling of receptors,35 and in prodrug design.18b A starting point for this approach is the identification of insensitive sites on the drug molecule, suitable for covalent attachment of a chemically functionalized chain. The chain may be modified in a stepwise fashion to optimize biological, physical, and spectroscopic properties, resulting in high affinity receptor probes.18a For example, an amine-functionalized adenosine antagonist has been converted into an isothio-cyanate-bearing affinity label for A1 adenosine receptors.35 Fluorescent and biotinylated probes for adenosine receptors18a have also been prepared using this approach.
We have previously explored sites for functionalization of the muscarinic agonist, oxotremorine,17a resulting in analogues that mostly lack agonist activity but retain receptor affinity. The approach is now extended to muscarinic antagonists. A structurally versatile class of high affinity receptor probes would be useful for characterizing muscarinic receptors. Recently, an isothiocyanate derivative of aprophen was reported to be an irreversible antagonist for muscarinic receptors.36
To summarize the effects on receptor affinity by incorporating a functionalized chain into pirenzepine: chain substitution at the C-8 or C-9 position essentially abolishes muscarinic receptor binding, whereas substitution of the endocyclic amide NH (position 5) results in weak affinity. Alkyl substitution of the distal N-methyl group of pirenzepine results in either intermediate affinity (short chains) or relatively high affinity (long chains of >4 methylenes having terminal amino or acylamino substituents). Acylation of the distal piperazine amino group abolishes receptor binding.
This study demonstrates clearly that the receptor affinity within a series of analogues may be increased by using a functionalized congener approach, although most of the analogues synthesized did not distinguish among muscarinic receptor subtypes. For example, relative to short-chain analogues (e.g. 33a,b and 34a,b) there was a constant increase in the affinity as the chain length of the straight alkyl derivatives was increased, at least up to ten methylenes. The increasing hydrophobicity may be a factor in this trend of higher affinity as the chain is lengthened, but it is not sufficient to account for all of the differences. Among free amine derivatives, a relatively large jump in potency was achieved at the point of seven methylene units. The decylamino analogue 34h was the most potent in the series of primary amines, which suggested that longer straight alkyl chains may result in even higher affinities. Although 34h is nonselective, related secondary and tertiary amine analogues 36 and 37 were selective for m1 receptors by factors of 5 and 7, respectively, suggesting that further elaboration of the chain structure may alter selectivity. In general, the affinity of the more potent analogues compared well with that of pirenzepine, although the m1-selectivity characteristic of pirenzepine was diminished or lost completely. For example, compound 37 was 9-fold and 29-fold more potent than pirenzepine at m1 and m2 receptors, respectively.
There is no explanation for the relatively weak activity of the analogues in which the chain contains an amide link. Compounds 29a and 32d both contain straight chains of six atoms between the piperazine amine and the (tert-butyloxycarbonyl)amino group, and, therefore may possibly attain similar conformations in the extended state, yet they differ in affinity by factors of 360 at m1 receptors and 280 at m2 receptors. Overall hydrophobicity (substitution of an amide bond for two methylene groups adds approximately 2 log P units) is not a sufficient explanation, as evidenced by the inactivity of the lipophilic Boc derivative 29a versus the corresponding amine 30a. Conformational factors or possibly distal sites of interaction between the antagonists and muscarinic receptor molecules remain as possible explanations. Other muscarinic ligands are thought to span distances on the receptor protein while in the bound conformation. By analogy, the m2-selective muscarinic antagonist methoctramine3 in the bound state has been proposed to bridge two vicinal receptor sites. A study of the affinity as a function of chain length separating two 2-(methyloxy)benzylamino pharmacophores indicated that there was an optimal chain length, which consisted of 24 atoms. Although there is no evidence to indicate that we have reached an optimal chain length, there is a leveling trend in the affinity beyond six methylenes.
In conclusion, we have located a site on the pirenzepine molecule for chain derivatization that provides the opportunity to synthesize potential spectroscopic or other affinity probes, or affinity columns for receptor purification. Furthermore, we may alter the overall hydrophobicity of the molecule, which might favorably affect the biodistribution of the analogues. The loss of selectivity in the pirenzepine derivatives may yet be overcome through further structure–activity studies. Similar chain derivatization may prove to be useful for other members of the pyridobenzodiazepine class and closely related classes of muscarinic antagonists.
Experimental Section
General
1H NMR spectra were recorded on a Varian XL-300 FT-NMR spectrometer and all values are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). For kinetic studies, a Varian 500-MHz spectrometer was used. Chemical ionization MS using ionized NH3 gas were recorded on a Finnigan 1015D mass spectrometer modified with EXTREL electronics. Fast atom bombardment MS were carried out on a JEOL JMS-SX102 mass spectrometer. Thin-layer chromatography (TLC) analyses were carried out on EM Kieselgel 60 F254, DC-Alufolien 200-μm plates and were visualized in an iodine chamber and/or with 1% ninhydrin in ethanol. Silica gel columns used MN-Kieselgel 60 230–400-mesh silica gel. Elemental analyses were performed by Atlantic Microlabs, Inc., Atlanta, GA. The term in vacuo refers to a water aspirator (15–30 mmHg) rotary evaporator. Percent yields are rounded to the nearest whole number.
Pirenzepine hydrochloride was obtained from Research Bio-chemicals, Inc. (Natick, MA). Telenzepine was the generous gift of Dr. M. Eltze of Byk Gulden Research Laboratories, Konstanz, West Germany.
5,11-Dihydro-8-(chlorosulfonyl)-6H-pyrido[2,3-,b][1,4]-benzodiazepin-6-one (7)
Compound 6 (1.04 g, 4.9 mmol) was added in small portions to 6 mL of chlorosulfonic acid. The mixture was heated to 65–70 °C for 30 min and then poured over 300 g of ice. A precipitate consisting of the 8-chlorosulfonyl derivative 7 was collected by filtration and immediately dissolved in the solvent used for the preparation of compounds 8, 9, and 10.
5,11-Dihydro-8-(methylthio)-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (8)
Intermediate 7, freshly prepared from 6 (0.5 g, 2.4 mmol), was reduced with triphenylphosphine (2.8 g, 11.0 mmol) in dioxane (20 mL) for 2 h at 90 °C.23 The crude 8-thio derivative was quenched (10 min, 25 °C) with excess methyl iodide (0.5 mL, 8.0 mmol) and crystallized from toluene to yield 50 mg (8% from 6) of 8 as a waxy solid: 1H NMR (CDCl3) δ 2.40 (s, 3 H, CH3), 6.25 (br s, 1 H, NH), 6.66 (d, J = 8.3,1 H), 6.86 (dd, J = 7.7,4.4 Hz, 1 H), 7.06 (d, J = 8.2 Hz, 1 H), 7.24 (dd, J = 8.3, 2.3 Hz, 1 H), 7.79 (d, J = 2.3 Hz, 1 H), 7.91 (d, J = 4.5 Hz, 1 H); MS (CI/NH3) m/e 258 (MH+, base), 243, 211,113.
5,11-Dihydro-8-(aminosulfonyl)-6H-pyrido[2,3-,b][1,4-benzodiazepine (9)
Intermediate 7, freshly prepared from 6 (1.0 g, 4.7 mmol), was dissolved in 25 mL of DMF and 25 mL of NH4OH. The mixture was stirred at room temperature for 20 min, and the precipitate was filtered, washed with water, and dried to yield 0.76 g (55%) of 9 as a white waxy solid: 1H NMR (DMSO-d6) δ 7.01 (dd, J = 7.7,4.6 Hz, 1 H), 7.23 (d, J = 7.6 Hz, 1 H), 7.25 (d, J = 8.5 Hz, 1 H), 7.73 (dd, J = 8.5, 2 Hz, 1 H), 7.94 (d, J = 4.6 Hz, 1 H), 8.23 (d, J = 2 Hz, 1 H), 9.15 (s, 1 H, NH), 10.10 (s, 1 H, NH); MS (CI/NH3) m/e 291 (MH+, base), 232.
5,11-Dihydro-8-[[[2-(Boc-amino)ethyl]amino]sulfonyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (10)
Intermediate 7, freshly prepared from 6 (1.04 g, 4.9 mmol), was dissolved in 6 mL of ethylenediamine and stirred for 15 min at room temperature. The crude product was precipitated by addition of excess petroleum ether, washed several times with ether and water, and dried overnight to yield 794 mg (48.6%) of 5,11-dihydro-8-[[(2-aminoethyl)amino]sulfonyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one as a white crystalline solid: mp >250 °C dec; 1H NMR (DMSO-d6) δ 2.51 (t, J = 6.3 Hz, 2 H), 2.72 (t, J = 6.3 Hz, 2 H), 6.98 (dd, J = 5.0, 7.8 Hz, 1 H), 7.25 (d, J = 8.5 Hz, 1 H), 7.32 (d, J = 7.8 Hz, 1 H), 7.69 (dd, J = 8.5,1.5 Hz, 1 H), 7.92 (d, J = 5.0 Hz, 1 H), 8.16 (d, J = 1.5 Hz, 1 H), 9.19 (s, 1 H); MS (CI/NH3) m/e 334 (MH+), 291,212,180. This intermediate (3.4 g, 10 mmol) was then dissolved in 30 mL of DMF, and tri-ethylamine (1.0 g, 10 mmol) and di-tert-butyl dicarbonate (2.4 g, 11 mmol) were added. The mixture was allowed to stir for 1 h before it was poured into 300 mL of water and filtered. The filtrate was washed with water and dried in vacuo to yield 3.9 g (90%) of 10 as a white solid: mp 195–198 °C: 1H NMR (DMSO-d6) δ 1.34 (s, 9 H, (CH3)3), 2.70 (m, 2 H, CH2), 2.90 (m, 2 H, CH2), 6.74 (br s, 1 H, NH), 7.00 (dd, J = 7.7, 4.6 Hz, 1 H), 7.23 (d, J = 7.6 Hz, 1 H), 7.3 (d, J = 8.5 Hz, 1 H), 7.55 (br t, 1 H, NH), 7.68 (dd, J = 8.6, 2.3 Hz, 1 H), 7.92 (dd, J = 4.7,1.3 Hz, 1 H), 8.15 (d, J = 2.2 Hz, 1 H); MS (CI/NH3) m/e 451 (MH+), 161 (base), 334, 212, 104.
General Procedure A. Reaction of Substituted 5,11-Di-hydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-ones with 2-Chloroacetyl Chloride and N-Methylpiperazine
The substituted 5,11-dihydro-6H-pyrido[2,3-b] [1,4]benzodiazepin-6-one (compounds 6,8,9,10,15, or 36) (1 equiv) was dissolved in dioxane containing triethylamine (2 equiv) and 2-chloroacetyl chloride (1.5 equiv) and refluxed for 5 h (complete TLC). The solution was cooled to room temperature, N-methylpiperazine (5 equiv) was added, and the solution was refluxed for 1 h. After cooling, the solvent was removed in vacuo and the crude mixture was purified on a silica gel column (95–20% CHCl3, 5–80% of 10/1 MeOH/NH4OH). This procedure was used to make compounds 11a, 11b, 11c, 15, and 36. A modified procedure was used to prepare compound 18, using piperazine in place of N-methyl-piperazine.
5,11-Dihydro-8-(methylthio)-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (11a)
Compound 11a was made from 8 according to general procedure A and was crystallized from methanol (≈68%): mp 205 °C dec; 1H NMR (CDCl3) δ 2.30 (s, 3 H, NCH3), 2.52 (s, 3 H, SCH3), 3.25 (d, J = 14.0·Hz, 1 H), 3.50 (m, 1 H), 7.31 (dd, J = 7.9, 3.2 Hz, 1 H), 7.47 (dd, J = 8.5, 2.2 Hz, 1 H), 7.53 (d, J = 8.5 Hz, 1 H), 7.61 (dd, J = 7.9,1.2 Hz, 1 H), 7.78 (d, J = 2.1 Hz, 1 H); MS (CI/NH3) m/e 398 (MH+, base), 241, 113.
5,11-Dihydro-8-(aminosulfonyl)-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (11b)
Compound 11b was made from 9 according to general procedure A: 1H NMR (CD3OD) δ 2.21 (s, 3 H, CH3), 2.10–2.50 (m, 8 H), 3.10 (m, 1 H), 3.61 (m, 1 H), 7.44 (dd, J = 8.0,4.5 Hz, 1 H), 7.65 (m, 1 H), 7.70 (m, 1 H), 8.11 (dd, J = 8.0, 2.2 Hz, 1 H), 8.28 (br s, 1 H), 8.38 (br s, 1 H); MS (CI/NH3) m/e 431 (MH+, base), 384, 308, 113.
5,11-Dihydro-8-[[[2-(Boc-amino)ethyl]amino]sulfonyl]-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]-benzodiazepin-6-one (11c)
Compound 11c was made from 10 according to general procedure A and was purified by crystallization of its oxalate salt from 2-propanol: mp 210–215 °C dec; lH NMR (D2O) δ 1.28 (s, 9 H, (CH3)3), 2.82 (s, 3 H, CH3), 2.40–3.60 (m, 16 H), 7.60 (m, 1 H), 7.70–7.95 (m, 2 H), 8.18 (m, 1 H), 8.29–8.45 (m, 2 H); free base (CDCl3) δ 1.41 (s, 9 H, (CH3)3), 2.18 (s, 3 H, CH3), 2.10–2.56 (m, 8 H), 3.10–3.35 (m, 6 H), 7.35 (dd, J = 8.0, 4.4, Hz, 1 H), 7.69 (dd, J = 7.9,1.2 Hz, 1 H), 7.76 (d, J = 8.5 Hz, 1 H), 8.07 (dd, J = 8.4,2.3 Hz, 1 H); MS (CI/NH3) m/e 575 (MH+), 518, 500, 476 (base), 431, 241.
General Procedure B. Removal of N-Boc Protecting Group
Excess TFA was added slowly to the neat Boc-protected amine derivative, and the reaction was stirred for 1 h or until complete by TLC. The excess TFA was removed under a stream of N2, and the residue was dried for 24 h at 50 °C under high vacuum (0.1 mmHg) to yield the TFA salts. For spectral analysis, an aliquot was neutralized with 2 M Na2CO3 solution, extracted into EtOAc (3× equal volume), dried over Na2SO4, and evaporated in vacuo. This procedure was used to make compounds 11d, 24c, 30a, and 30b.
5,11-Dihydro-8-[[(2-aminoethyl)amino]sulfonyl]-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-6][1,4]benzodiazepin-6-one (11d)
Compound 11d was made from 11c, using general procedure B: mp 155–158 °C; 1H NMR (D2O) oxalate salt δ 2.92 (s, 3 H, CH3), 2.60–3.65 (m, 14 H), 7.60 (m, 1 H), 7.81 (br d, J = 8.4 Hz, 1 H), 7.85 (br d, J = 8.1 Hz, 1 H), 8.32 (br s, 1 H), 8.38 (d, J = 4.1 Hz, 1 H); MS (CI/NH3) m/e 474 (MH+), 384, 352, 244, 241 (base).
General Procedure C. N-Acylation
The amine intermediate was dissolved in CH3CN, and 1.5 equiv of triethylamine and 1.2 equiv of acetic anhydride were added. After 15 min, or when judged complete using TLC, the CH3CN was removed under a stream of N2. The residue was dissolved in EtOAc and washed with 2 M Na2CO3 saturated with NaCl (3× equal volume). The organic phase was dried over Na2SO4 and all volatile materials were removed in vacuo. The crude product was chromatographed (CHCl3/CH3OH/NH4OH, 40/10/1) to give the corresponding N-acetylamino compound (11e, 19a, 28, 31a,b, 35a–h).
5,11-Dihydro-8-[[[2-(acetylamino)ethyl]amino]-sulfonyl]-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido-[2,3-b][1,4]benzodiazepin-6-one Oxalate (11e)
Compound 11e was made from 11d, using general procedure C. The oxalate salt was crystallized from methanol: mp 190–195 °C; 1H NMR (D2O) δ 1.69 (s, 3 H, CH3), 2.88 (s, 3 H, CH3), 2.60–3.60 (m, 14 H), 7.60 (m, 1 H), 7.76 (br d, J = 8.3 Hz, 1 H), 7.85 (br d, J = 7.8 Hz, 1 H), 8.16 (br s, 1 H), 8.30 (s, 1 H), 8.40 (m, 1 H); MS (CI/NH3) m/e 516 (MH+), 243, 212, 159, 103 (base).
Methyl 2-Amino-4-[[(dimethyl-tert-butylsilyl)oxy]-methyl]benzoate (13)
Methyl 2-amino-4-carbomethoxybenzoate (12, 4.0 g, 19.0 mmol) was dissolved in dry THF and cooled in a dry ice–acetone bath unde an inert atmosphere. Super-Hydride (65 mL, 1.0 M) was added slowly by syringe over a period of 1 h. A small aliquot of the solution was quenched with water and purified on a silica gel column to yield methyl 2-amino-4-(hydroxymethyl)benzoate as a white solid: mp 100–102 °C; 1H NMR (CDCl3) δ 3.82 (s, 3 H, CH3), 4.64 (s, 2 H, CH2), 5.82 (br s, 2 NH2), 6.58 (d, J = 8.1 Hz, 1 H), 6.66 (br s, 1 H), 7.81 (d, J = 8.0 Hz, 1 H); MS (CI/NH3) m/e 182 (MH+, base), 167,150. Compound 13 was prepared from the remaining solution above using tert-butyldimethylchlorosilane.24 A small amount of the resulting solution was purified on a silica gel column to yield pure 13: 1H NMR (CDCl3) δ 0.15 (s, 6 H, Si(CH3)2), 1.00 (s, 9 H, t-Bu), 3.85 (s, 3 H, CH3), 4.65 (s, 2 H, CH2O), 6.55 (dd, J = 8.3,1.0 Hz, 1 H), 6.66 (br s, 1 H), 7.80 (d, J = 8.0 Hz, 1 H); MS (CI/NH3) m/e 296 (MH+) base, 186, 164.
5,11-Dihydro-9-[[(dimethyl-tert-butylsilyl)oxy]-methyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (14)
Intermediate 13 (2.95 g, 10 mmol) was added to solution of 3-amino-2-chloropyridine (1.4 g, 11 mmol) and potassium tert-butoxide (1.7 g, 15 mmol) in 20 mL of dry THF. After 2 h the solvent was evaporated and the crude product was crystallized from petroleum ether to yield 1.4 g (36%) of N-(2′-chloro-3-pyridyl)-2-amino-4-[[(dimethyl-tert-butylsilyl)oxy]methyl]benzamide as a white waxy solid: 1H NMR (CDCl3) δ 0.15 (s, 6 H, Si(CH3)2), 1.00 (s, 9 H, t-Bu), 4.80 (s, 2 H, CH2O), 5.80 (br s, 2 H, NH2), 6.75 (d, J = 8.0 Hz, 1 H), 6.80 (s, 1 H), 7.35 (dd, J = 8.2,4.7 Hz, 1 H), 7.56 (d, J = 8.2,1 H), 8.20 (dd, J = 4.7,1.7 Hz, 1 H), 8.45 (br s, 1 H, NH), 8.80 (dd, J = 8.2,1.7 Hz, 1 H) [Irradiation at δ 7.56 collapsed the doublet at δ 6.75 to a singlet, irradiation at δ 8.20 collapsed each doublet of doublets at δ 7.35 and δ 8.80 to a doublet (J = 8.2 and 8.2 Hz), and irradiation at δ 8.80 collapsed each doublet of doublets at δ 7.35 and δ 8.20 to a doublet (J = 4.7 and 4.7 Hz).]; MS (CI/NH3) m/e 392 (MH+, base), 334, 281, 264. This intermediate (750 mg, 1.9 mmol) and p-toluenesulfonic acid (25 mg, 0.13 mmol) were suspended in 20 mL of toluene. The toluene was removed, leaving a waxy solid, which was heated at 215 °C for 15 min. Part of the resulting brown solid was purified on a silica gel column to give pure 14: 1H NMR (CDCl3) δ 0.15 (s, 6 H, Si(CH3)2), 1.00 (s, 9 H, t-Bu), 4.75 (s, 2 H, CH2O), 6.75 (s, 1 H), 6.92 (m, 2 H), 7.15 (d, J = 7.7 Hz, 1 H), 7.92 (m, 2 H) [Irradiation at δ 7.92 collapsed the signal at δ 6.92 to a doublet (J = 7.7 Hz) and a singlet.]; MS (CI/NH3) m/e 356 (MH+, base), 315, 251.
5,11-Dihydro-9-[[(dimethyl-tert-butylsilyl)oxy]-methyl]-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (15)
Compound 15 was made from crude compound 14 according to general procedure A. The crude product was crystallized from chloroform/hexane and further purified on a column to yield 68 mg (7.1% based on 13) of 15: mp 245–248 °C; 1H NMR (CDCl3) δ 0.15 (s, 6 H, Si(CH3)2), 0.95 (s, 9 H, t-Bu), 1.50–2.50 (m, 8 H), 2.15 (s, 3 H, CH3), 3.20 (d, J = 14 Hz, 1 H), 3.50 (m, 1 H), 4.82 (s, 2 H, CH2O), 7.32 (dd, J = 7.9, 3.2 Hz, 1 H), 7.41 (d, J = 8.0 Hz, 1 H), 7.60 (dd, J = 7.9,1.3 Hz, 1 H), 7.94 (d, J = 8.1 Hz, 1 H), 8.20 (br s, 1 H), 10.17 (br s, 1 H); MS (CI/NH3) m/e 496 (MH+, base), 463, 396, 356, 113.
5,11-Dihydro-9-(hydroxymethyl)-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (16)
Compound 15 (15 mg, 0.03 mmol) was dissolved in 1 mL of CHCl3, and a solution of 1 M tetrabutylammonium fluoride in THF (40 μL) was added. The solution was allowed to stand at room temperature overnight. The solvent was removed in vacuo, and the remaining residue was dissolved in saturated NaHCO3, washed with ether to remove nonpolar impurities, and extracted with CHCl3. The CHCl3 extracts were combined, evaporated, and purified on a short silica gel column to yield 10 mg (86%) of 16 as a waxy solid: 1H NMR (CDCl3) δ 2.05–2.50 (m, 60 H), 2.20 (s, 3 H, CH3), 3.22 (d, J = 14 Hz, 1 H), 3.60 (m, 1 H), 4.80 (s, 2 H, CH2O), 7.30 (dd, J = 7.8, 4.7 Hz, 1 H), 7.44 (d, J = 8.1 Hz, 1 H), 7.50 (dd, J = 1.6,7.8 Hz, 1 H), 7.62 (s, 1 H), 7.96 (d, J = 8.1 Hz, 1 H), 8.02 (m, 1 H); MS (CI/NH3) m/e 382 (MH+, base), 242, 141, 113.
5,11-Dihydro-5-ethyl-11-[(4-methyl-1-piperazinyl)-acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (17a)
5-Ethyl-11-hydro-6H-pyrido[2,3Hb][1,4]benzodiazepin-6-one was prepared according to literature26 and condensed according to general procedure A to yield compound 17a: 1H NMR (CDCl3) (HCl salt) 1.30–1.40 (bs, 3 H), 2.05–2.40 (m, 3 H), 2.75 (s, 3 H, CH3), 2.90–3.60 (m, 8 H), 3.85 (m, 1 H), 4.40 (m, 1 H), 7.30–7.60 (m, 4 H), 7.78 (d, J = 7.7 Hz, 1 H), 7.81 (d, J = 7.5 Hz, 1 H); MS (CI/NI3) m/e 380 (MH+ base), 240, 113, 101.
5,11-Dihydro-11-[(1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (18)
Method A
Compound 18 was made from 6, 2-chloroacetyl chloride, and excess piperazine, using a modified procedure A.
Method B
Pirenzepine di-hydrochloride (5.2 g, 11.3 mmol), α-chloroethyl chloroformate (10 mL), and N,N,N-diisopropylethylamine (20 mL) were suspended in 100 mL of anhydrous CHCl3, and the mixture was refluxed for 1 h. The CHCl3 was removed in vacuo and the solid was dissolved in 50 mL of methanol and 5 mL of 1 M HCl in ether (ph 1–2). The solution was refluxed for another 3 h before it was basified with aqueous sodium carbonate and washed with ether. The aqueous layer was saturated with NaCl and extracted into CHCl3 to yield 4.2 g (69%) of 18: 1H NMR (CDCl3) δ 2.00–2.80 (m, 8 H), 3.18 (br d, J = 14.0 Hz, 1 H), 3.71 (d, J = 14.0 Hz, 1 H), 7.30 (dd, J = 4.7, 7.9 Hz, 1 H), 7.45 (m, 1 H), 7.63 (m, 3 H), 7.80 (m, 3 H), 7.97 (d, J = 7.6 Hz, 1 H), 8.25 (br s, 1 H); MS (CI/NH3) m/e 338 (MH+ base), 212, 127.
General Procedure D. Alkylation of 18
A solution of 18 (1 equiv), the appropriate halide (1.2 equiv), and triethylamine (2 equiv) in THF/MeOH (10/1) or DMF was stirred at 25–40 °C until reaction was complete by TLC. All volatile materials were removed in vacuo, and the remaining residue was dissolved in 1 N HCl. The aqueous layer was washed with three equal portions of CH2Cl2 and then made basic (≈pH 9) by addition of aqueous sodium carbonate. The product was extracted into CH2Cl2 (3 × 10 mL), and the combined organic layers were dried over Na2SO4, filtered, and evaporated in vacuo to give the crude product. The crude product was purified on a silica gel column (gradient mixture of 80 to 90% CHCl3, 10 to 20% MeOH, and 1 to 2% NH4OH) to give the corresponding N-alkyl derivatives (22–24).
General Procedure for Acylation of Compound 18
Compound 18 (23.3 mg, 0.69 mmol) and methanesulfonyl chloride (30 μL) were dissolved in 2 mL of chloroform. The solvent was evaporated and the residue was recrystallized from chloroform/ether to give the monohydrochloride of 5,11-dihydro-11-[(4-methanesulfonyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b]-[1,4]benzodiazepin-6-one (20) in 50% yield.
5,11-Dihydro-11-[(4-propargyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (22)
Compound 22 was made by using general procedure D using intermediate 18 (42.3 mg, 0.125 mmol), propargyl bromide (22.4 mg, 0.188 mmol), and triethylamine (35 μL, 0.251 mmol) in THF/MeOH. The product was recrystallized from CH2Cl2/MeOH to give 31.4 mg of pure 22 (67%): mp 249–250 °C; 1H NMR (CDCl3) δ 2.30–2.60 (br m, 8 H), 3.21–3.31 (m, 4 H), 3.46 (m, 1 H), 7.33 (m, 1 H), 7.44 (m, 1 H), 7.54–7.67 (m, 3 H), 7.93 (d, J = 7.8 Hz, 1 H), 8.27 (br s, 1 H); MS (CI/NH3) m/e 376 (MH+, base), 254, 212.
5,11-Dihydro-11-[(4-benzyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (23)
Compound 23 (41.1 mg, 98%) was made by using general procedure D [18 (33.0 mg, 0.098 mmol), benzyl bromide (20.1 mg, 0.117 mmol), and triethylamine (27.3 μL, 0.196 mmol) in THF/MeOH]: 1H NMR (CDCl3) δ 1.93–2.51 (br m, 8 H), 3.11 (d, J = 14.4 Hz, 1 H), 3.32 (br s, 2 H), 3.54 (br d, J = 14.4 Hz, 1 H), 7.16 (br m, 6 H), 7.36 (m, 1 H), 7.47 (d, J = 6.9 Hz, 1 H), 7.56 (s, 1 H), 7.92 (d, J = 7.8 Hz), 8.19 (br s, 1 H), 9.81 (br s, 1H); MS (CI/NH3) m/e 428 (MH+, base), 254, 212, 177. The oxalate salt was prepared for pharmacological testing.
5,11-Dihydro-11-[[4-[p-[[[2-(Boc-amino)ethyl]amino]-carbonyl]benzyl]-1-piperazinyl]acetyl]-6H-pyrido[2,3-b]-[1,4]benzodiazepin-6-one (24b)
Compound 24b (197 mg, 72%) was made by using general procedure D [18 (150 mg, 0.445 mmol), p-(bromomethyl)-N-[2-(Boc-amino)ethyl]benzamide37 (175 mg, 0.489 mmol), and triethylamine (155 μL, 1.11 mmol) in THF/ MeOH].
5,11-Dihydro-11-[[4-(2-hydroxyethyl)-1-piperazinyl]-acetyl]-6H-pyrido[1,3-b][1,4]benzodiazepin-6-one (25)
Compound 18 (103 mg, 0.31 mmol) and 2-iodoethanol (0.6 g, 3.5 mmol) were dissolved in 4 mL of ethanol and heated at 95 °C for 3 h. The volatile material was removed in vacuo and the crude product was crystallized from ethyl acetate/2-propanol to give 77 mg of 25 (65%): mp 197–200 °C; 1H NMR (CDCl3, free base) δ 2.00–2.70 (m, 10 H), 3.30 (d, 1 H), 3.50–3.70 (m, 4 H), 7.95 (d, 1 H), 8.30 (s, 1 H), 9.50 (br s, 1 H); MS (CI/NH3) m/e 382 (MH+ base), 252, 212.
5,11-Dihydro-11-[[4-(2-chloroethyl)-1-piperazinyl]-acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one Dihydro-chloride (26)
Compound 25 (110 mg, 0.22 mmol) was refluxed for 1 h with thionyl chloride (1 mL) in chloroform (4 mL). The volatile material was removed in vacuo, and the residue was basified with aqueous Na2CO3 (pH 10) and washed immediately with ether to completely remove the nonpolar impurity (TLC: CHCl3/CH3OH/NH4OH, 9/1/0.1, Rf 0.4). The product was extracted into CHCl3 from the cold aqueous phase and the combined CHCl3 layers were concentrated in vacuo and acidified using ether/ HCl. The precipitate was dried in vacuo to yield 29 mg of 26 (25%) as a white solid: mp 188–190 °C; 1H NMR (D2O, HCl salt) δ 3.00–4.00 (m, 14 H), 7.50–7.70 (m, 3 H), 7.70–7.90 (m, 3 H), 8.40 (s, 1 H); MS (CI/NH3) m/e 400 (MH+), 212 (base), 364, 338; FAB mass spectroscopy showed peaks at mass 438 (M + K), 422 (M + Na), 400 (M + H), 364, 329,307,289,211,154, and 136. The salt was stable upon storage at −20 °C for at least 6 months.
5,11-Dihydro-11-[[4-(2-aminoethyl)-1-piperazinyl]-acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (27)
Compound 26 (8.6 mg, 0.017 mmol) was dissolved in concentrated NH4OH (0.4 mL) and stirred overnight. The crude product was extracted into CHCl3, and the organic layer was evaporated to dryness. The remaining residue was recrystallized from chloroform/petroleum ether to yield 7.2 mg of 27 (97%): mp 188–190 °C; 1H NMR (CDCl3) δ 2.00–2.70 (m, 12 H), 3.20 (d, J = 14 Hz, 1 H), 3.60 (br d, J = 14 Hz, 1 H), 7.26 (dd, J = 7.8, 4.7 Hz, 1 H), 7.40 (m, 1 H), 7.55 (dd, J = 7.9,1.4 Hz, 1 H), 7.58–7.62 (br s, 1 H), 7.92 (d, J = 7.7 Hz, 1 H), 8.26 (br s, 1 H), 9.35 (br s, 1 H); MS (CI/NH3) m/e 381 (MH+, base), 338, 252, 212.
5,11-Dihydro-11-[[4-[2-(acetylamino)ethyl]-1-piperazinyl]acetyl]-6H-pyrido[2,3-b][1,4-]benzodiazepin-6-one (28)
Compound 28 was made from 27 using general procedure C: mp 93–95 °C; 1H NMR (CDCl3) δ 2.00 (s, 3 H, CH3), 2.05–2.50 (m, 10 H), 3.00 (m, 2 H), 3.20 (m, 1 H), 3.70 (d, J = 14 Hz, 1 H), 6.00 (br s, 1 H, NH), 7.30 (dd, J = 7.9, 4.7 Hz, 1 H), 7.40 (m, 1 H), 7.60 (m, 2 H), 7.95 (d, J = 7.6 Hz, 1 H), 8.25 (br s, 1H), 10.30 (br s, 1 H, NH); MS (CI/NH3) m/e 423 (MH+, base), 252, 212.
5,11-Dihydro-11-[[4-[2-[[3-(Boc-amino)-1-oxopropyl]-amino]ethyl]-1-piperazinyl]acetyl]-6H-pyrido[2,3-b][1,4]-benzodiazepin-6-one (29a)
Compound 27 (100 mg, 0.28 mmol) was dissolved in 10 mL of CHCl3, N-t-Boc-2-alanine N-hydroxysuccinimide ester (70 mg, 0.27 mmol) was added in one portion, and the reaction mixture was stirred at room temperature for 6 h. The mixture was basified (aqueous K2CO3) and extracted with CHCl3. The crude product was purified on a silica gel column (gradient mixture of 80 to 90% CHCl3, 10 to 20% MeOH, and 1 to 2% NH4OH) to yield 135.9 mg of 29a (90%) as a white solid: mp 110–115 °C dec; 1H NMR (CDCl3) δ 1.40 (s, 9 H, 3 CH3), 2.0–2.55 (m, 14 H), 3.0 (m, 1 H), 3.5 (s, 2 H), 3.7 (m, 1 H), 7.30 (dd, J = 7.9, 4.7 Hz, 1 H), 7.40 (m, 1 H), 7.58–7.64 (m, 2 H), 7.93 (d, J = 7.7 Hz, 1 H), 8.26 (bs, 1 H), 9.80 (br s, 1 H); MS (CI/NH3) m/e 552 (MH+), 478, 452, 301, 211.
5,11-Dihydro-11-[[4-[2-[[4-(Boc-amino)-1-oxobutyl]-amino]ethyl]-1-piperazinyl]acetyl]-6H-pyrido[2,3-b][1,4]-benzodiazepin-6-one (29b)
Compound 27 (100 mg, 0.28 mmol) was dissolved in 5 mL of DMF. N-t-Boc-4-aminobutyric acid (58 mg, 0.28 mmol) were added followed by 1,3-dicyclohexylcarbodiimide (59 mg, 0.28 mmol) and 1-hydroxybenzotriazole (58 mg, 0.35 mmol). The mixture was stirred for 48 h at room temperature, diluted with water, extracted with CHCl3, dried, and chromatographed on a silica gel column (CHCl3/MeOH/NH4OH 80/20/2) to yield 95 mg (64%) of 29b: mp 135 °C dec; 1H NMR (CDCl3) δ 1.42 (s, 9 H, (CH3)3), 3.50–3.70 (m, 4 H, NH), 7.30 (dd, J = 7.9, 4.7 Hz, 1 H), 7.40 (m, 1 H), 7.58–7.68 (m, 2 H), 7.95 (d, J = 7.8 Hz, 1 H), 8.25 (br s, 1 H), 9.30 (br s, 1 H, NH); MS (EI) m/e 565 (MH+), 491, 350 (base), 253, 211.
5,11-Dihydro-11-[[4-[2-[[6-(Boc-amino)-1-oxohexyl]-amino]ethyl]-1-piperazinyl]acetyl]-6H-pyrido[2,3-b][1,4]-benzodiazepin-6-one (32d)
6-(tert-Butyloxycarbonylamino)-hexanoic acid was converted to the corresponding N,O-di-methylhydroxamate in 94% yield, using DCC/(dimethyl-amino)pyridine in methylene chloride by the procedure of Martinez et al.34 The hydroxamate was then treated with lithium aluminum hydride at 0 °C in THF to give 6-(tert-butyloxy-carbonylamino)hexanal in 90% yield. Compound 18 (35 mg, 0.1 mmol) and 6-(tert-butyloxycarbonylamino)hexanal (20 mg, 0.09 mmol) were dissolved in THF containing a minimum of MeOH and treated with 4-Å molecular sieves and sodium cyanoborohydride (6.6 mg, 0.1 mmol). After 24 h, the mixture was filtered and the solvent evaporated. The residue was taken up in ethyl acetate and extracted into 1 M citric acid. The aqueous layer was washed with ethyl acetate and basified, and the crude product was recovered upon evaporation of the organic layer. The product was chromatographed on a silica gel column (CHCl3/MeOH/ NH4OH 80/20/2) to yield 11 mg (20%) of 32d.
General Procedure E. Alkylation of 18 with (Bromo-alkyl)phthalimides (33a–h)
5-(Bromohexyl)phthalimide (280 mg, 0.87 mmol), prepared by alkylation of potassium phthalimide with 1,5-dibromohexane in DMF, compound 18 (203 mg, 0.60 mmol), and triethylamine (0.21 mL) were combined in DMF and stirred for several days at room temperature. As the reaction proceeded, additional quantities of 5-(bromohexyl)phthalimide and triethylamine were added. The DMF was removed by azeotropic distillation with benzene and petroleum ether, leaving an orange oil, which crystallized overnight to yield 257 mg (75%) of 5,11-dihydro-11-[[4-[2-[[6-(phthaloylamino)-1-oxohexyl]-amino]ethyl]-1-piperazinyl]acetyl]-6H-pyrido[2,3-b][1,4]benzo-diazepin-6-one (33d). The phthaloyl group was removed by using hydrazine hydrate in EtOH/MeOH.
General Procedure F. Alkylation/Amination of 18 with Dibromoalkanes and Ammonium Hydroxide (34a–h)
Compound 18 (1 equiv), a 1,n-dibromoalkane (n = 3–10, 10 molar equiv), and triethylamine (5 molar equiv) were dissolved in methanol and stirred at room temperature (12–24 h). The mixture was then acidified (aqueous HCl) and washed with ether. The resulted aqueous phase was basified and extracted with CHCl3, and the chloroformic extracts were combined and evaporated. The residue was dissolved in concentrated NH4OH/methanol and stirred at room temperature (40–72 h). The solvent was then removed, and the crude product was chromatographed on a silica gel column, using 60–90% CHCl3 and 10–40% MeOH/NH4OH (10/1, v/v) as eluent.
5,11-Dihydro-11-[[4-(10-aminodecyl)-1-piperazinyl]-acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (34h)
mp 95–97 °C; 1H NMR (CDCl3) δ 1.10–1.50 (m, 16 H), 2.10–2.70 (m, 12 H), 3.15 (d, J = 14 Hz, 1 H), 3.60 (br d, J = 14 Hz, 1 H), 7.30 (dd, J = 7.9, 4.7 Hz, 1 H), 7.40 (m, 1 H), 7.58 (dd, J = 7.8,1.4 Hz, 1 H), 7.70 (d, J = 3.8 Hz, 1 H), 7.96 (d, J = 7.7 Hz, 1 H), 8.28 (br s, 1 H); MS (CI/NH3) m/e 493 (MH+, base), 254, 212.
Detection of Aziridinium Intermediate
For the 1H NMR studies of reaction kinetics, the hydrochloride salt of 26 (4 mg) was dissolved in 0.5 mL of 100 mM K2DPO4/NaD2PO4 buffer (pD 7.4). The NMR tube was immediately inserted in the Varian 500 MHz proton NMR probe, which had been heated to 37 °C, and spectra were recorded at regular intervals. Relative amounts of the chloroethyl derivative 26 and its conversion products at various times were obtained by integration or by measuring relatives height (for the aziridinium salt) and assuming that 26 was converted quantitatively to 25 and the O-phosphate ester, via the aziridinium ion. Rate constants for the cyclization of 26 and for the formation of 25 were obtained from the first-order rate equations as described above. The concentration of the aziridinium ion as a function of time was fitted by nonlinear regression analysis.
Binding Assay
Inhibition of [3H]NMS binding in membranes from rat heart cells, in NG108-15 cells having m4 receptors, or in transfected rat A9L cells (from connective tissue) expressing m1, m3, or m4 muscarinic receptors (obtained from Dr. M. Brann, NIH) was measured. A crude membrane fraction was obtained as follows. Confluent cultures were rinsed three times with phosphate-buffered saline and lysed in a solution of 2 mM Tris-HCl (pH 7.1) and 1 mM EDTA for 30 min at 2 °C. The cells were harvested by scraping and homogenized on a Polytron (10 s, 75% mix). Nuclei were removed in a low speed centrifugation (400g, 5 min) and a crude membrane preparation was obtained by centrifugation of the supernatant at 50000g for 20 min. The resulting pellet was resuspended in the lysis buffer and recentrifuged at 50000g for 20 min. Membranes were stored frozen at −70 °C until needed. An aliquot of the membrane fraction (150–300 μg of protein) was incubated for 90 min at 37 °C with 0.5 nM [3H]NMS and various concentrations of the unlabeled analogue in DMEM-Hepes. The total volume was 1 mL. The incubation was terminated by rapid filtration over GF/B filters using a Brandel cell harvester. The filters were washed three times with ice-cold 0.9% NaCl, equilibrated in scintillation counting fluid, and counted on a Beckmann LS 5801 liquid scintillation counter at 47% efficiency. Nonspecific binding was determined by co-incubation with 1 μM atropine and amounted to less than 15% of total counts. It was routinely subtracted from the total counts.
Saturation by [3H]NMS before and after treatment of the membranes with the potentially affinity label was measured to assay for irreversible inhibition. Aliquots of brain membranes were incubated with 1.0 mM of freshly dissolved 26 in phosphate-buffered saline (pH 8.0,10 mM phosphate, 0.15 M NaCl) for 60 min at room temperature and then centrifuged at 15 000 rpm for 10 min. The resulting pellet was resuspended in 25 mL of fresh phosphate-buffered saline (pH 7.2) and centrifuged as above. The pellet was again resuspended in fresh buffer and centrifuged, and aliquots were taken for [3H]NMS saturation binding experiments.
Acknowledgments
We thank Dr. Herman Yeh of NIH for assistance with NMR studies and Naftali Malka for technical assistance.
Footnotes
Registry No. 1, 28797-61-7; 3, 107831-56-1; 4, 6298-19-7; 5, 134-20-3; 6, 885-70-1; 7, 133727-44-3; 8, 133727-45-4; 9, 133727-46-5; 10, 133727-47-6; 11a·HCl, 133727-48-7; 11b, 133727-49-8; 11c, 133727-50-1; 11c·oxalate, 133727-51-2; 11d·oxalate, 133727-53-4; 11e·oxalate, 133727-55-6; 13, 133727-56-7; 14, 133727-57-8; 15, 133727-58-9; 16, 133727-59-0; 17a·HCl, 133727-60-3; 17b, 133727-61-4; 18, 63257-31-8; 19a, 96448-78-1; 19b·HCl, 133727-62-5; 20·HCl, 133727-63-6; 21, 133727-64-7; 22, 69628-58-6; 23·oxalate, 133727-65-8; 24a·oxalate, 133727-67-0; 24b·oxalate, 133727-69-2; 24c·oxalate, 133727-71-6; 24d·oxalate, 133727-73-8; 25, 28781-43-3; 26·HCl, 133727-74-9; 27, 133727-75-0; 28, 133727-76-1; 29a, 133727-77-2; 29b, 133727-78-3; 30a·TFA, 133727-80-7; 30b·TFA, 133727-82-9; 31a, 133727-83-0; 31b, 133727-84-1; 32a, 133727-85-2; 32b, 133727-86-3; 32c, 133727-87-4; 32d·oxalate, 133727-89-6; 32e, 133727-90-9; 32f, 133727-91-0; 32g, 133727-92-1; 32h, 133727-93-2; 33a, 133727-94-3; 33b·oxalate, 133727-96-5; 33c, 133727-97-6; 33d, 133727-98-7; 33e, 133727-99-8; 33f, 133728-00-4; 33g, 133728-01-5; 33h, 133728-02-6; 34a, 133728-03-7; 34b, 133728-04-8; 34c, 133728-05-9; 34d, 133728-06-0; 934e, 133728-07-1; 34f, 133728-08-2; 34g, 133728-09-3; 34h, 133728-10-6; 35a, 133728-11-7; 35b, 133728-12-8; 35c, 133728-13-9; 35d, 133728-14-0; 35e, 133728-15-1; 35f, 133728-16-2; 35g·oxalate, 133728-18-4; 35h, 133728-19-5; 36·oxalate, 133728-21-9; 37·oxalate, 133728-23-1; NH2(CH2)2NH2, 107-15-3; C6H5COCl, 98-88-4; CH3SO2Cl, 124-63-0; C6H5NCS, 103-72-0; BrCH2C≡CH, 106-96-7; C6H5CH2Br, 100-39-0; p-BrCH2C6H4CONHCH3, 118507-18-9; p-BrCH2C6H4CONH-(CH2)2NHBOC, 120131-81-9; p-BrCH2C6H4CONH(CH2)4NHBOC, 120131-82-0; ICH2CH2OH, 624-76-0; BOC(CH2)5CON(Me)OMe, 133728-24-2; Br(CH2)3Pht, 5460-29-7; Br(CH2)4Pht, 5394-18-3; Br(CH2)5Pht, 954-81-4; Br(CH2)6Pht, 24566-79-8; Br(CH2)7Pht, 52824-42-7; Br(CH2)8Pht, 17702-83-9; Br(CH2)9Pht, 93667-91-5; Br(CH2)10Pht, 24566-80-1; Br(CH2)3Br, 109-64-8; Br(CH2)4Br, 110-52-1; Br(CH2)5Br, 111-24-0; Br(CH2)6Br, 629-03-8; Br(CH2)7Br, 4549-31-9; Br(CH2)8Br, 4549-32-0; Br(CH2)9Br, 4549-33-1; Br(CH2)10Br, 4101-68-2; CHO(CH2)6NHBOC, 84766-90-5; CHO(CH2)4NHBOC, 94136-78-4; CHO(CH2)6NHBOC, 133728-25-3; CHO(CH2)7NHBOC, 133728-26-4; CHO(CH2)8NHBOC, 133728-27-5; CHO(CH2)9NHBOC, 133728-28-6; CHO(CH2)10NHBOC, 133728-29-7; 5,11-dihydro-8-[2-aminoethylsulfenamido]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one, 133728-30-0; 2-chlorooctyl chloride, 79-04-9; N-methylpiperazine, 109-01-3; piperazine, 110-85-0; α-chloroethyl chloroformate, 50893-53-3; methyl 2-amino-4-carbomethoxybenzoate, 5372-81-6; methyl 2-amino-4-(hydroxymethyl)benzoate, 133728-31-1; 5-ethyl-11-hydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one, 4937-79-5; N-(2′-chloropyrid-3-yl)-2-amino-4-[[(dimethyl-tert-butylsilyl)oxy]methyl]-benzamide, 133728-32-2; N-t-BOC-2-alanine succinimido ester, 32703-87-0; N-t-BOC-4-aminobutyric acid, 57294-38-9; 6-(tert-butyloxycarbonylamino)hexanoic acid, 6404-29-1; (dimethyl-amino)pyridine, 1122-58-3; 6-(tert-butyloxycarbonylamino)hexanal, 80860-42-0.
References
- 1.Frotscher M, Misgeld U, editors. Central Cholinergic Synaptic Transmission; Birkhauser Verlag; Basel: 1989. [Google Scholar]
- 2.Kromer W, Gönne S. Int J Exp Clin Pharmacol. 1988;37(suppl 1):48. doi: 10.1159/000138506. [DOI] [PubMed] [Google Scholar]
- 3.(a) Melchiorre C, Cassinelli A, Quaglia W. J Med Chem. 1987;30:201. doi: 10.1021/jm00384a034. [DOI] [PubMed] [Google Scholar]; (b) Melchiorre C, Cassinelli A, Angeli P, Giardina D, Gulini U, Quaglia W. Trends Pharm Sci (supplement) 1988:55. [PubMed] [Google Scholar]
- 4.Goyal R. New Engl J Med. 1989;321:1022. doi: 10.1056/NEJM198910123211506. [DOI] [PubMed] [Google Scholar]
- 5.Maclagan J, Barnes P. Trends Phar Sci. 1989:88–92. Suppl. “Subtypes of Muscarinic Receptors IV”. [PubMed] [Google Scholar]
- 6.(a) Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]; (b) Bonner TI, Young AC, Brann MR, Buckley NJ. Neuron. 1988;1:403. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 7.Peralta EG, Ashkenazi A, Winslow JW, Ramachandran J, Capon DJ. Nature. 1988;334:434. doi: 10.1038/334434a0. [DOI] [PubMed] [Google Scholar]
- 8.Maeda A, Kubo T, Mishina M, Numa S. FEBS Lett. 1988;239:339. doi: 10.1016/0014-5793(88)80947-5. [DOI] [PubMed] [Google Scholar]
- 9.Levine RR, Birdsall NJM. Trends in Pharmacological Science, Supplement. Vol. 10. Elsevier Trends Journals; Cambridge, UK: 1989. Subtypes of Muscarinic Receptors IV; p. VII. [Google Scholar]
- 10.Hughes AR, Martin MW, Harden TK. Proc Natl Acad Sci USA. 1984;81:5680. doi: 10.1073/pnas.81.18.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams PR, Brown DA, Constanti AJ. Physiol. 1982;332:223. doi: 10.1113/jphysiol.1982.sp014411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yatani A, Hamm H, Codina J, Mazzoni MR, Birnbaumer L. Science. 1988;241:828. doi: 10.1126/science.2457252. [DOI] [PubMed] [Google Scholar]
- 13.Fisher A, Brandeis R, Karton I, Pittel Z, Dachir S, Sapir M, Grunfeld Y, Levy A, Heldman E. Novel Approaches to the Treatment of Alzheimer’s Disease. In: Meyer EM, Simpkins JW, Yamamoto J, editors. Advances in Behavioral Biology. Vol. 36. Plenum; New York: 1989. pp. 11–16. [Google Scholar]
- 14.Baker R, Saunders J. Ann Rep Med Chem. 1989;24:31. [Google Scholar]
- 15.Hammer RB, Berrie CP, Birdsall NJM, Burgen ASV, Hulme EC. Nature. 1980;283:90. doi: 10.1038/283090a0. [DOI] [PubMed] [Google Scholar]; (b) Hammer R, Giachetti A. Life Sci. 1982;31:2991. doi: 10.1016/0024-3205(82)90066-2. [DOI] [PubMed] [Google Scholar]
- 16.Mutschler E, Feifel R, Moser U, Tacke R, Wess J, Lambrecht G. Eur J Pharmacol. 1990;183:117. [Google Scholar]
- 17.(a) Bradbury BJ, Baumgold J, Jacobson KA. J Med Chem. 1990;33:741–748. doi: 10.1021/jm00164a044. [DOI] [PubMed] [Google Scholar]; (b) Bradbury BJ, Baumgold J, Paek R, Kammula U, Zimmet J, Jacobson KA. J Med Chem. 1991;34:1073. doi: 10.1021/jm00107a029. [DOI] [PubMed] [Google Scholar]
- 18.(a) Jacobson KA, Daly JD. Nucleosides Nucleotides. 1991;10:1029. [Google Scholar]; (b) Barone S, Churchill PC, Jacobson KA. J Pharm Exp Therap. 1989;250:79. [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Eberlein WG, Trummlitz G, Engel WW, Schmidt G, Pelzer H, Mayer N. J Med Chem. 1987;30:1378. doi: 10.1021/jm00391a019. [DOI] [PubMed] [Google Scholar]; (b) Eberlein WG, Engel WW, Trummlitz G, Schmidt G, Hammer R. J Med Chem. 1988;31:1169. doi: 10.1021/jm00401a016. [DOI] [PubMed] [Google Scholar]
- 20.Eberlein WG, Engel W, Mihm G, Rudolf K, Wetzel B, Entzeroth M, Mayer N, Doods HN. Trends Pharm Sci. 1989;10(supplement):50. [PubMed] [Google Scholar]
- 21.Engel WW, Eberlein WG, Mihm G, Hammer R, Trummlitz G. J Med Chem. 1989;32:1718. doi: 10.1021/jm00128a008. [DOI] [PubMed] [Google Scholar]
- 22.Eltze M, Gönne S, Riedel R, Schlotke B, Schudt C, Simon WA. Eur J Pharmacol. 1985;112:211. doi: 10.1016/0014-2999(85)90498-4. [DOI] [PubMed] [Google Scholar]
- 23.Oae S, Togo H. Bull Chem Soc Jpn. 1983;56:3802. [Google Scholar]
- 24.Ireland RE, Thompson WJ. Tetrahedron Lett. 1979:4706. [Google Scholar]
- 25.Ogilvie KK, Beaucage SL, Schifman AL, Theriault NY, Sadana KL. Can J Chem. 1978;56:2768. [Google Scholar]
- 26.Schmidt G. U.S. Patent 3, 406, 168. 1968
- 27.Thomae K. Belgian Patents 867, 638 and 867, 638. 1978
- 28.Hammer R, Kaubisch N, Kopitar Z, Proz A, Zimmer A, Koss FW. Therapiewoche. 1977;27:1567. [Google Scholar]
- 29.Olofson RA, Martz JT, Senet JP, Piteau M, Malfroot T. J Org Chem. 1984;49:2081. [Google Scholar]
- 30.Wheatley M, Hulme EC, Birdsall NJM, Curtis CAM, Eveleigh P, Pedder EK, Poyner D. Trends Pharm Sci. 1988;(supplement):19. [PubMed] [Google Scholar]
- 31.Ringdahl B, Mellin C, Ehlert FJ, Roch M, Rice KM, Jenden DJ. J Med Chem. 1990;33:281. doi: 10.1021/jm00163a046. [DOI] [PubMed] [Google Scholar]
- 32.(a) Winkler JD, Thermos K, Weiss B. Psychopharmacology. 1987;92:286. doi: 10.1007/BF00210832. [DOI] [PubMed] [Google Scholar]; (b) Hait WN, Glazer L, Kaiser K, Cross J, Kennedy KA. Mol Pharmacol. 1987;32:404. [PubMed] [Google Scholar]
- 33.Jones SV, Barker JL, Buckley NJ, Bonner TI, Brann MR. Mol Pharmacol. 1988;34:421. [PubMed] [Google Scholar]
- 34.Martinez J, Bali JP, Rodriguez M, Castro B, et al. J Med Chem. 1985;28:1874. doi: 10.1021/jm00150a020. [DOI] [PubMed] [Google Scholar]
- 35.Stiles GL, Jacobson KA. Mol Pharmacol. 1988;34:724. [PMC free article] [PubMed] [Google Scholar]
- 36.Newman AH, Covington J, Oleshansky M, Jackson BW, Weissman BA, Leader H, Chiang PK. Biochem Pharmacol. 1990;40:1357. doi: 10.1016/0006-2952(90)90404-9. [DOI] [PubMed] [Google Scholar]
- 37.Shai Y, Kirk KL, Channing MA, Dunn BB, Lesniak MA, Eastman RC, Finn RD, Roth J, Jacobson KA. Biochemistry. 1989;28:4801. doi: 10.1021/bi00437a042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olah ME, Jacobson KA, Stiles GL. Arch Biochem Biophys. 1991;283:440. doi: 10.1016/0003-9861(90)90665-l. [DOI] [PMC free article] [PubMed] [Google Scholar]