

Abstract
Assessing the risk profiles of potentially sensitive populations requires a “tool chest” of methodological approaches to adequately characterize and evaluate these populations. At present, there is an extensive body of literature on methodologies that apply to the evaluation of the pediatric population. The Health and Environmental Sciences Institute Subcommittee on Risk Assessment of Sensitive Populations evaluated key references in the area of pediatric risk to identify a spectrum of methodological approaches. These approaches are considered in this article for their potential to be extrapolated for the identification and assessment of other sensitive populations. Recommendations as to future research needs and/or alternate methodological considerations are also made.
Keywords: sensitive populations, pharmacokinetics, pharmacodynamics, genetic variability, pediatric population, risk assessment, exposure assessment
INTRODUCTION
Inherent within the risk assessment paradigm is the need to understand the relationship of exposure and response and how that defines risk. In order to accomplish this evaluation, consideration of how specific exposure and response factors can change under different risk assessment contexts and for different populations is needed. To protect public health, part of this focus must consider factors that impact our risk estimates for vulnerable populations, especially if the vulnerable population under consideration is a substantial proportion of the overall population. For the purposes of this discussion, the following definitions have been adopted from Makri et al. (2004) and the National Environmental Justice Advisory Council (2004) and can be applied to the metrics used to formulate the risk assessment problem as outlined by Daston et al. (2004):
Susceptibility is defined as a capacity characterized by biological (intrinsic) factors that can modify the effect of a specific exposure, leading to higher health risk at a given relevant exposure level. The term sensitivity is used to describe the capacity for higher risk due to the combined effect of susceptibility (biological factors) and differences in exposure. Vulnerability incorporates the concepts of susceptibility and sensitivity, as well as additional factors that include social and cultural parameters (e.g., socio-economic status and location of residence) that can contribute to an increased health risk.
The probability of identifying potential risks and adequately defining quantitative responses across diverse human populations is increased by evaluating mechanisms that define sensitive or vulnerable populations. If risk assessments consider factors such as age, genetics, environment, exposure, or combinations of these and other factors, then the underlying assumption is that overall populations can be adequately protected by protecting vulnerable populations.
An increasingly important component of research evaluations has been the recognition that both exposure and response factors can vary widely across ages and that because of unique developmental considerations these factors need to be examined in children. Hence, the potential for sensitivity in the pediatric population can be defined by age-related differences in both exposure and response. (For the purposes of this discussion, the “pediatric population” is defined as individuals from birth to 18 years of age.) This recognition has sparked significant recent research focused on improving our understanding of when and how such developmental factors can make a difference in defining the potential for adverse health outcomes.
In 2006, the Health and Environmental Sciences Institute (HESI) of the International Life Sciences Institute (ILSI) identified risk assessment for sensitive populations as a priority emerging issue. A subcommittee was formed to broaden and increase knowledge about the characterization of sensitive populations, identify opportunities to apply current and proposed methods for assessing risks to sensitive populations, and integrate results from applications and lessons learned to improve risk assessment for diverse sensitive or vulnerable populations in the future. The objective of this HESI effort was aimed at identifying lessons learned from the extensive body of literature on pediatric health and evaluating when it might be used, in part or in total, for identifying and assessing other vulnerable populations. A thorough literature search encompassing inclusive search terms was conducted and, as an initial step, the Subcommittee came to a consensus opinion regarding 50 of the most relevant published papers. (The literature search was restricted to the time frame from January 1996 to October 2007 and used the following search terms: Disease Susceptibility AND environmental exposure OR environmental pollutants OR xenobiotics OR chemical toxicity OR hazardous substances OR risk assessment OR risk factor OR pharmaceutical OR pharmaceutical preparation OR chemistry, pharmaceutical OR alternative medicine OR complementary therapy. The search was then limited by the MESH terms, Age Factors OR Aging OR Human Development OR Children.) Other key references were incorporated into the effort by individual contributing authors at their discretion. However, it should be emphasized that this effort was not intended to be a complete review of all applicable literature on this topic.
Recognizing that this was a subjective process, each study selected for further review was evaluated for (1) general utility for evaluating pediatric subjects as a sensitive population; (2) perceived or recognized gaps in the study that would preclude definitive conclusions on life-stage sensitivity; (3) how “sensitivity” was (or was not) defined; and (4) how the study and the approaches therein might be suitable for extrapolation to other populations or life-stage–defined groups. It was expected that this review of selected papers from the literature on the topic of pediatric health and sensitivity would clarify the following issues:
Identification of critical biological, toxicological, and exposure-related factors that should be examined when evaluating sensitivity among subpopulations, in this case, the pediatric population.
Identification of methodological approaches, models, and experimental designs that have been used when evaluating pediatric subjects as a population and which of these may be useful in the evaluation of other populations.
Identification of those parameters that are unique to the pediatric population and, therefore, not useful in extrapolating to other groups.
Identification of key gaps in the pediatric/vulnerability literature that need to be addressed for determining relevancy of applying lessons learned from this literature to other potentially vulnerable groups.
CONSIDERATION OF PEDIATRIC SUBJECTS AS A SENSITIVE POPULATION MODEL WHEN EVALUATING OTHER POTENTIALLY VULNERABLE GROUPS
Within the context of identifying and assessing populations relative to susceptibility, sensitivity, or vulnerability to exogenous agents (e.g., chemicals, pharmaceuticals, or natural substances), there have been multiple lessons already learned from pediatric research. These lessons may lead to insights on other populations of interest. The bases for this opinion and perspective follow.
The Pediatric and Adult Populations Are Different
A perspective has emerged that essentially states “children are not little adults.” This perspective infers that pediatric biological systems, detoxification processes, and exposures, among other factors, are not those of adults, and, therefore, the pediatric population may have greater or less sensitivity to exogenous agents. Based on this precept, considerable research and effort has been expended in evaluating this population. As a result, there is a considerable, and still growing, amount of information and data to bring to bear when discussing potential age-dependent sensitivity.
Regulatory Initiatives Concerning the Pediatric Population
The increased attention to potential pediatric sensitivity from exposure to exogenous agents has translated into several legislative initiatives. Most notably, the 1996 Food Quality Protection Act was enacted to address concerns resulting from pesticide exposures not considered to be adequately covered via existing Federal Insecticide, Fungicide, and Rodenticide Act regulations. This specific focus on pesticides was followed in 1997 by a more general requirement via an executive order, “The Protection of Children from Environmental Health Risks and Safety Risks.” The attention on the pediatric population, safety, and risk eventually moved beyond agricultural chemicals (e.g., pesticides) and extended itself into other programs, encompassing other xenobiotics (e.g., industrial chemicals), such as the U.S. Voluntary Children's Chemical Evaluation Program; the European Union's Science, Children, Awareness Raising, Legal instruments and Evaluation (SCALE) program; the U.S. Environmental Protection Agency’s (EPA's) Cancer Guidelines (USEPA, 2003a); and more specifically, the U.S. EPA's Supplemental Guidance for Assessing Susceptibility from Early-Life Exposure to Carcinogens (USEPA, 2005a).
Legislation has also been enacted that addresses the testing and use of therapeutics in pediatric subjects. Unfortunately, it took several therapeutic misadventures to highlight the differences in drug disposition and response between pediatric and adult patients (e.g., Weiss et al., 1960). These events were the major drivers for legislative changes to encourage pediatric clinical trials both in the United States (the 1997 FDA Modernization Act; the 2002 Best Pharmaceuticals for Children Act; and the 2007 FDA Revitalization Act) and in Europe (Regulation EC No. 1901/2006 on Medicinal Products for Paediatric Use).
Synthesis of Available Information on the Pediatric Population for Utility in Evaluating Other Populations
From the above examples, it is clear that the pediatric population has been, and will continue to be, a sentinel population relative to evaluations involving sensitivity, health effects, and risk. As such, the database that continues to evolve for this population should be considered when evaluating other populations. Distinguishing which factors, parameters, and approaches are unique to pediatrics versus those representing a common denominator germane to most, if not all, populations will be important.
The following sections summarize, with further detail, the information on the pediatric population reviewed by the HESI Subcommittee and elaborate on the potential to extrapolate this information to the identification and assessment of other sensitive populations. An overview of parameters that have been used to define pediatric subjects as a vulnerable population is presented, followed by a more detailed discussion of three critical factors that have been most informative with regards to characterizing risk in this population, i.e., pharmacokinetics, pharmacodynamics, and genetics. The promises and challenges of biomarkers of susceptibility in the pediatric and other sensitive populations are also discussed, as well as how all of these factors are being incorporated into risk assessment models.
In addition to the intrinsic factors outlined below that have been used to define pediatric subjects as a susceptible population, this population group often inhabits environments or is subject to environments that may alter the exposure paradigm relative to adults and, as such, the pediatric population also may be considered a sensitive group. Extrapolating to the identification and/or definition of other sensitive populations, the routes of exposure, population-specific environments, and/or population-specific therapeutics clearly must be taken into consideration. Certainly, such scenarios have been taken into consideration in the past when considering exposures restricted to a particular occupational setting, but this concept has not necessarily been extrapolated to the process of defining other sensitive populations.
Host Factors and Windows of Susceptibility/Sensitivity
For the purposes of this discussion, critical windows of susceptibility/sensitivity are defined as intervals of time when a defined subpopulation is more sensitive to a chemical exposure than is the average population (Selevan et al., 2000). The existence of such time intervals in the developing human has been well documented and is generally an accepted concept. The underlying mechanisms responsible for such sensitive windows of time are not as well understood and can involve differences in pharmacokinetics, pharmacodynamics, behavior, and/or exposure. Increased knowledge regarding these factors will lead to a more robust risk assessment process.
There have been several recent reviews on the many pharmacokinetic factors that contribute to windows of susceptibility in children, each potentially important because of substantial differences from what is observed in the adult population (Alcorn and McNamara, 2002; Ginsberg et al., 2004a; Hines, 2008). These will be discussed more fully under pharmacokinetics. Although an understanding of a chemical's full mechanism of action might be desirable with regards to defining pharmacodynamics, for the purposes of risk assessment, knowledge regarding mode of action (MOA) is usually sufficient. (MOA is defined as the sequence of key cellular and biochemical events [measurable parameters] that result in a toxic effect, while mechanism of action implies a more detailed understanding of the molecular basis of the toxic effect [Seed et al., 2005].) More importantly, such knowledge often can inform the prediction of a toxic response at untested doses, the response from exposure to multiple chemicals that act via the same MOA, and assist in predicting critical windows of exposure. For example, as discussed by Euling and Kimmel (2001), if a chemical's MOA is through androgen receptor binding, developmental periods that depend on androgen action are likely candidates for critical windows of susceptibility. Because of the difficulties in testing nontherapeutic chemicals in human volunteers and especially sensitive populations, the risk assessment process can take advantage of the knowledge gained regarding the pharmacokinetics and pharmacodynamics of a particular drug or toxicant that shares the same MOA and apply these same parameters (Ginsberg et al., 2004b; McCarver, 2004).
Do windows of susceptibility exist for other populations and can we use the lessons learned from our study of the pediatric population to assist in defining other vulnerable populations? Application of these same principles would appear to apply. Thus, if key pharmacokinetic and/or pharmacodynamic factors are different during a particular time interval for a given population and/or there is a dependent relationship between the chemical's MOA and that particular time interval, then any one or all three of the above parameters can contribute to defining a window of susceptibility.
Ginsberg et al. (2004b) outlined a three-phased process for assessing the contribution of pharmacokinetics to defining windows of susceptibility in the pediatric population: problem formulation, data analysis, and risk characterization. As shown in later guidance issued by the USEPA (2006a), this three-step process can be expanded to incorporate pharmacodynamics and MOA, which then assists in defining windows of susceptibility in other populations (Fig. 1). More recently, a series of review articles by Brown et al. (2008), Cohen Hubal et al. (2008), and Makris et al. (2008) demonstrates further expansion and description of this process.
FIG. 1.
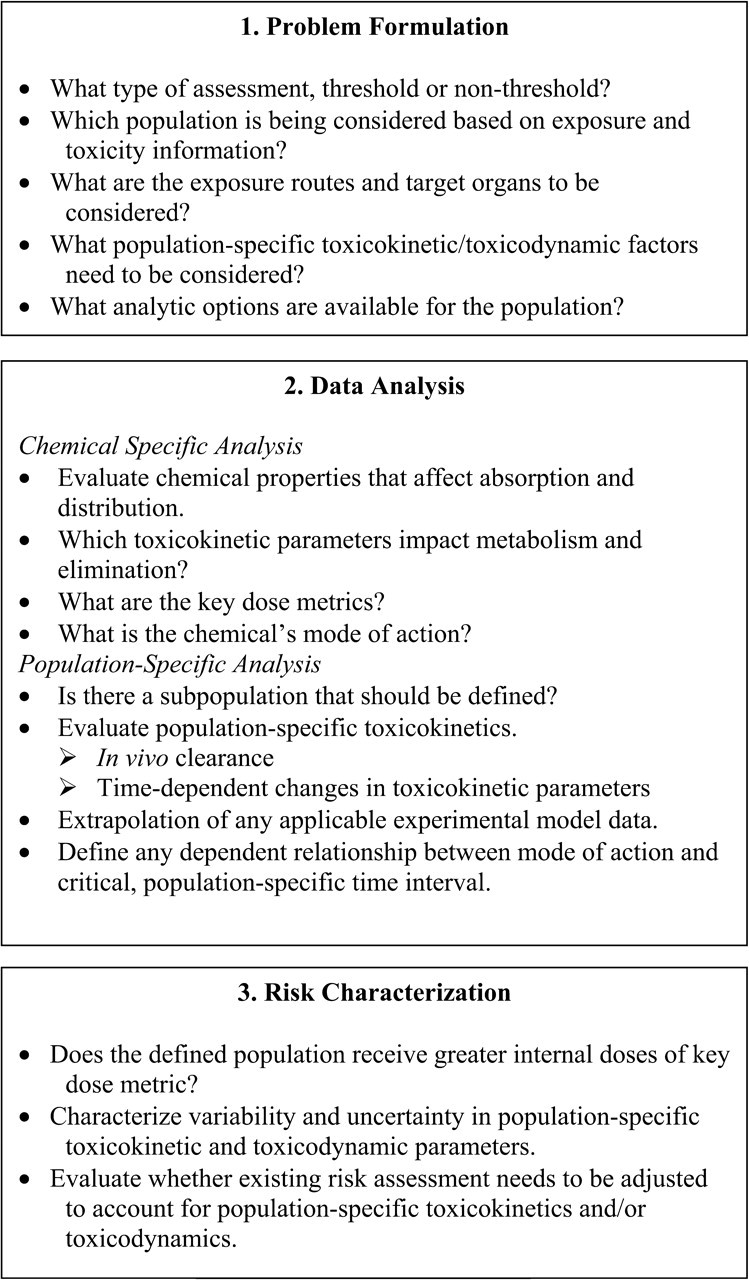
Three-phased process for assessing the contribution of toxicokinetics to defining windows of susceptibility in children (Ginsberg et al., 2004b).
Physiological Considerations
There are a number of excellent reviews available that have evaluated the impact of physiological changes as they relate to potential risk in the pediatric population from exposure to xenobiotics (Alcorn and McNamara, 2002; Ginsberg et al., 2004a,b). In general, and not surprisingly, the research has shown that a thorough understanding of the changes that occur during development, maturation, and eventual senescence of physiological systems is essential to the determination of potential susceptibility of a population of interest.
For the pediatric population, the process of physiological development and maturation has been identified as critical to the risk assessment process. The need to better understand the effect of physiological factors on the dynamics resulting from xenobiotic exposure has been spurred by the lack of pediatric-specific pharmacological data, leading to large uncertainty in the practice of using adult-tested pharmaceuticals in the treatment of pediatric patients. Investigations have shown that some of the largest variability between potential effects of drugs and chemicals on adults versus pediatric patients occurs in the first few weeks/months of life. Some examples include changes in body composition, functional and structural maturation of major organ systems (e.g., gastrointestinal, vascular, hepatic, renal, and respiratory systems), and the resulting effects on kinetics and dynamics.
While this paper will not try to replicate the exhaustive reviews concerning the importance of evaluating physiological factors for a potential population of concern, some brief highlights that illustrate the impact these factors can have on the assessment of risk are worth noting.
For example, intragastric pH is elevated in the neonate relative to later life stages, resulting in lower bioavailability of weakly acidic chemicals. In contrast, pinocytic activity is more active in infants and intestinal motor activity has not yet matured, both increasing the potential for chemical absorption.
There are also age-dependent changes in body composition that will impact volume of distribution. Thus, infants up to approximately 3 months of age have lower lipid content, reducing the retention of lipophilic chemicals, and greater water content, increasing the volume of distribution of hydrophilic chemicals. In older infants, i.e., 3 months to 2 years of age, lipid content relative to adults is increased, resulting in greater retention of lipophilic drugs.
With regard to organ system maturation, the ratio of liver to body mass is not constant and in fact, is considerably greater in infants and young children than in adults. This difference results in a greater potential for hepatic extraction and metabolic clearance, accounting for some, but not all, of the developmental differences observed in clearance between adults and pediatric patients (Murry et al., 1995; Noda et al., 1997). Scaling by normalizing to a 70-kg individual using the 3/4 power allometric rule [activity70 kg = activity/(weight/70 kg)0.75] can be used to adjust for the differences in liver size relative to body mass in pediatric versus adult individuals for the purposes of comparisons (e.g., Zaya et al., 2006). In addition, recent studies have determined that microsomal protein content also changes with age. Although there is considerable interindividual variability, at birth, mean microsomal content was 28 mg/g liver and increased to a maximum of 40 mg/g liver around 28 years of age with a subsequent decline to 29 mg/g liver for the average 65 year old (Barter et al., 2007, 2008). Data from Hines (2008) are consistent with this observation and, furthermore, would suggest significantly less microsomal content in the fetal liver. For chemicals undergoing microsomal enzyme–dependent metabolism, the age-dependent changes in microsomal content between pediatric and adult patients would affect drug disposition in a direction opposite to that of the changes in liver size relative to body mass. Thus, both parameters need to be taken into consideration when assessing pharmacokinetics in the pediatric population relative to adults.
The maturation of kidney structure and function also has a profound impact on the effect of chemicals that depend on renal clearance for elimination and/or termination of biological action. Nephrogenesis begins as early as 9 weeks gestation and is complete by 36 weeks. However, vasoconstriction and reduced renal blood flow result in a substantially diminished glomerular filtration rate (GFR) in the term infant versus the adult. With parturition and the resulting decrease in vascular resistance and increase in cardiac output and renal blood flow, GFR increases rapidly and approaches adult levels by the first year of life. Despite these parturition-associated events, GFR is more tightly correlated with postconceptional age than postnatal age, clearly suggesting that maturation of renal structure continues to influence GFR in the postnatal period. Tubular secretion and reabsorption also play an important role in overall renal clearance of chemicals. At birth, the renal tubules are not yet mature, either structurally or functionally, leading to clearance that is only 20–30% of adult values. Increases to adult levels of tubular secretion are attained by 7–8 months. Information on the ontogeny of specific renal transport enzymes in the human remains deficient and would greatly aid in our understanding of early life-stage differences in response and risk for adverse events from chemical exposure. Clearly, similar changes in renal function in different age groups, in response to disease or as a result of genetic variation, also would impact susceptibility and should be considered when evaluating potentially sensitive populations.
These are just a few examples of physiological factors, which once investigated and incorporated into the risk assessment process proved to have significant impact on the identification of potential susceptibility for the pediatric population. Clearly, consideration of physiological factors should be assessed when evaluating other populations of concern.
Behavioral Aspects That Influence Exposure in the Pediatric Population
Interactions with their environment and resulting exposures can be different in pediatric subjects versus adults (Bearer, 1995; Cohen Hubal et al., 2000a; Goldman, 1995; National Academies of Sciences, 1993). These differences in potential exposure have been reviewed and are primarily due to changes in physiology (discussed above) and behavior across developmental life stages (Cohen Hubal et al., 2000a; Firestone et al., 2007; USEPA, 2005b).
Behavioral factors important for characterizing critical windows of exposure include locations (immediate environment), activity, diet, and product use. These will all vary with developmental life stage and may have a significant influence on exposure.
In considering the behavioral characteristics of the pediatric population and how they might inform regarding the sensitivity or vulnerability of other populations, clearly one must consider many of the same parameters enumerated above when attempting to both identify and characterize other potentially sensitive populations. Thus, unique locations wherein a given population spends a significant portion of their time, unique activities that might influence exposure, and/or diets or product use all might have a substantial influence on exposure and as such, be important when determining and managing risk for a given population.
Ultimately, as identified within this review, determination of vulnerability for any population is dependent on many factors, prominent of which appear to be physiology, genetic influences, pharmacokinetic and pharmacodynamic factors, as well as differences in exposure and resultant internal dosimetry. The remainder of this paper provides greater details on the factors that had the most impact on defining and assessing sensitivity in the pediatric population and our determination of the applicability of these factors to defining and assessing other populations of concern.
RELEVANT FACTORS IN THE IDENTIFICATION AND ASSESSMENT OF SENSITIVE POPULATIONS
Critical Factor 1: Pharmacokinetics and Vulnerable Populations
Differences in pharmacokinetic parameters between pediatric and adult patients are recognized as having significant impact on the risk for adverse drug effects (e.g., chloramphenicol [Weiss et al., 1960] and cisapride [(Kearns et al., 2003; Pearce et al., 2001; Treluyer et al., 2001]), and similarly will impact risk for other chemical toxicities. Considerable work has been done on the ontogeny of drug-metabolizing enzymes in liver (Hines, 2008), but little in extrahepatic tissue. The limited studies that have been done in the intestinal tract would suggest age-dependent changes in oxidative enzymes that are different than those observed in liver. Very few studies are available on the potential for age-dependent changes in transporter expression in either hepatic or extrahepatic tissue. However, a single relatively recent study would suggest that at least for ABCB1 (MDR1 or p-glycoprotein), there is little or no change in activity in the elderly (Brenner and Klotz, 2004). Changes in the levels of the major chemical-binding plasma proteins do occur with age and will influence pharmacokinetics.
Several groups demonstrated low-level expression of one or more cytochromes P450 early in fetal liver development (Cresteil et al., 1982; Hakkola et al., 1994; Kitada et al., 1991; Lee et al., 1991; Pasanen et al., 1987). However, most, if not all, of these studies utilized experimental designs that were limited by their specificity, sample size, and/or range of ages covered. With the development of highly specific antibody probes, more sensitive detection techniques, and the greater understanding of gene complexity as a benefit of the human genome project, a more complete knowledge of developmental expression patterns has been achieved.
An overview of the existing knowledge regarding hepatic xenobiotic metabolizing enzyme ontogeny was recently published by Hines (2008). This review revealed common developmental expression patterns, permitting the categorization of the various enzymes into one of three classes. As typified by CYP3A7, FMO1, and SULT1E1, class 1 enzymes are expressed at their highest level during the first trimester and remain at high concentrations, or decrease during gestation, but are silenced or expressed at low levels within 1–2 years after birth. CYP3A5 and SULT1A1 are examples of enzymes that can be categorized as belonging to class 2. These enzymes are expressed at relatively constant levels throughout gestation. Moderate postnatal increases in expression are observed for some of these enzymes (e.g., CYP2C19 [Koukouritaki et al., 2004]), but not all. CYP3A4, CYP2E1, FMO3, and SULT2A1 are examples of class 3 enzymes that are not expressed or are expressed at low levels in the fetus. For many, the onset of expression can be seen in either the second or third trimester. However, substantial increases in expression are observed within the first 1–2 years after birth. This third category of ontogeny represents the largest number of xenobiotic metabolizing enzymes. Whether this same classification can be used for extrahepatic tissues remains to be determined.
For those class 3 xenobiotic metabolizing enzymes, i.e., those that undergo a perinatal onset or significant increase in hepatic expression, most if not all exhibit greater interindividual variability during this time frame. As an example, both CYP2C9 (Koukouritaki et al., 2004) and 2E1 (Johnsrud et al., 2003) exhibited an approximately 100-fold range of expression in the perinatal period, which was approximately two times greater than that observed within any other age bracket. This is largely explained by what appears to be variability in the postnatal onset or increase in expression for many of the class 3 enzymes. Thus, during the neonatal period, nearly 50% of the tissue samples exhibited CYP2C9 and 2E1 expression levels that were no different than those observed in the fetal third-trimester samples, while the remaining samples exhibited CYP2C9 and 2E1 expression levels that were similar or approached the maximum observed over the entire sample set. In the case of FMO3, interindividual differences in the onset of expression during the first years of life are likely a major cause for the case reports of transient trimethylaminuria in children (Mayatepek and Kohlmüeller, 1998). However, the observation of perinatal hypervariability also can be extended to some class 1 enzymes. The largest variation in CYP3A7 expression (>100-fold) was observed in infant samples, likely explained by variation in the silencing or suppression of this gene. Thus, there are windows of hypervariability during the ontogeny of many of the xenobiotic metabolizing enzymes that would have a significant impact on the risk for adverse events in this population, but would not be predicted based on pharmacogenetic studies in adults.
Despite recent advances in our understanding of xenobiotic metabolizing enzyme ontogeny, several important knowledge gaps remain. Additional studies are needed to define the true ontogeny of many of the enzyme systems, particularly in extrahepatic tissues. Too much of our current knowledge is based on in vitro or in vivo studies that utilized tissue samples or recruited patients, respectively, representing narrow windows of time or omitting what would appear to be critical time windows. Conclusions drawn from such studies can be contradictory and misleading. Finally, the mechanisms regulating xenobiotic metabolizing enzyme ontogeny remain poorly understood. Increased knowledge regarding ontogeny regulatory mechanisms would be informative in defining the MOA for chemicals that might disrupt this process. Despite these knowledge gaps, the field has progressed to a point that has permitted the development of robust physiologically based pharmacokinetic models that provide a much improved means of predicting age-specific chemical disposition (Ginsberg et al., 2004b; Johnson et al., 2006; Nong et al., 2006) (see “Implementation of Population-Specific Factors in Risk Assessment/Modeling” section below).
The lessons learned in using pharmacokinetics to define the pediatric population as a susceptible or sensitive population also can be used effectively in defining other such populations. Clearly, any population or subpopulation in which significant pharmacokinetic deficiencies exist is potentially at greater risk for a chemical exposure that is dependent on that physiological process or metabolic pathway for detoxification and/or clearance. Although its impact has not been well defined, a recent example is that described above for microsomal content (Barter et al., 2007, 2008). On average, the hepatic microsomal content of a 65-year-old is the same as a neonate, although the spectrum of enzymes being expressed may be both quantitatively and qualitatively different. Nevertheless, given that many of the oxidative enzymes involved in chemical detoxication are localized within this cellular compartment, the resulting decrease in metabolic ability simply due to a reduction in microsomal content would be predicted to impact risk in the elderly. Of interest, expression of the major drug transporter ABCB1 does not appear to change in the elderly (Brenner and Klotz, 2004). In contrast, GSTP1 expression in the prostate has been shown to decrease with increasing age and is associated with increased methylation of key CpG islands in the GSTP1 promoter (Kwabi-Addo et al., 2007). Genetic factors that negatively impact pharmacokinetic parameters also could be incorporated into any risk assessment model that uses pharmacokinetics as an analytic tool and assist in defining sensitive populations. Many such genetic variants are common, existing at frequencies greater than 5% (i.e., high frequency), but are tolerated in the gene pool because a phenotype is only observed in exposed individuals (i.e., low penetrance). Completion and refinement of the HapMap project and the development of tools for genome-wide association studies and in-depth pathway analysis has resulted in the capability of identifying multiple genetic variants, each of which contributes to the biological response to a given chemical exposure. Although the potential for this approach is just beginning to be realized, examples of its use to develop therapeutic dosing algorithms that take into account differences in sensitivities among populations have been reported (Caldwell et al., 2008; Sconce et al., 2005; Tham et al., 2006). Similar approaches should be applicable to defining populations sensitive to specific chemical exposures and quantifying relative risk.
Critical Factor 2: Pharmacodynamics: Investigated Areas of Susceptibility
In looking at the available data for the pediatric population, several critical areas of pharmacodynamic research have become the major focus for assessing susceptibility. Of these, an attempt has been made to review three significant areas with regard to their impact on assessing risk to pediatric subjects. Whether these specific end points of concern are of value in assessing other potentially sensitive populations is discussed as well. These areas include neurotoxicity, oncogenicity, and immunotoxicity.
Neurotoxicity
Human studies, including clinical case reports, have been responsible for identifying well over 20 human developmental toxicants. Of these, over half are known to affect the developing nervous system. Various animal models have been used to confirm the developmental neurotoxicity that results from exposure to these agents and, along with clinical evidence, have implicated several chemical classes such as vitamins, selected solvents, psychoactive drugs, polyhalogenated hydrocarbons, insecticides, and antimitotics (Bellinger, 2007; Boersma and Lanting, 2000; Cory-Slechta, 2005). For the cases where human data are available, comparisons may be made to data generated in animal models. Although there is ample evidence that animal models can be predictive of human outcome, it is important to select animal models carefully, and use them under specific study conditions to maximize cross-species extrapolation (Slikker and Chang, 1998, and references therein). In the context of this discussion and adding to this complexity, model selection also should consider whether it reflects a sensitive subpopulation. Furthermore, the nature and extent of developmental neurotoxic effects often are dependent on the timing of exposure to a toxic agent or combinations of agents and environmental conditions, i.e., organisms exhibit distinct temporal windows of susceptibility. Variations in neurotoxic outcomes across species are expected because stages of nervous system development can vary significantly between species in relation to the time of birth. Thus, the time and duration of exposure in animal models also must be selected carefully to match the window of exposure in the human situation and allow cross-species extrapolation.
Two primary assumptions are fundamental to developing a sound strategy for understanding developmental neurotoxicity: (1) the developing nervous system may be more or less susceptible to neurotoxic insult than the adult depending on the stage of development and agent used and (2) neurobiological, neurochemical, neurophysiological, neuropathological, and behavioral evaluations are necessary and complementary approaches for determining the type and degree of nervous system toxicity (Slikker and Chang, 1998, and references therein).
One of the important advances in understanding possible mechanisms of action for a developmental neurotoxicant has been the elucidation of the underlying biology of programed cell death or apoptosis. The importance of apoptosis to the normal development and function of the nervous system has been demonstrated through the understanding of the developmental role of the brain's major excitatory neurotransmitter, glutamate. Glutamate, an amino acid, modulates neurotransmission, neuroplasticity, neuronal outgrowth, and survival via two classes of receptors: fast ligand-gated ionotropic receptors and slower metabotropic (mGlu) receptors. Ionotropic glutamate receptors consist of three subtypes: N-methyl-D-aspartate; α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; and kainate receptors (Conn and Pin, 1997; Dingledine et al., 1999; Lea and Faden, 2006).
An excellent example that illustrates many of the above points is the antagonism of the NMDA receptor system. NMDA-type glutamate receptors are widely distributed throughout the central nervous system (CNS). The NMDA receptor regulates a calcium channel, and the receptor subunit composition is the variable that determines function. The NMDA receptor system and the intracellular signaling processes it modulates play major roles in the normal development of the CNS by controlling a variety of critical steps in creating and arranging neuronal architecture.
Another primary function of the NMDA receptor complex is in learning and memory processes. Long-lasting changes in the excitability of several associated neurons as a result of repeated release of L-glutamate is associated with the phenomenon of long-term potentiation (LTP). The involvement of the glutamate receptor system and LTP is strongly linked to new learning and memory in animal models. As developing neuronal inputs increase in strength and number, postsynaptic Ca++ ion influx through NMDA receptors increases. This Ca++ ion influx is postulated to trigger changes in neuronal metabolism and gene expression (Scheetz and Constantine-Paton, 1994).
Along with these central roles as “brain sculptor” and “memory maker,” the NMDA receptor system also has the potential to do harm. During development, especially during postnatal days 7–14 in the rat, the CNS exhibits an enhanced susceptibility to the toxic effects of deranged NMDA system function. This enhanced susceptibility has been suggested to result from the increased expression of specific glutamate receptor subunits (Miyamoto et al., 2001).
Because of the critical role of the NMDA receptor system in brain development, antagonism of this system can have profound, long-lasting detrimental effects. If stimulation of glutamate release reinforces neuronal connections, then blockade of that stimulation by NMDA antagonists may result in fewer or nonfunctional connections. Several developmental neurotoxicants, including selected anticonvulsants, are reported to produce their toxicity on the developing nervous system via antagonism of the NMDA receptor system (Ikonomidou et al., 1999; Popke et al., 2001a,b). Other agents whose toxicity is thought to be mediated by interaction with the NMDA receptor include methylmercury, ethanol, and selected anesthetic agents (Guilarte, 1997; Kumari and Ticku, 1998; Miyamoto et al., 2001).
Data generated in several developmental animal models support the fundamental concepts that dose and/or duration of exposure, stage of development, and underlying mechanisms are all important for understanding the potential of neurologically active agents or combinations of such agents and other environmental conditions to produce harm to pediatric subjects. However, until we learn more about the specific mechanisms, or at least MOA, involved, it remains difficult to extrapolate these findings and general approaches to precisely defining and characterizing risk in humans, let alone other sensitive populations.
Oncogenicity
The topic of oncogenic risks from exposures to mutagenic and other environmental toxicants is relatively complex for several reasons: there are multiple causes of cancer, and many of the animal and human studies only focus on one of the causes or one of the mechanisms. In human adult and pediatric subjects, the following factors can contribute to the oncogenic etiology or MOA:
spontaneous mutations (chromosomal abnormalities, point mutations at the molecular level)
genetic predisposition; inherited “cancer” genes
increased accumulation of mutations in populations of cells due to aging or with an increased rate of proliferation as the result of inflammatory processes (Cohen et al., 1995, 1998)
genotoxic and mutagenic drugs, chemicals, and physical agents
endocrine receptor agonists and antagonists; other receptor agonists and antagonists
increased mutations in hamartomas and other forms of displaced tissues (i.e., columnar epithelium in the lining of the vagina from intrauterine diethylstilbestrol [DES] exposure)
immunological suppression from genetic diseases or environmental exposures to immunosuppressive toxicants.
Cancer is a leading cause of death in childhood and adolescence (Napier, 2003). In the age group of 1–4 years, cancer is the third highest cause of death. From age 5–9 and 10–14, cancer is the second highest cause of death, and from age 15–19, cancer is the fourth highest cause of death. During adulthood, cancer is the second highest cause of death (American Cancer Society, 2008). Environmental oncogenic exposures can occur during preconception, pregnancy, childhood, adolescence, or adulthood. While there are many causes of cancer, environmental factors overall (as opposed to genetic factors) are thought to account for 75–80%.
What is the impact of chemicals and drugs on the prevalence of cancer in these specific age groups? There has been a perception among some that pediatric subjects are at greater risk from exposure to all environmental oncogenic chemicals. Yet, existing data indicate that developing organisms may be less susceptible or at least equally susceptible to some environmental toxicants (Brent and Weitzman, 2004; Brent et al., 2004; Done, 1964; Scheuplein et al., 2002). If the oncogenic effect is deterministic and therefore has a threshold, then the threshold for pediatric subjects may be lower.
However, mutagenic toxicants are considered to exhibit a stochastic mechanism of action, and theoretically there may not be a threshold. There is not unanimity of opinion concerning the universal application of the linear-no-threshold hypothesis for risk assessment of mutagenic oncogenic agents. Nevertheless, in the absence of available chemical-specific data or MOA, current EPA guidelines recommend additional default adjustments be applied in cancer risk assessment for infants and children. When exposures to mutagenic carcinogens occur before 2 years of age, a 10-fold adjustment factor is applied. If exposure occurs between 2 and 16 years of age, a threefold adjustment factor is applied (USEPA, 2005b).
When evaluating the potential risk from exposures to the developing organism, the following life stages have to be considered.
Preconceptal exposure to environmental toxicants.
The risk of mutagenic exposures to the gametocytes of adults and the risk of mutations in the ova or sperm that would increase the risk of cancer in the F1 offspring have been studied in animal models and large human populations exposed to environmental toxicants. High exposures of some cytotoxic drugs and chemicals can produce sterility in animal models, as well as an increase in the incidence of pregnancy loss from unbalanced chromosome abnormalities. However, the frequency of chromosome abnormalities in the offspring that are viable is low. In studies on human populations exposed to environmental toxicants, the incidence of cancer in the viable offspring is not measurably increased. Collectively, these data suggest that the risk for cancer in offspring following preconceptal toxicant exposure is low (Ames and Gold, 1990; Boice et al., 2003; Brent, 1994, 1999, 2007; Brent et al., 2004; Byrne, 1999; Mulvihill et al., 1987; Neel, 1999; Neel and Lewis, 1990; Nygaard et al., 1991a,b; Winther et al., 2004).
In utero exposures to environmental toxicants.
There is literature that indicates that in utero embryonic or fetal exposures to environmental toxicants can increase the risk of cancer in the offspring. Two widely studied intrauterine toxicants are DES and ionizing radiation. In the case of the former toxicant, an increased risk of cancer in the offspring has been well documented. Thus, Herbst et al. (1971) reported the cluster of cases of clear-cell adenocarcinoma of the vagina (CCACV) in young women whose mothers had been administered DES during pregnancy. As the data were collected over many years, the incidence of CCACV from DES intrauterine exposure diminished and is estimated to be between 1:1000 and 1:10,000. The mechanism of action is most likely due to malformations of the genital tract following intrauterine exposure to DES, resulting in the displacement of uterine columnar epithelium in the vagina. The displaced vaginal columnar epithelium is more susceptible to the development of cancer in this abnormal site. A mutagenic effect of DES is a less tenable explanation.
Studies of the exposure of ionizing radiation to pregnant animals and to pregnant women have not resulted in consistent results with regard to the risk of cancer in the offspring. Stewart and colleagues reported that the embryo was much more susceptible to the oncogenic effects of ionizing radiation than the child or adult (Stewart, 1973; Stewart et al., 1958; Stewart and Kneale, 1970). A mechanism hypothesized by many to explain an increased risk of cancer in offspring was that irradiation in utero would increase the prevalence of chromosome abnormalities. However, Nakano et al. (2007) irradiated mice in utero with 1 or 2 Gy of x-rays and 6-week-old mice with the same exposures. The mice irradiated at 6 weeks of age had a 5% incidence of translocations, while the mice that were irradiated in utero had a 0.8% incidence of translocations. The authors found that the embryos were susceptible to the induction of chromosome aberrations, but that the aberrant cells did not persist because fetal stem cells tend to be free of aberrations, and their progeny replace the preexisting cell populations in the postnatal period. Similarly, Boice and Miller (1999) published their interpretation of the data pertaining to the oncogenic risks of intrauterine radiation, noting “evidence for a causal association derives almost exclusively from case-control studies, whereas practically all cohort studies find no association, most notably the series of atomic bomb survivors exposed in utero.”
The most recent report from the Radiation Effects Research Foundation supports the conclusions of Boice and Miller (1999). Preston et al. (2008) compared the oncogenic effect of in utero and childhood radiation exposure and concluded “the lifetime risks following in utero exposure may be considerably lower than following childhood exposure,” although further follow-up will be needed to address this question more definitively because the oldest surviving in utero exposed cohort members were only 55 years of age. Thus, the most recent data suggest that the embryo is less susceptible to the oncogenic effects of ionizing radiation and that there may even be a threshold for the oncogenic effects.
Environmental toxicant exposures in children and adolescents.
An approximate 10-fold increase in carcinogenic risks has been suggested for children exposed to high doses of ionizing radiation (Hall, 2002). However, the analysis from these authors failed to show the relative susceptibility of different-aged individuals to the oncogenic effect of radiation, but rather the decreasing ability of older individuals to manifest the full extent of the oncogenic risks and the greater risk that the older population will die from nonradiation causes. Forty to 50 years are required to manifest the full extent of the risk of whole-body ionizing radiation.
While there are some investigators who believe that the developing embryo, child, and adolescent are more susceptible to the oncogenic effects of mutagenic toxicants, there clearly are many exceptions to such a generalization. There is very little discussion in the literature about the dose-response curve of mutagenic toxicants, whether there is a threshold for mutagenic effects at low-level exposures to environmental chemicals, and whether the no-observed adverse effect level is the same or lower relative to other age groups. Thus, the risk to the pediatric population from low-dose chemical exposure remains an important knowledge gap.
Most publications refer to the variable risk of cancer in the fetus, child, and adolescent as the result of variable susceptibility. However, there are two other explanations for an increased life-long cancer risk after exposure during infancy. A developing organism has a greater proportion of its cells undergoing division, and therefore the cells may not be more susceptible per se, but the proportion of susceptible cells may be greater. More important is the fact that the child that is exposed has a lifetime to manifest the genotoxic effect in a clinical malignancy (waiting for that second mutation to occur).
Potential for extrapolation to other populations
Because children are still developing, it is believed that children may be more susceptible to the effects of environmental toxicants, and more specifically, that they may be more susceptible to oncogenic and mutagenic chemicals. For protection purposes, the supplemental cancer risk assessment guidance (e.g., USEPA, 2005a) recommends a linear, nonthreshold model and application of default adjustment factors to account for increased susceptibility for children. What we do not know is whether there is, in fact, a threshold for chemically induced oncogenesis and, if there is a threshold, whether the threshold is lower, higher, or the same for children than adults. If such a threshold exists and the underlying mechanisms can be defined, such a parameter would aid in the extrapolation of differential susceptibility in other populations.
Studies dealing with the survivors of childhood leukemia indicate that boys who receive chemotherapy are four times more likely to be infertile than the girl survivors. We do not know the extent of infertility of males in adult populations treated with chemotherapy when compared to females similarly exposed, and, as such, whether this pharmacodynamic outcome might extrapolate to other populations.
Therapeutic and unintended immunosuppression is associated with an increased risk of cancer in both the pediatric and adult populations. The relationship between modulated immune function, genotoxicity, and neoplasia is complex. Genotoxic chemicals may increase cancer risk via oncogenic mutations and suppression of immune mechanisms that recognize and destroy neoplastic cells, although prolonged use of nongenotoxic immunosuppressive drugs also increases cancer risk. However, immunosuppression and its impact on risk would extrapolate to the definition and characterization of other sensitive populations.
An increased susceptibility to oncogenic/mutagenic agents due to an increased percentage of proliferating cells may well apply to other populations in which a similar phenomenon occurs, e.g., individuals in whom tissues are undergoing repair or experiencing an inflammatory response.
Immunotoxicity
Infectious and allergic diseases are more common prior to immune system maturation, which occurs around puberty (Dietert and Piepenbrink, 2006a; Luebke et al., 2006). A number of factors are thought to account for increased susceptibility, including functional immaturity of the immune system, leading to constitutive immunosuppression, a lack of prior immunological experience with most common pathogens, and age-related differences in the integrity of the host's anatomical and functional barriers. Studies in laboratory animals also indicate that the immature immune system is more susceptible or, in some cases, more sensitive to a variety of chemicals (Dietert and Piepenbrink, 2006a; Luebke et al., 2006).
Immunotoxicant susceptibility is a product of critical maturational events that are required for the immune system to function properly. Qualitative or quantitative changes in immune system cells and tissues translate into potentially long-lasting changes in immune function, some of which may persist for life following developmental exposure (Dietert and Piepenbrink, 2006a; Luebke et al., 2006). This MOA is an unlikely cause of immunotoxicity in adults as histogenesis of immune tissues and establishment of immune system cell lineages are complete before birth. Sensitivity of the developing immune system may reflect the additive or synergistic product of constitutive and chemically induced immunosuppression, expressed as altered function at lower doses. Thus, results obtained in developmental immunotoxicity (DIT) studies may predict potential adverse effects of chemical exposure in sensitive subpopulations of adults with risk factors for decreased immunocompetence (e.g., advanced age, pregnancy, therapeutic and recreational drug use, stress), although the predictive value of DIT data for sensitive subpopulations has not been systematically evaluated.
Inherited or acquired severe immunosuppression is typically associated with a dramatic increase in the incidence of infectious and neoplastic disease, although opportunistic infections (those that are rare in the general population) and less common tumors are the norm in severely suppressed individuals. However, mild to moderate changes in immune function in both young and aged adults are associated with lower responses to vaccination as well as increased risk of community-acquired infections (Luebke et al., 2004). Furthermore, a significantly elevated adjusted odds ratio for increased infections (inner ear, respiratory, chicken pox) was reported for breast-fed children of mothers with elevated milk polychlorinated biphenyl levels, even though laboratory values for lymphocyte end points fell within the normal range (Weisglas-Kuperus et al., 2000). Detecting an increase in the incidence of common infections at the population level is complicated by the background rate of one to two infections per year per individual; thus, epidemiological studies that attempt to associate widespread human exposure to a potentially immunotoxic chemical with a change in the rate of infectious disease is difficult. Nevertheless, the greater susceptibility of the developing organism to immunotoxicants may provide the most sensitive screening method to detect potentially adverse effects on cellular, humoral, and innate immune function at doses that have minimal effects on normal, healthy adults.
Severe immunosuppression is linked to increased rates of neoplastic disease, but evidence for increased cancer risk at mild to moderate levels of immunosuppression is sparse (Luebke et al., 2004), and most neoplastic diseases in young adult humans appear to be independent of exposure to environmental agents or inherited risk factors (Bleyer et al., 2006). However, perinatal exposure of mice to DES reduces the number of natural killer (NK) cell progenitors in the bone marrow, and tumor cell killing by splenic NK cells in mice for up to 18 months (essentially for the normal lifetime of a mouse). Although inhibition of antitumor cell activity was the focus of early studies, it has become apparent that NK cells are a critical source of proteins (cytokines) that stimulate T cells to kill infected cells, and a source of costimulatory signals required for T-cell activation. Thus, the relationship between developmental exposure to DES in humans and increased self-reported rates of infection in children of women who took DES (Vingerhoets et al., 1998) may be a result of indirect effects of T-cell immunity caused by a reduction in a pool of required stimulatory cells. However, it has not been established that this is the underlying MOA for immune system effects in DES-exposed humans or that similar effects occur following exposure to other estrogenic compounds in humans.
Adverse immune system effects are not limited to reduced resistance to infections or neoplastic disease and may include an increased risk of autoimmune disease (Holladay and Smialowicz, 2000) or allergy. For example, the recent increase in allergic asthma in highly developed, less agrarian countries has been attributed in part to reduced contact with infectious agents in the environment at an early age (Chang and Pan, 2008). Pregnancy-associated changes in maternal hormone levels drive a shift away from production of cytokines associated with the default adult proinflammatory, cell-mediated response toward increased antibody response. These changes reduce the chance of rejecting the histoincompatible fetus (Piccinni et al., 2000). Unfortunately, the latter response profile includes upregulated production of immunoglobulin E and eosinophil production, prime participants in allergic disease. Infants are born with a similar, proantibody response phenotype that, with exposure to infectious agents in the environment, shifts to the default adult profile. However, recent evidence indicates that various environmental chemicals (e.g., lead) (Dietert and Piepenbrink, 2006b) can delay or perhaps partially prevent the normal shift to the adult response phenotype if exposure occurs during development, potentially increasing the risk of allergy and asthma and concurrently decreasing resistance to certain types of infections. Other environmental agents, including fine particulate air pollutants and diesel exhaust, appear to have similar effects on the balance of immune function (D'Amato et al., 2005). Thus, the developmental lead exposure data suggest that these pollutants may have similar effects on the immature immune system and may contribute to excess allergic diseases, including asthma.
As noted above, disruption of immune organ histogenesis is an unlikely MOA for adult immunotoxicity, even in potentially susceptible subpopulations exposed to chemicals as adults. However, other MOAs are associated with immunotoxicants, including shifts in cytokine production and defects in cellular activation or function (Dietert and Piepenbrink, 2006a; Holladay and Smialowicz, 2000), an apparent MOA that may be shared by susceptible subpopulations of adults (Luebke et al., 2006). Thus, greater sensitivity of the developing organism may enhance prediction of adverse chemical effects at lower doses in sensitive subpopulations of adults with risk factors for decreased immunocompetence (e.g., advanced age, pregnancy, therapeutic and recreational drug use, stress).
Summary from Pharmacodynamic Examples
These few examples of investigations into the pharmacodynamic differences in children, with regard to xenobiotic effects on important organ systems, clearly demonstrate the complexity of assessing the potential for dynamic-related susceptibility, sensitivity, and vulnerability. In reviewing these data, most of the identified susceptibilities and sensitivities were related to the unique attributes of children's rapidly developing life-stage and exposure potential. Therefore, the applicability of these data to other populations of concern is somewhat limited. One lesson, however, that should not be overlooked is the importance of identifying and understanding target organs and MOA. When pharmacodynamic differences were found to play a role in the susceptibility, sensitivity, and/or vulnerability of children, they almost always were intimately correlated with other critical factors such as kinetics, genetics, and/or exposure. Therefore, pharmacodynamics should remain an integral part of identifying and assessing potentially sensitive populations.
Critical Factor 3: Genetics
Genetic factors are well recognized, though poorly understood, components contributing to individual variability in developmental responses to environmental exposures (Makri et al., 2004; Stephenson, 2005). Although associations linking specific genetic makeup (genotype) and enzyme function or protein activity (phenotype) to disease outcome have been explored extensively for cancer, research has been less focused on noncancer end points. However, where research has been conducted, it is highly suggestive that individual genotype–phenotype relationships can play a role in disease susceptibility (Faustman et al., 2000; Kimmel, 2005; Neri et al., 2006).
DNA is a dynamic structure and is subject to constant modifications: Mistakes occur during normal cellular processes of replication and recombination, while various lesions can be induced by environmental genotoxic agents. Because DNA represents a target for spontaneous or induced modifications (Lewin, 1997), functional divergence from the original DNA blueprint must be dealt with and corrected. Organisms have evolved and developed a network of repair systems to safeguard the integrity of their genomes, which guarantees fidelity in the face of replication and/or recombination errors; induced genotoxic damages caused by exogenous and/or endogenous agents of physical, chemical or viral nature; and the cumulative effects of aging (Wood, 1997). Such repair mechanisms behave toward the DNA molecule as biological sentinels that watch, identify, and then repair detected damages. A deficiency in the activity and/or efficacy of any of these systems leads to the accumulation of lesions that may alter potentially three fundamental DNA functions: replication, transcription, and recombination. When such dysfunction is not immediately lethal for the cell, the accumulation of induced lesions will result in gene mutations and/or chromosomal exchanges or rearrangements increasing genomic instability and, under some circumstances, the risk of cancers or genetic diseases (Reichrath, 2006; Wang et al., 2008; Wei et al., 2007).
Genetics and Susceptibility
Because of the biological and genetic heterogeneity in human populations, the occurrence of broad differences in disease susceptibility/sensitivity among individuals is not surprising. Despite the emergence of many papers on biomonitoring of individuals exposed to genotoxicants, few molecular epidemiological data exist for neonates, infants, and children, making it difficult to define the role of genetics in determining possible differential susceptibility in this population (Pohl et al., 2005).
A large number of genetic and enzymatic components contribute to interindividual differences in response to genotoxic agent exposure. Thus, genetic variation among these components is highly relevant in determining the ultimate occurrence of genetic-based diseases. Genetic susceptibility factors are closely associated with the integrity and activity of genes involved in absorption, distribution, and the different steps of metabolism (activation and/or detoxification) (i.e., pharmacokinetics), thereby altering the effective biological dose. Of these, genetic polymorphisms within genes involved in xenobiotic metabolism have proven highly relevant to disease susceptibility (Anderson et al., 2000; Hong and Yang, 1997). Although less well studied, variability within genes encoding pharmacodynamic targets is receiving increasing attention.
It is important to consider that the expression of a particular genetic polymorphism may vary from tissue to tissue (Estrada et al., 2000) and also that the incidence or frequency often varies with respect to ethnicity and/or race. For example, in the Caucasian population, 43–52% of individuals are characterized by a null glutathione-S-transferase M1 genotype due to the deletion of the whole gene. The incidence of this same variant is somewhat higher in individuals of Asian descent (48–60%), but significantly lower in African Americans (Eaton, 2000). Other examples of ethnic differences in polymorphism frequencies between Caucasian and Asian populations include CYP2D6 poor metabolizers (8% vs. 1%), CYP2C19 poor metabolizers (2.5% vs. 15%), and N-acetyltransferase 2 deficiency (40–70% vs. 10–20%) (Lin et al., 1993; Wedlund, 2000).
Several examples of gene-environment interactions within a temporal context have been described. For example, infants deficient in methemoglobin reductase (MR) and fed with preparations containing tap water contaminated with nitrates from agricultural practices may exhibit anemia. However, similar symptoms may present due to an inherited genetic deficiency in MR or in structurally modified hemoglobin M or H chains. As another example, the human paraoxonase gene (PON1), which is involved in the inactivation of many organophosphate pesticides, has a reduced capability to hydrolyze the organophosphate paraoxon when arginine 192 is substituted by glutamine. A highly variable deficiency in PON1 also is observed in neonates and infants such that activity at birth is two- to sevenfold less than that observed at 2 years of age (Cole et al., 2003). Because acetylcholinesterase activity and cholinergic systems are essential for learning and memory, further research on paraoxonase genetic variation within the context of aging and the MOA of xenobiotics is important (Faustman et al., 2000). To this latter point, recent research has noted the importance of dose relative to PON1 activity (Cole et al., 2005; Timchalk et al., 2002). Even PON1 knock-out mice have a high degree of tolerance to organophosphate exposure until exposures are very high relative to environmental levels (Cole et al., 2005). Thus, the toxicological principle that dose affects mechanism (Slikker et al., 2004) needs careful attention for the interpretation of real-world significance of genetic variability. These examples clearly illustrate the uncertainties associated with the identification of genetic polymorphisms and the need to assess their impact within the context of age, living conditions, exposure level, disease status, and comedication.
DNA Repair and Susceptibility
In adults, failure in DNA fidelity during replication can lead to somatic mutations that increase the risk of disease, most notably cancer. In addition, direct DNA damage to germ cells can lead to birth defects in offspring. The same effects are probable in the postnatal pediatric population, but to a much greater degree due to the increased rate of cellular replication during development offering a greater possibility of DNA replication infidelity. In addition, because one mutated cell occurring early in development could lead to a large fraction of a tissue carrying the same mutation with a reduced ability to carry out the function of the tissue, the potential for disease or disability resulting from DNA infidelity in pediatric subjects is broader than it is for adults.
If the probability of failure to repair DNA damage is fixed for each cellular replication, then the risk of a mutation occurring is increased by introducing an agent that increases the rate of DNA damage. This is true in adults and pediatric subjects, and the difference in their susceptibility in each tissue and organ is predominantly driven by the differences in cellular replication rates and the added years that the pediatric subject has to develop a cancer following the mutational event. If a chemical alters the probability of repairing DNA damage by reducing the effectiveness of the repair processes, then adults may be at substantially reduced risk compared to infants due to less cellular replication. What is learned and applied to the pediatric population is applicable to populations with inflammatory conditions due to genetic polymorphisms or those who have chronic exposures that increase cellular replication (e.g., occupational exposure to a cytotoxic agent that induces chronic regenerative hyperplasia). These populations would face similar susceptibility concerns as children.
The breadth of susceptibility in subpopulations due to differences in DNA repair processes is unknown, but the examples that exist suggest this is an area that is in need of further study and should be considered when identifying and characterizing potential sensitive population groups.
ENVIRONMENTAL EXPOSURE
Life-Stage Classification for Exposure Assessment and Identification of Sensitive Populations
A significant challenge associated with monitoring and assessing individual- and population-level exposure to environmental chemicals is developing methods capable of rigorously assessing age and life-stage–related changes in behavior and physiology. Age-related and life-stage–related differences will determine the appropriate distribution of exposure factors required to address specific exposure scenarios.
Identifying the most highly exposed age range or life stage for a particular population and exposure scenario requires a better scientific basis than is currently available. Approaches used today are limited in scope and potentially in applicability to the full range of geographic, cultural, ethnic, and economic diversity in populations worldwide. In addition, systematic approaches for linking/coordinating hazard and exposure assessment are required to ensure that critical windows of susceptibility are integrated with windows of highest exposure to identify sensitive populations.
Several recent activities focused on assessing exposures to the pediatric population have considered how to categorize life stages for exposure assessment (Table 1). The U.S. EPA document titled “Guidance on Selecting Age Groups for Monitoring and Assessing Childhood Exposures to Environmental Contaminants” published in 2005 (Firestone et al., 2007; USEPA, 2005b) recommends age bins for the pediatric population based on physiology and behavior. The scope of this document focuses on birth through 18 years of age and is designed specifically to promote a more uniform approach for exposure assessments conducted across U.S. EPA program offices and regions. Prenatal and preconceptal life stages were identified as important periods for consideration in assessing health risks from early-life exposures, and these life stages were added to the U.S. EPA–recommended age bins in the USEPA (2006a) document titled “Framework for Assessing Health Risks of Environmental Exposures to Children.” The International Programme on Chemical Safety Environmental Health Criteria (EHC) document titled “Principles for Evaluating Health Risks in Children Associated with Exposure to Chemicals” (World Health Organization [WHO], 2006) cites the U.S. EPA guidance document in the exposure section. In a few instances, life stages defined at the beginning of the EHC consistent with WHO terminology are slightly different than the EPA-recommended exposure bins that were used in the exposure chapter of the EHC document. The U.S. Food and Drug Administration (FDA) has issued guidance consistent with the International Conference on Harmonisation with age categories that are different from those suggested by either WHO or EPA (USFDA, 2000). Even with the focus on the pediatric population in these four documents, there is not a uniform approach for identifying the important life stage (age range) based on characteristics of a particular population and on the exposure/risk assessment question of interest.
TABLE 1.
Pediatric Life Stage Category Definition by Different Agencies
| Age bracket | Descriptor |
| U.S. EPAa | |
| Birth to < 1 month | |
| 1 month to < 3 months | |
| 3 months to < 6 months | |
| 6 months to < 1 year | |
| 1 to < 2 years | |
| 2 to < 3 years | |
| 3 to < 6 years | |
| 6 to < 11 years | |
| 11 to < 16 years | |
| 16 to < 21 years | |
| U.S. FDAb | |
| Preterm newborn infant | |
| 0 to 27 days | Term newborn infant |
| 28 days to 23 months | Infants and toddlers |
| 2 to 11 years | Children |
| 12 to 16 or 18 years | Adolescents |
| WHO (developmental stages)c | |
| Birth to 28 days | Neonate |
| 28 days to 1 year | Infant |
| 1 to 4 years | Young child |
| 2 to 3 years | Toddler |
| 4 to 12 years | Older child |
| 12 to 18 years | Adolescent |
Exposure Sources and Pathways
Exposure sources and pathways may change significantly as a function of developmental life stage. For example, sources may be identified in (1) residence and workplace for pregnant and lactating women; (2) residence, daycare, and outdoor play areas for infants and toddlers; (3) residence, school, and locations of after-school activities for school-age children; and (4) residence, school, and locations of after-school activities and workplace for adolescents. For a given source, exposure media (e.g., water, soil/dust/sediments, food, and objects/surfaces) and exposure routes (i.e., inhalation, ingestion, dermal absorption, and indirect ingestion) define the pathway of exposure (Cohen Hubal et al., 2000b). Relevant exposure media may also change with life stage. For example, the fetus will be exposed to cord blood and amniotic fluid, the infant to breast milk, the teething child to many objects (both intended and unintended) from mouthing, and school-age children and adolescents to recreational and/or vocational environments.
Time Frame of Exposure
Potential health risks resulting from environmental exposures during early life stages are often difficult to recognize and assess due to a potential time lag between the relevant timing of exposure and of outcomes that may be expressed at any subsequent life stage including those far removed from that of exposure (USEPA, 2006a).
In addition, as for any population, exposure patterns will vary both spatially and temporally. Tracking short-term, intermittent acute exposures may be particularly important for assessing risks during critical windows of development. Yet, these also are among the most challenging exposures to characterize (Özkaynak et al., 2005).
Other Vulnerability Factors
Exposure factors and resulting effects during developmental stages may be a function of additional individual and population characteristics. Limited data on gender-related activity patterns in the pediatric population have been reviewed previously (Cohen Hubal et al., 2000a). Differences in behaviors, activities, and locations have been identified in children as young as preschool age. Characteristics of the communities in which pediatric subjects live may also be important for identifying vulnerable populations based on potential for exposure. These may include socioeconomic status, family structure, ethnicity, cultural setting, geographic location, and season. Other factors specific to the individual include genetic differences (see above), nutritional status, and health status. Mechanisms of vulnerabilities associated with individual and community characteristics include differential preparedness and differential ability to recover. These mechanisms have been defined and discussed in the context of cumulative and community-based risk assessment (deFur et al., 2007; Kyle et al., 2006; USEPA, 2003b).
All the guidance developed over the years for assessing pediatric risk has emphasized the importance of understanding exposure. Methods for monitoring, modeling, and analysis of exposure continue to evolve, and their use is critical to our ability to identify and characterize vulnerable populations.
BIOMARKERS OF SUSCEPTIBILITY
Biomarkers may be defined as indicators signaling events in a biological system and are classified into three categories: biomarkers of exposure, effect, and susceptibility (National Research Council, 1987). Exposure biomarkers may include either the quantitation of exogenous agents or complexes of endogenous substances and exogenous agents within the biological system. Biomarkers of effect may be indicators of an endogenous component of a biological system or an altered state of a system that is recognized as an alteration or disease. A biomarker of susceptibility is an indicator that a biological system is especially vulnerable to toxic insult by an exogenous agent (National Research Council, 1987).
Excellent examples of recognized biomarkers specific for disease risk in the pediatric population are those in the highly successful newborn screening programs that have been implemented in many countries. Although the number of tests included in the newborn screen varies from country to country, and within the United States from state to state, all the tests meet the WHO's criteria for population screening that also would have to be met by any biomarker screen that might be proposed for the identification of a susceptible population. Thus, the disease outcome must be both serious and avoidable by treatment, the screening process must be both reliable and inexpensive, and the identification of individuals at a presymptomatic stage must be enabled (Wilson and Jungner, 1968). As an example, the newborn screen for congenital hypothyroidism usually involves a relatively inexpensive and highly reliable radioimmune assay for thyrotropin and/or thyroxine that normally exhibits a surge in the immediate perinatal period. Identification of individuals suffering from congenital hypothyroidism triggers hormone replacement therapy, which effectively prevents the subsequent development of cretinism (Djemli et al., 2006). There is considerable ongoing effort to identify robust genotypic or phenotypic biomarkers of disease risk that could effectively identify susceptible populations for which prevention programs might be implemented that would prevent or minimize exposure.
IMPLEMENTATION OF POPULATION-SPECIFIC FACTORS IN RISK ASSESSMENT/MODELING
There are numerous methods for assessing potential risk to a given group or population. The two main constituents of risk remain the same, exposure and hazard. The more one knows about each of these elements the more detailed, and hopefully, the more accurate will be the assessment of risk. Risk assessment methods generally range from simplistic deterministic approaches to probabilistic distributions of measured variables.
In the most basic form of risk assessment (deterministic), many variables will be based on default assumptions and single-point estimates used in linear algorithms, which then calculate a single risk value for a given subpopulation. In the absence of measured data, these general assumptions are used to estimate duration and frequency of exposures; internal dose resulting from various routes of exposure; and inter- and intraspecies differences, which try to account for kinetics and dynamics specific to the subpopulation of interest. In more complex types of risk assessment (probabilistic), default point estimates are replaced with distributions of variables based on measured data from exposure and pharmacokinetic studies, as well as pharmacodynamic information. The more data that can be generated from the population of interest, the more accurate the estimation of risk. However, most of the data are still predominately generated using animal models and require some extrapolation assumptions.
Both basic and complex risk assessment have value in evaluating potential human health effects related to environmental exposure to xenobiotics. Basic assessments provide a general sense (first pass view) to regulators as to whether a chemical poses a possible risk. Unfortunately, since they rely on generic assumptions that could over- or underestimate risk, the degree of uncertainty in these assessments is usually quite large. More complex risk assessment can greatly improve the confidence in the estimated risk by using actual measured values to lower the uncertainty in the assessment, but are data intensive.
The development of data and methodologies for assessing risk has made significant progress in the last 10 years, especially for the pediatric population. In reviewing the literature that describes the progression of methodological development, several key lessons become apparent. In assessing risk to a population of interest, the first hurdle is in having enough reliable data specific to that population, which can accurately estimate exposure and characterize hazard. When concerns regarding potential risk to the pediatric population from environmental exposures first caught the attention of the public and legislators, there was very little information available regarding how much and how often pediatric subjects were being exposed, and a highly variable amount of mostly adult animal data with regard to the hazard profile of the chemicals of interest. There has been a concerted effort to better characterize risk to the pediatric population from chemicals in commerce, pharmaceuticals, and pesticides. As these three general groups of chemicals are regulated under separate government agencies and regulations, significant differences in available or required data and assessment methodologies still exist. However, review of the literature has shown a rapidly growing body of data that address some of the most critical areas of risk modeling. These areas include investigations into the potential effects, both short and long term, of exposure at different developmental stages and on key physiological systems such as the nervous, immune, and endocrine systems; studies looking at the differences in kinetics and dynamics between and within species and populations (animal vs. humans, and adults vs. pediatric subjects), which help to evaluate the relevance and more accurate extrapolation of data; and lastly, studies that attempt to measure real environmental exposures and the factors that may affect exposure estimates for a given population (e.g., behavioral activities and diet).
Of the literature evaluated by this working group, a large amount of the data was generated to evaluate pediatric pharmacokinetic capabilities. Pharmacokinetic data can play an important role in risk assessment. Kinetic information, specific to a subpopulation of interest, can be applied in estimating internal dose and target tissue concentrations from various routes of exposure, can infer potential susceptibility based on decreased or increased metabolism and elimination, and can help to extrapolate data from other species or populations to the population of concern.
Pharmacokinetic Models
The application of pharmacokinetic models to inform the appropriate therapeutic regimen, when extrapolating from animal models to humans, has been employed in pharmaceutical risk evaluation for many years. However, initial use of these models was based almost exclusively on kinetic information generated from adult animals and humans. In addition, as has been described, route, frequency, and duration of exposure in a sensitive population can vary widely from the general population. These factors, compounded with differences in basic kinetics (absorption, distribution, metabolism, and elimination), can lead to gross uncertainties in the overall estimation of risk. Only within the last 5–10 years has there been significant emphasis placed on the appropriate modeling of potentially susceptible life stages. As was discussed in detail in previous sections, a plethora of data has been developed recently to better define pediatric kinetic parameters (e.g., physiological scaling, characterization of the development of enzyme and other metabolic systems, and characterization of the developmental changes in critical target organ systems over continuous life stages) (Clewell et al., 2004; Edginton et al., 2006; Ginsberg et al., 2004b; Hines, 2008). This information has had a significant impact on the ability to more accurately estimate internal dose levels, and more importantly target tissue concentrations, resulting in better assessments of potential adverse health effects. Current modeling for the pediatric population has produced significant changes in the use, regulation, and/or remediation of chemicals such as caffeine, theophylline, perchlorate, lead, arsenic, bisphenol A, and chloroform (Beck et al., 2001; Bellinger, 2007; Clewell et al., 2007; Ginsberg et al., 2004a; Liao et al., 2007; Willhite et al., 2008). Recently, the U.S. EPA released a database containing human physiological parameter values specific to older adults (USEPA, 2008). These values, obtained from published scientific literature, are intended to provide a scientific basis for selection of several key physiological parameters for dosimetry modeling in older adults.
Thus, evaluation of the pediatric population as a sensitive group has clearly demonstrated the value of the appropriate parameterization and application of physiologically based pharmacokinetic models. Such models should not be overlooked in the identification and assessment of potentially sensitive populations.
Exposure Assessment Tools: Guidance, Measurement, Modeling, and Data
Several recently published documents present frameworks for considering pediatric sensitivity to environmental exposures (Brown et al., 2008; Reiss et al., 2003; USEPA, 2006a; WHO, 2006). In general, these documents provide a comprehensive overview of the issues for assessing exposures to the pediatric population across developmental life stages, provide the general approach for assessing exposure, and advocate for systematic consideration of important windows of vulnerability. The need for a life-stage approach to risk assessment is highlighted in the recently released report “A Framework for Assessing Health Risks of Environmental Exposures to Children” (USEPA, 2006a) and the publications that further discuss and expand upon the report (Brown et al., 2008; Cohen Hubal et al., 2008; Makris et al., 2008). The Framework emphasizes the need to account for potential exposures to environmental agents during all stages of development and consideration of the relevant adverse health outcomes that may occur as a result of such exposures. A report developed in parallel by the WHO, Principles for Evaluating Health Risks in Children Associated with Exposure to Chemicals (WHO, 2006), also focuses on the potential vulnerability of the pediatric population to chemical exposures and the potential for increased risk of adverse effects from early-life exposure. Many of the issues highlighted in these frameworks could be generalized to address other sensitive populations.
Four major types of information are used to characterize exposure: questionnaire-based metrics, surrogate exposure metrics, personal exposure measures, and biomonitoring data (Cohen Hubal et al., 2008; Needham et al., 2005). The availability of various data types will then determine the approaches that can be used to estimate/calculate exposure for risk assessment. Three main approaches are currently used: point-of-contact, scenario evaluation, and dose reconstruction (USEPA, 2006b). The point-of-contact or direct approach requires measurements of chemical concentrations or radiation levels at the point where exposure occurs (at the interface between the person and the environment), and records, or at least estimates, the length of contact with each agent. This approach, often used in occupational settings, does not take into account an individual's characteristics or behaviors. Using the dose reconstruction approach, estimates of exposure are developed from population-level biomonitoring data. Methodologies for using this approach with the pediatric population are discussed in the next section (“Biomonitoring for Life-Stage Exposure Assessment”).
The scenario evaluation approach, sometimes referred to as the indirect approach, requires data on chemical concentration, frequency, and duration of exposure, as well as information on the exposed life stage. In this approach, models are required to link the different data to estimate individual- or population-level exposures or distributions of exposures. Currently, the scenario evaluation approach is often used to conduct risk assessments required to make regulatory decisions. As such, many of the available exposure assessment tools developed to address the pediatric population as sensitive are designed to be used to conduct scenario-based exposure assessments.
Biomonitoring for Life-Stage Exposure Assessment
Biomonitoring data include measurement of chemical, metabolite, or molecular markers in biological fluid and tissues. Exposure biomonitoring data are increasingly being used in epidemiology and in public health surveillance and have the advantage of providing an integrated measure of total exposure from all routes and sources. Methods that have been applied to measure exposure in the pediatric population are generally applicable to other sensitive populations.
Challenges associated with using biomonitoring data for exposure and risk assessment have been discussed by Albertini et al. (2006); most of these will be generally of concern for any sensitive group. Specifically, there are significant challenges associated with estimating and interpreting toxicant exposures and health risks from biomonitoring data. The science of detecting contaminants has outpaced the science of interpreting public health implications of measured internal exposure. Though low levels of environmental contaminants can be measured in tissues of children and fetuses, it is not always known whether the measured exposure leads to an adverse health outcome. One of the most difficult questions remains—There exists a multiplicity of contaminants, which may interact in an additive, synergistic, or possibly antagonistic manner. Assessing one chemical at a time ignores this reality. In addition, information on exposure pathways is often required to link biomonitoring results to contaminant sources and to reduce exposures and risks.
Barr et al. (2005) prioritize for each life stage preferred biological matrices for assessing exposure to different classes of environmental chemicals. This prioritization scheme is based on matrix availability, time period of concern for a particular exposure or health effect, and properties of the environmental chemicals to be monitored. The authors point out that the scheme was developed specifically to address the chemicals and developmental time points of interest to the National Children's Study. However, this type of prioritization scheme is well conceived, and could be extended to address chemicals and time points of interest for any sensitive population.
DISCUSSION
The focus of this paper is to use our knowledge regarding pediatric subjects as a sensitive population to guide the discussion of three fundamental questions: (1) What scientific issues need to be considered in defining and characterizing a sensitive subpopulation? (2) What scientific data gaps exist in the procedures and approaches for assessing risk in sensitive populations? and (3) How well can existing procedures and approaches for assessing risk in the pediatric population as a sensitive subpopulation be extrapolated to other population groups?
The major scientific factors relating to pediatric sensitivity have been categorized into five groupings: physiological and behavioral parameters, pharmacokinetics, pharmacodynamics, genetics, and exposure. As a general statement, in all five categories used to characterize the pediatric population, lack of baseline data (as a function of life stage) on physiological processes, constitutive activity of key metabolic enzymes, DNA repair capacity and fidelity, gene expression and protein characterization, and behaviors related to exposure reduce the ability to qualitatively and quantitatively predict differences in sensitivity. In addition, lack of these same data also makes it difficult to interpret and quantify genetic, cultural, and environmental factors that alter baseline response and increase vulnerability. While acknowledging these limitations in each category, there are also lessons to be learned that translate to a broader description of what approaches could be used to identify and characterize other sensitive populations. In the following discussion, each category is evaluated for the three key questions that served as the impetus for this investigation.
There are lessons extracted from the pediatric population that inform the implications of physiological and kinetic differences between pediatric subjects and other life stages. This has been demonstrated through direct measurement of physiologic and metabolic capacity, adverse reactions to drugs due to modified phase 1 and phase 2 metabolism, and, in the laboratory, age-dependent differences in physiological capacities and enzyme content within human tissues and differences between adult-only exposed and fetal/perinatally exposed animals in toxic response for a variety of chemicals.
To characterize the potential differences in risk between subgroups of the general population, it is important to understand how these populations differ in the ontogeny of metabolizing enzymes in key metabolizing organs, as well as the rate of metabolism for each of the enzymes likely to alter a chemical in the body. This is not an easy task. First, defining the “average” response from which to develop baseline information between groups is problematic because metabolic capacity can be altered by genetic and cultural differences, as well as concomitant exposures (e.g., drugs) that can impact pharmacokinetics and pharmacodynamics through multiple mechanisms. The genetic differences between individuals can lead to complicated differences in metabolism depending upon whether that genetic difference is in the promoter region of a gene or in the transcribed region (Portier and Bell, 1998). The cultural influences on metabolism arise through differences in diets and other exposures that can serve to inhibit, induce, or compete for key enzymatic activity, and these would need to be quantified in each population.
Given these observations, it is reasonable to presume that in the absence of scientific data, the pediatric and other subpopulations could be differentially sensitive to any drug or chemical. This presumption has led to the development of scientific databases that guide the evaluation of risks to the pediatric population for given exposures, and in the absence of that data, suggest default approaches that appropriately protect this population. By extrapolation, such scientific databases to guide evaluation of risk to other populations would be equally helpful. In addition, physiologically based pharmacokinetic models are seeing increased use in risk modeling and assessment, and the lessons learned from the kinetic differences observed in the pediatric population can and are being applied to other potentially sensitive populations. Filling in the knowledge gaps enumerated above will improve this process.
Pharmacodynamic susceptibilities seen in the pediatric population derive predominantly from the fact that their bodies are still in a developmental state with a different physiology from adults (e.g., higher breathing rates), higher cellular replication rates, ongoing tissue organization, and highly specialized cellular differentiation occurring only during development. Combining these processes with certain environmental insults that interact with them leads to the potential for sensitivity. Key among these processes are those governing DNA fidelity, gene expression and transcription, developmental differentiation (e.g., processes governing maturation of the immune system, processes governing apoptosis during development), and cellular migration. For all these processes, there are parallels in adults that can be informed by what we know from studies in the pediatric population. In addition, there can be long-term consequences of pharmacodynamic changes during development that create lifetime susceptibilities that can also be used to inform risk assessments.
There are genetic polymorphisms that result in increased risk of disease. In addition, there are genetic polymorphisms that, when linked with environmental exposures, alter disease risk from the exposure. These gene-environment interactions can affect both pediatric and adult subjects, but children may exhibit differential sensitivity relative to adults for a given gene-environment interaction based on expression levels of the gene product in question. Much of the knowledge available on genetic polymorphisms, environmental exposure, and disease in humans has focused on polymorphisms of key metabolizing enzymes for which the polymorphism results in altered metabolism of a xenobiotic resulting in a change in risk from exposure. The greatest focus in this area for the pediatric population has been in the area of genetic polymorphisms that alter the susceptibility of pediatric patients to the environmental triggers that initiate asthma attacks.
Information on the alteration of risks in the pediatric population from gene-environment interactions is limited. The majority of data in this area have been generated in adults and have focused on key metabolism genes. Hence, in this case, extrapolation from the adult to the pediatric subject is required, but can be problematic. However, with good baseline data, it should be possible to make a reasonable scientific judgment of the magnitude of the pediatric risk relative to the adult based on changes in the activity of the enzyme. Data quantifying such changes are becoming increasingly available. The implications for pediatric disease could be very different for a given genetic polymorphism and exposure due to differential expression of genes in different organs in the developing human as compared to the adult.
Lastly, pediatric behaviors and environments are different than those of adults. Given detailed models of pediatric and adult behaviors that allow for accurate exposure analyses, it is possible to more fully understand the exposure-driven differences in sensitivity between populations, by using their individual susceptibility linked to the detailed exposure analyses. Scientifically, the challenges here are in evaluating the behavior patterns, which can be quite varied and may be modified by a large number of additional factors such as socioeconomic status. In addition, the predominant route of exposure may be different for pediatric subjects versus adults, leading to differential sensitivities in organs and tissues.
Geographical, cultural, ethnic, and economic factors are all likely to play a role in behavior and in environmental exposures. Because these behaviors also are linked with age and because the vulnerability is also likely to change with age, understanding this interaction is key to determining vulnerability. Finally, as noted earlier, because the actual routes of exposure could change with life stage, a better understanding of the risks from exposures to environmental agents through different routes is needed, as are tools that will allow for extrapolations from one route to another in a life-stage–dependent manner.
In summary, this review of approaches and methods for assessing risk to the pediatric population has outlined a number of general issues pertaining to the identification and evaluation of vulnerable human populations. Below are key points:
The pediatric population has been studied from a number of scientific perspectives relative to their potential vulnerability to environmental exposures, and this knowledge should be considered when evaluating potential vulnerability of other age groups and populations.
Windows of sensitivity exist within the pediatric population that are defined by transient differences in pharmacokinetics, pharmacodynamics, physiology, behavior, and/or exposure. Transient differences in these same parameters may be useful in defining windows of sensitivity in other populations. Knowing the MOA of a given toxicant can help define a window of susceptibility for different life stages.
Determination of sensitivity to any age group or population is dependent on multiple factors including genetic influences, pharmacokinetics and pharmacodynamics, behavior, exposure, and internal dosimetry. Collectively, these key determinants, and perhaps others yet to be defined, should be considered when evaluating potential vulnerability and impact on health risk.
The MOA of many neurotoxicants appears to be highly dependent on specific properties of the developing nervous system. The extrapolation of these data to assist in the definition and characterization of other sensitive populations will require more knowledge about MOA and the specifics of developmental susceptibilities of the nervous system.
The developing immune system is more susceptible and more sensitive to the effects of immunotoxicants, and adverse effects of toxicant exposure are often more persistent than in the mature immune system. Better understanding of the greater susceptibility of the developing organism may enhance the ability to predict adverse effects in susceptible subpopulations of adults with risk factors for decreased immunocompetence (e.g., advanced age, pregnancy, therapeutic and recreational drug use, stress).
The pediatric population may be more susceptible to the mutagenic effects of oncogenic exposures, and this differential susceptibility could result in different effect levels at different exposures. Furthermore, pediatric cancers and their cures can make this population group susceptible to other exposures and increase disease risk (including for second cancers) in adulthood.
The integration of life-stage–dependent pharmacokinetic and pharmacodynamic data into computational models has improved the utility of these tools for risk assessment. Similar knowledge regarding other potentially sensitive populations should be of equal value and improve our ability to assess risk for these subpopulations.
FUNDING
HESI Emerging Issues Subcommittee.
Acknowledgments
All authors declare they have no competing financial interest. HESI is a global branch of ILSI, a public, nonprofit scientific foundation with branches throughout the world. HESI provides an international forum to advance the understanding and application of scientific issues related to human health, toxicology, risk assessment, and the environment. HESI is widely recognized among scientists from government, industry, and academia as an objective, science-based organization within which important issues of mutual concern can be discussed and resolved in the interest of improving public health. As part of its public benefit mandate, HESI's activities are carried out in the public domain, generating data and other information for broad scientific use and application. Like all HESI committees, the HESI Subcommittee on Risk Assessment for Sensitive Populations was composed of a strong group of interdisciplinary, multisector, international scientists from government, academia, and industry. HESI's “tripartite” approach to scientific consensus is a proven mechanism for addressing public health issues, particularly when those issues are of mutual concern to multiple sectors and are of international importance. More information about HESI and the Subcommittee can be accessed at www.hesiglobal.org.
References
- Albertini R, Bird M, Doerrer N, Needham L, Robison S, Sheldon L, Zenick H. The use of biomonitoring data in exposure and human health risk assessment. Environ. Health Perspect. 2006;114:1755–1762. doi: 10.1289/ehp.9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcorn J, McNamara PJ. Ontogeny of hepatic and renal systemic clearance pathways in infants: part I. Clin. Pharmacokinet. 2002;41:959–998. doi: 10.2165/00003088-200241120-00003. [DOI] [PubMed] [Google Scholar]
- American Cancer Society. Cancer Facts and Figures. Atlanta, GA: American Cancer Society; 2008. [Google Scholar]
- Ames BN, Gold LS. Chemical carcinogenesis: too many rodent carcinogens. Proc. Natl. Acad. Sci. U.S.A. 1990;87:7772–7776. doi: 10.1073/pnas.87.19.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LM, Diwan BA, Fear NT, Roman E. Critical windows of exposure for children's health: cancer in human epidemiological studies and neoplasms in experimental animal models. Environ. Health Perspect. 2000;108(Suppl. 3):573–594. doi: 10.1289/ehp.00108s3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr DB, Wang RY, Needham LL. Biologic monitoring of exposure to environmental chemicals throughout the life stages: requirements and issues for consideration for the National Children's Study. Environ. Health Perspect. 2005;113:1083–1091. doi: 10.1289/ehp.7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter ZE, Bayliss MK, Beaune PH, Boobis AR, Carlile DJ, Edwards RJ, Houston JB, Lake BG, Lipscomb JC, Pelkonen OR, et al. Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr. Drug Metab. 2007;8:33–45. doi: 10.2174/138920007779315053. [DOI] [PubMed] [Google Scholar]
- Barter ZE, Chowdry JE, Harlow JR, Snawder JE, Lipscomb JC, Rostami-Hodjegan A. Covariation of human microsomal protein per gram of liver with age: absence of influence of operator and sample storage may justify interlaboratory data pooling. Drug Metab. Dispos. 2008;36:2405–2409. doi: 10.1124/dmd.108.021311. [DOI] [PubMed] [Google Scholar]
- Bearer CF. How are children different from adults? Environ. Health Perspect. 1995;103:7–12. doi: 10.1289/ehp.95103s67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck BD, Mattuck RL, Bowers TS, Cohen JT, O'Flaherty E. The development of a stochastic physiologically-based pharmacokinetic model for lead. Sci. Total Environ. 2001;274:15–19. doi: 10.1016/s0048-9697(01)00728-8. [DOI] [PubMed] [Google Scholar]
- Bellinger DC. Children's cognitive health: the influence of environmental chemical exposures. Altern. Ther. Health Med. 2007;13:S140–S144. [PubMed] [Google Scholar]
- Bleyer A, Viny A, Barr R. Cancer in 15- to 29-year-olds by primary site. Oncologist. 2006;11:590–601. doi: 10.1634/theoncologist.11-6-590. [DOI] [PubMed] [Google Scholar]
- Boersma ER, Lanting CI. Environmental exposure to polychlorinated biphenyls (PCBs) and dioxins. Consequences for long-term neurological and cognitive development of the child lactation. Adv. Exp. Med. Biol. 2000;478:271–287. [PubMed] [Google Scholar]
- Boice JD, Jr, Miller RW. Childhood and adult cancer following intrauterine exposure to ionizing radiation. Teratology. 1999;59:227–233. doi: 10.1002/(SICI)1096-9926(199904)59:4<227::AID-TERA7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Boice JD, Jr, Tawn EJ, Winther JF, Donaldson SS, Green DM, Mertens AC, Mulvihill JJ, Olsen JH, Robison LL, Stovall M. Genetic effects of radiotherapy for childhood cancer. Health Phys. 2003;85:65–80. doi: 10.1097/00004032-200307000-00013. [DOI] [PubMed] [Google Scholar]
- Brenner SS, Klotz U. p-Glycoprotein function in the elderly. Eur. J. Clin. Pharmacol. 2004;60:97–102. doi: 10.1007/s00228-004-0733-4. [DOI] [PubMed] [Google Scholar]
- Brent RL. Biological factors related to male mediated reproductive and developmental toxicity. In: Olsah AF, Mattison DR, editors. Male-Mediated Developmental Toxicity. New York: Plenum Press; 1994. pp. 209–242. [Google Scholar]
- Brent RL. Utilization of developmental basic science principles in the evaluation of reproductive risks from pre- and post-conception environmental radiation exposures. 1999 doi: 10.1002/(SICI)1096-9926(199904)59:4<182::AID-TERA2>3.0.CO;2-H. Paper presented at: Thirty-third Annual Meeting of the National Council on Radiation Protection and Measurements: the Effects of Pre- and Post-conception Exposure to Radiation, April 2–3, 1997, Arlington, VA. Teratology 59, 182–204. [DOI] [PubMed] [Google Scholar]
- Brent R. Lauriston S. Taylor lecture: fifty years of scientific research: the importance of scholarship and the influence of politics and controversy. Health Phys. 2007;93:348–379. doi: 10.1097/01.HP.0000282111.66056.c2. [DOI] [PubMed] [Google Scholar]
- Brent RL, Tanski S, Weitzman M. A pediatric perspective on the unique vulnerability and resilience of the embryo and child to environmental toxicants: the importance of rigorous research concerning age and agent. Pediatrics. 2004;113:935–944. [PubMed] [Google Scholar]
- Brent RL, Weitzman M. The vulnerability, sensitivity, and resiliency of the developing embryo, infant, child, and adolescent to the effects of environmental chemicals, drugs, and physical agents as compared to the adult. Pediatrics. 2004;113:933–1172. [Google Scholar]
- Brown RC, Barone S, Jr, Kimmel CA. Children's health risk assessment: incorporating a lifestage approach into the risk assessment process. Birth Defects Res. (Part B) 2008;83:511–521. doi: 10.1002/bdrb.20172. [DOI] [PubMed] [Google Scholar]
- Byrne J. Long-term genetic and reproductive effects of ionizing radiation and chemotherapeutic agents on cancer patients and their offspring. Teratology. 1999;59:210–215. doi: 10.1002/(SICI)1096-9926(199904)59:4<210::AID-TERA4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, Gardina P, Hubbard J, Turpaz Y, Langaee TY, Eby C, et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111:4106–4112. doi: 10.1182/blood-2007-11-122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TW, Pan AY. Cumulative environmental changes, skewed antigen exposure, and the increase of allergy. Adv. Immunol. 2008;98:39–83. doi: 10.1016/S0065-2776(08)00402-1. [DOI] [PubMed] [Google Scholar]
- Clewell HJ, Gentry PR, Covington TR, Sarangapani R, Teeguarden JG. Evaluation of the potential impact of age- and gender-specific pharmacokinetic differences on tissue dosimetry. Toxicol. Sci. 2004;79:381–393. doi: 10.1093/toxsci/kfh109. [DOI] [PubMed] [Google Scholar]
- Clewell RA, Merrill EA, Gearhart JM, Robinson PJ, Sterner TR, Mattie DR, Clewell HJ., 3rd Perchlorate and radioiodide kinetics across life stages in the human: using PBPK models to predict dosimetry and thyroid inhibition and sensitive subpopulations based on developmental stage. J. Toxicol. Environ. Health A. 2007;70:408–428. doi: 10.1080/15287390600755216. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Anderson TA, de Oliveria LM, Arnold LL. Tumorigenicity of sodium ascorbate in male rats. Cancer Res. 1998;58:2557–2561. [PubMed] [Google Scholar]
- Cohen SM, Garland EM, Cano M, St. John MK, Khachab M, Wehner JM, Arnold LL. Effects of sodium ascorbate, sodium saccharin and ammonium chloride in the male rat urinary bladder. Carcinogenesis. 1995;16:2743–2750. doi: 10.1093/carcin/16.11.2743. [DOI] [PubMed] [Google Scholar]
- Cohen Hubal EA, Moya J, Selevan SG. A lifestage approach to assessing children's exposure. Birth Defects Res. (Part B) 2008;83:522–529. doi: 10.1002/bdrb.20173. [DOI] [PubMed] [Google Scholar]
- Cohen Hubal EA, Sheldon LS, Burke JM, McCurdy TR, Berry MR, Rigas ML. Children's exposure assessment: a review of factors influencing children's exposure, and the data available to characterize and assess that exposure. Environ. Health Perspect. 2000a;108:475–486. doi: 10.1289/ehp.108-1638158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Hubal EA, Sheldon LS, Zufall MJ, Burke JM, Thomas K. The challenge of assessing children's residential exposure to pesticides. J. Exp. Anal. Environ. Epidemiol. 2000b;10:638–649. doi: 10.1038/sj.jea.7500128. [DOI] [PubMed] [Google Scholar]
- Cole TB, Jampsa RL, Walter BJ, Arndt TL, Richter RJ, Shih DM, Tward A, Lusis AJ, Jack RM, Costa LG, et al. Expression of human paraoxonase (PON1) during development. Pharmacogenetics. 2003;13:357–364. doi: 10.1097/00008571-200306000-00007. [DOI] [PubMed] [Google Scholar]
- Cole TB, Walter BJ, Shih DM, Tward AD, Lusis AJ, Timchalk C, Richter RJ, Costa LG, Furlong CE. Toxicity of chlorpyrifos and chlorpyrifos oxon in a transgenic mouse model of the human paraoxonase (PON1) Q192R polymorphism. Pharmacogenet. Genom. 2005;15:589–598. doi: 10.1097/01.fpc.0000167327.08034.d2. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Cory-Slechta DA. Studying toxicants as single chemicals: does this strategy adequately identify neurotoxic risk? Neurotoxicol. 2005;26:491–510. doi: 10.1016/j.neuro.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Cresteil T, Beaune P, Kremers P, Flinois JP, Leroux JP. Drug-metabolizing enzymes in human foetal liver: partial resolution of multiple cytochromes P450. Pediatric Pharmacol. 1982;2:199–207. [PubMed] [Google Scholar]
- D'Amato G, Liccardi G, D'Amato M, Holgate S. Environmental risk factors and allergic bronchial asthma. Clin. Exp. Allergy. 2005;35:1113–1124. doi: 10.1111/j.1365-2222.2005.02328.x. [DOI] [PubMed] [Google Scholar]
- Daston G, Faustman E, Ginsberg G, Fenner-Crisp P, Olin S, Sonawane B, Bruckner J, Breslin W, McLaughlin TJ. A framework for assessing risks to children from exposure to environmental agents. Environ. Health Perspect. 2004;112:238–256. doi: 10.1289/ehp.6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deFur PL, Evans GW, Cohen Hubal EA, Kyle AD, Morello-Frosch RA, Williams D. Vulnerability as a function of individual and group resources in cumulative risk assessment. Environ. Health Perspect. 2007;115:817–824. doi: 10.1289/ehp.9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietert RR, Piepenbrink MS. Perinatal immunotoxicity: why adult exposure assessment fails to predict risk. Environ. Health Perspect. 2006a;114:477–483. doi: 10.1289/ehp.8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietert RR, Piepenbrink MS. Lead and immune function. Crit. Rev. Toxicol. 2006b;36:359–385. doi: 10.1080/10408440500534297. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Djemli A, Van Vliet G, Delvin EE. Congenital hypothyroidism: from paracelsus to molecular diagnosis. Clin. Biochem. 2006;39:511–518. doi: 10.1016/j.clinbiochem.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Done AK. Developmental pharmacology. Clin. Pharmacol. Ther. 1964;5:432–479. doi: 10.1002/cpt196454432. [DOI] [PubMed] [Google Scholar]
- Eaton DL. Biotransformation enzyme polymorphism and pesticide susceptibility. Neurotoxicology. 2000;21:101–112. [PubMed] [Google Scholar]
- Edginton AN, Schmitt W, Voith B, Willmann S. A mechanistic approach for the scaling of clearance in children. Clin. Pharmacokinet. 2006;45:683–704. doi: 10.2165/00003088-200645070-00004. [DOI] [PubMed] [Google Scholar]
- Estrada L, Kanelakis KC, Levy GN, Weber WW. Tissue- and gender-specific expression of N-acetyltransferase 2 (Nat2*) during development of the outbred mouse strain CD-1. Drug. Metab. Dispos. 2000;28:139–146. [PubMed] [Google Scholar]
- Euling SY, Kimmel CA. Developmental stage sensitivity and mode of action information for androgen agonists and antagonists. Sci. Total Environ. 2001;274:103–113. doi: 10.1016/s0048-9697(01)00736-7. [DOI] [PubMed] [Google Scholar]
- Faustman EM, Silbernagel SM, Fenske RA, Burbacher TM, Ponce RA. Mechanisms underlying children's susceptibility to environmental toxicants. Environ. Health Perspect. 2000;108(Suppl. 1):13–21. doi: 10.1289/ehp.00108s113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestone M, Moya J, Cohen Hubal E, Zartarian V. Identifying childhood age groups for exposure assessments and monitoring. Risk Anal. 2007;27:701–714. doi: 10.1111/j.1539-6924.2007.00918.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg G, Hattis D, Russ A, Sonawane B. Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: implications for assessing children's risks from environmental agents. J. Toxicol. Environ. Health A. 2004a;67:297–329. doi: 10.1080/15287390490273550. [DOI] [PubMed] [Google Scholar]
- Ginsberg G, Slikker W, Jr, Bruckner J, Sonawane B. Incorporating children's toxicokinetics into a risk framework. Environ. Health Perspect. 2004b;112:272–283. doi: 10.1289/ehp.6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman LR. Children—unique and vulnerable environmental risks facing children and recommendations for response. Environ. Health Perspect. 1995;103(Suppl. 6):13–18. doi: 10.1289/ehp.95103s613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilarte TR. PB2+ inhibits NMDA receptor function at high and low affinity sites: developmental and regional brain expression. Neurotoxicology. 1997;18:43–51. [PubMed] [Google Scholar]
- Hakkola J, Pasanen M, Purkunen R, Saarikoski S, Pelkonen O, Mäenpää J, Rane A, Raunio H. Expression of xenobiotic-metabolizing cytochrome P450 forms in human adult and fetal liver. Biochem. Pharmacol. 1994;48:59–64. doi: 10.1016/0006-2952(94)90223-2. [DOI] [PubMed] [Google Scholar]
- Hall E. Lessons we have learned from our children: cancer risks from diagnostic radiology. Pediatr. Radiol. 2002;32:700–706. doi: 10.1007/s00247-002-0774-8. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina: an association of maternal stilbestrol therapy with tumor appearing in young women. N. Engl. J. Med. 1971;284:878–881. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- Hines RN. Ontogeny of drug metabolism enzymes and implications for adverse drug reactions. Pharmacol. Ther. 2008;118:250–267. doi: 10.1016/j.pharmthera.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Holladay SD, Smialowicz RJ. Development of the murine and human immune system: differential effects of immunotoxicants depend on time of exposure. Environ. Health Perspect. 2000;108(Suppl. 3):463–473. doi: 10.1289/ehp.00108s3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JY, Yang CS. Genetic polymorphism of cytochrome P450 as a biomarker of susceptibility to environmental toxicity. Environ. Health Perspect. 1997;105(Suppl. 4):759–762. doi: 10.1289/ehp.97105s4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 2006;45:931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- Johnsrud EK, Koukouritaki SB, Divakaran K, Brunengraber L, Hines RN, McCarver DG. Human hepatic CYP2E1 expression during development. J. Pharmacol. Exp. Ther. 2003;307:402–407. doi: 10.1124/jpet.102.053124. [DOI] [PubMed] [Google Scholar]
- Kearns GL, Robinson PK, Wilson JT, Wilson-Costello D, Knight GR, Ward RM. Cisapride disposition in neonates and infants: in vivo reflection of cytochrome P450 3A4 ontogeny. Clin. Pharmacol. Ther. 2003;74:312–325. doi: 10.1016/S0009-9236(03)00225-X. [DOI] [PubMed] [Google Scholar]
- Kimmel GL. An overview of children as a special population—relevance to predictive biomarkers. Toxicol. Appl. Pharmacol. 2005;206:215–218. doi: 10.1016/j.taap.2004.06.032. [DOI] [PubMed] [Google Scholar]
- Kitada M, Taneda M, Itahashi K, Kamataki T. Four forms of cytochrome P-450 in human fetal liver: purification and their capacity to activate promutagens. Jpn. J. Cancer Res. 1991;82:426–432. doi: 10.1111/j.1349-7006.1991.tb01866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koukouritaki SB, Manro JR, Marsh SA, Stevens JC, Rettie AE, McCarver DG, Hines R, N Developmental expression of human hepatic CYP2C9 and CYP2C19. J. Pharmacol. Exp. Ther. 2004;308:965–974. doi: 10.1124/jpet.103.060137. [DOI] [PubMed] [Google Scholar]
- Kumari M, Ticku MK. Ethanol and regulation of the NMDA receptor subunits in fetal cortical neurons. J. Neurochem. 1998;70:1467–1473. doi: 10.1046/j.1471-4159.1998.70041467.x. [DOI] [PubMed] [Google Scholar]
- Kwabi-Addo B, Chung W, Shen L, Ittmann M, Wheeler T, Jelinek J, Issa JP. Age-related DNA methylation changes in normal human prostate tissues. Clin. Cancer Res. 2007;13:3796–3802. doi: 10.1158/1078-0432.CCR-07-0085. [DOI] [PubMed] [Google Scholar]
- Kyle AD, Woodruff TJ, Axelrad DA. Integrated assessment of environment and health: America's children and the environment. Environ. Health Perspect. 2006;114:447–452. doi: 10.1289/ehp.8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea PM, IV, Faden AI. Metabotropic glutamate receptor subtype 5 antagonists MPEP and MTEP. CNS Drug Rev. 2006;12:149–166. doi: 10.1111/j.1527-3458.2006.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee QP, Fantel AG, Juchau MR. Human embryonic cytochrome P450S: phenoxazone ethers as probes for expression of functional isoforms during organogenesis. Biochem. Pharmacol. 1991;42:2377–2385. doi: 10.1016/0006-2952(91)90244-y. [DOI] [PubMed] [Google Scholar]
- Lewin B. Gene VI. Oxford: Oxford University Press; 1997. [Google Scholar]
- Liao KH, Tan YM, Conolly RB, Borghoff S, J, Gargas ML, Andersen ME, Clewell HJ., 3rd Bayesian estimation of pharmacokinetic and pharmacodynamic parameters in a mode-of-action-based cancer risk assessment for chloroform. Risk Anal. 2007;27:1535–1551. doi: 10.1111/j.1539-6924.2007.00987.x. [DOI] [PubMed] [Google Scholar]
- Lin HJ, Han CY, Lin BK, Hardy S. Slow acetylator mutations in the human polymorphic N-acetyltransferase gene in 786 Asians, blacks, Hispanics, and whites: application to metabolic epidemiology. Am. J. Hum. Genet. 1993;52:827–834. [PMC free article] [PubMed] [Google Scholar]
- Luebke RW, Chen DH, Dietert R, Yang Y, King M, Luster MI. The comparative immunotoxicity of five selected compounds following developmental or adult exposure. J. Toxicol. Environ. Health B Crit. Rev. 2006;9:1–26. doi: 10.1080/15287390500194326. [DOI] [PubMed] [Google Scholar]
- Luebke RW, Parks C, Luster MI. Suppression of immune function and susceptibility to infections in humans: association of immune function with clinical disease. J. Immunotoxicol. 2004;1:15–24. doi: 10.1080/15476910490438342. [DOI] [PubMed] [Google Scholar]
- Makri A, Goveia M, Balbus J, Parkin R. Children's susceptibility to chemicals: a review by developmental stage. J. Toxicol. Environ. Health B Crit. Rev. 2004;7:417–435. doi: 10.1080/10937400490512465. [DOI] [PubMed] [Google Scholar]
- Makris SL, Thompson CM, Euling SY, Selevan SG, Sonawane B. A lifestage-specific approach to hazard and dose-response characterization for children's health risk assessment. Birth Defects Res. (Part B) 2008;83:530–546. doi: 10.1002/bdrb.20176. [DOI] [PubMed] [Google Scholar]
- Mayatepek E, Kohlmüeller D. Transient trimethylaminuria in childhood. Acta Paediatr. 1998;87:1205–1207. doi: 10.1080/080352598750031257. [DOI] [PubMed] [Google Scholar]
- McCarver DG. Applicability of the principles of developmental pharmacology to the study of environmental toxicants. Pediatrics. 2004;113(Suppl. 4):969–972. [PubMed] [Google Scholar]
- Miyamoto K, Nakanishi H, Moriguchi S, Fukuyama N, Eto K, Wakamiya J, Murao K, Arimura K, Osame M. Involvement of enhanced sensitivity of N-methyl-D-aspartate receptors in vulnerability of developing cortical neurons to methylmercury neurotoxicity. Brain Res. 2001;901:252–258. doi: 10.1016/s0006-8993(01)02281-8. [DOI] [PubMed] [Google Scholar]
- Mulvihill J, Connelly RR, Austin DF, Bragg K, Cook JW, Hassinger DD, Holmes FF, Holmes DF. Cancer in offspring of long-term survivors of childhood and adolescent cancer. Lancet. 1987;2:813–817. doi: 10.1016/s0140-6736(87)91012-9. [DOI] [PubMed] [Google Scholar]
- Murry DJ, Crom WR, Reddick WE, Bhargava R, Evans WE. Liver volume as a determinant of drug clearance in children and adolescents. Drug Metab. Dispos. 1995;23:1110–1116. [PubMed] [Google Scholar]
- Nakano M, Kodama Y, Ohtaki K, Nakashima E, Niwa O, Toyoshima M, Nakamura N. Chromosome aberrations do not persist in the lymphocytes or bone marrow cells of mice irradiated in utero or soon after birth. Radiat. Res. 2007;167:693–702. doi: 10.1667/RR0718.1. [DOI] [PubMed] [Google Scholar]
- Napier K. Real risks vs hyped fears. In: Juberg DR, editor. Are Children More Vulnerable to Environmental Chemicals? New York: American Council on Science and Health; 2003. pp. 193–215. [Google Scholar]
- National Academies of Sciences. Pesticides in the Diets of Infants and Children. Washington, DC: National Academies Press; 1993. [PubMed] [Google Scholar]
- National Environmental Justice Advisory Council. Washington, DC.: US Environmental Protection Agency NEJAC Cumulative Risks/Impacts Work Group; 2004. Ensuring risk reduction in communities with multiple stressors: environmental justice and cumulative risks/impacts. Available at: http://www.epa.gov/compliance/resources/publications/ej/nejac/nejac-cum-risk-rpt-122104.pdf. Accessed 2008. [Google Scholar]
- National Research Council. Biological markers in environmental health research. Committee on Biological Markers of the National Research Council. Environ. Health Perspect. 1987;74:3–9. doi: 10.1289/ehp.74-1474499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham LL, Özkaynak H, Whyatt RM, Barr DB, Wang RY, Naeher L, Akland G, Bahadori T, Bradman A, Fortmann R, et al. Exposure assessment in the National Children's Study: introduction. Environ. Health Perspect. 2005;113:1076–1082. doi: 10.1289/ehp.7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel JV. Changing perspectives on the genetic doubling dose of ionizing radiation for humans, mice, and Drosophila. Teratology. 1999;59:216–222. doi: 10.1002/(SICI)1096-9926(199904)59:4<216::AID-TERA5>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Neel JV, Lewis SE. The comparative radiation genetics of humans and mice. Am. Rev. Genet. 1990;24:327–362. doi: 10.1146/annurev.ge.24.120190.001551. [DOI] [PubMed] [Google Scholar]
- Neri M, Ugolini D, Bonassi S, Fucic A, Holland N, Knudsen LE, Srám RJ, Ceppi M, Bocchini V, Merlo DF. Children's exposure to environmental pollutants and biomarkers of genetic damage. II. Results of a comprehensive literature search and meta-analysis. Mutat. Res. 2006;612:14–39. doi: 10.1016/j.mrrev.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Noda T, Todani T, Watanabe Y, Yamamoto S. Liver volume in children measured by computed tomography. Pediatr. Radiol. 1997;27:250–252. doi: 10.1007/s002470050114. [DOI] [PubMed] [Google Scholar]
- Nong A, McCarver DG, Hines RN, Krishnan K. Physiologically-based modeling of inter-child differences in pharmacokinetics on the basis of subject-specific data on hepatic CYP2E1 levels. Toxicol. Appl. Pharmacol. 2006;214:78–87. doi: 10.1016/j.taap.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Nygaard R, Clausen N, Simes MA, Márky I, Skjeldestad FE, Kristinsson JR, Vuoristo A, Wegelius R, Moe PJ. Reproduction following treatment for childhood leukemia: a population-based prospective cohort study of fertility and offspring. Med. Pediatr. Oncol. 1991a;19:459–466. doi: 10.1002/mpo.2950190603. [DOI] [PubMed] [Google Scholar]
- Nygaard R, Garwicz S, Haldorsen T, Hertz H, Jonmundsson GK, Lanning M, Moe PJ. Second malignant neoplasms in patients treated for childhood leukemia. A population-based cohort study from the Nordic countries. The Nordic Society of Pediatric Oncology and Hematology (NOPHO) Acta Paediatr. Scand. 1991b;80:1220–1228. doi: 10.1111/j.1651-2227.1991.tb11812.x. [DOI] [PubMed] [Google Scholar]
- Özkaynak H, Whyatt RM, Needham LL, Akland G, Quackenboss J. Exposure assessment implications for the design and implementation of the National Children's Study. Environ. Health Perspect. 2005;113:1108–1115. doi: 10.1289/ehp.7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasanen M, Pelkonen O, Kauppila A, Park SS, Friedman FK, Gelboin HV. Characterization of human fetal hepatic cytochrome P-450-associated 7-ethoxyresofufin O-deethylase and aryl hydrocarbon hydroxylase activities by monoclonal antibodies. Dev. Pharmacol. Ther. 1987;10:125–132. doi: 10.1159/000457737. [DOI] [PubMed] [Google Scholar]
- Pearce RE, Gotschall RR, Kearns GL, Leeder JS. Cytochrome P450 involvement in the biotransformation of cisapride and racemic norcisapride in vitro: differential activity of individual CYP3A isoforms. Drug Metab. Dispos. 2001;29:1548–1554. [PubMed] [Google Scholar]
- Piccinni MP, Scaletti C, Maggi E, Ramagnani S. Role of hormone-controlled Th1- and Th2-type cytokines in successful pregnancy. J. Neuroimmunol. 2000;109:30–33. doi: 10.1016/s0165-5728(00)00299-x. [DOI] [PubMed] [Google Scholar]
- Pohl HR, van Engelen JG, Wilson J, Sips AJ. Risk assessment of chemicals and pharmaceuticals in the pediatric population: a workshop report. Reg. Toxicol. Pharmacol. 2005;42:83–95. doi: 10.1016/j.yrtph.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Popke EJ, Allen RR, Pearson EC, Hammond TG, Paule MG. Differential effects of two NMDA receptor antagonists on cognitive-behavioral development in nonhuman primates I. Neurotoxicol. Teratol. 2001a;23:319–332. doi: 10.1016/s0892-0362(01)00156-8. [DOI] [PubMed] [Google Scholar]
- Popke EJ, Allen RR, Pearson EC, Hammond TG, Paule MG. Differential effects of two NMDA receptor antagonists on cognitive-behavioral performance in young nonhuman primates II. Neurotoxicol. Teratol. 2001b;23:333–347. doi: 10.1016/s0892-0362(01)00138-6. [DOI] [PubMed] [Google Scholar]
- Portier CJ, Bell DA. Genetic susceptibility: significance in risk assessment. Toxicol. Lett. 1998;102-103:185–189. doi: 10.1016/s0378-4274(98)00305-1. [DOI] [PubMed] [Google Scholar]
- Preston DL, Cullings H, Suyama A, Funamoto S, Nishi N, Soda M, Mabuchi K, Kodama K, Kasagi F, Shore RE. Solid cancer incidence in atomic bomb survivors exposed in utero or as young children. J. Natl. Cancer Inst. 2008;100:428–436. doi: 10.1093/jnci/djn045. [DOI] [PubMed] [Google Scholar]
- Reichrath J. DNA repair mechanisms: underestimated key players for cancer prevention and therapy. J. Mol. Histol. 2006;37:179–181. [Google Scholar]
- Reiss R, Anderson EL, Lape J. A framework and case study for exposure assessment in the Voluntary Children's Chemical Evaluation Program. Risk Anal. 2003;23:1069–1084. doi: 10.1111/1539-6924.00383. [DOI] [PubMed] [Google Scholar]
- Scheetz AJ, Constantine-Paton M. Modulation of NMDA receptor function: implications for vertebrate neural development. FASEB J. 1994;8:745–752. doi: 10.1096/fasebj.8.10.8050674. [DOI] [PubMed] [Google Scholar]
- Scheuplein R, Charnley G, Dourson M. Differential sensitivity of children and adults to chemical toxicity. Biological basis. Reg. Toxicol. Pharmacol. 2002;35:429–447. doi: 10.1006/rtph.2002.1558. [DOI] [PubMed] [Google Scholar]
- Sconce EA, Khan TI, Wynne HA, Avery P, Monkhouse L, King BP, Wood P, Kesteven P, Daly AK, Kamali F. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood. 2005;106:2329–2333. doi: 10.1182/blood-2005-03-1108. [DOI] [PubMed] [Google Scholar]
- Seed J, Carney EW, Corley RA, Crofton KM, DeSesso JM, Foster PM, Kavlock R, Kimmel G, Klaunig J, Meek ME, et al. Overview: using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit. Rev. Toxicol. 2005;35:663–672. doi: 10.1080/10408440591007133. [DOI] [PubMed] [Google Scholar]
- Selevan SG, Kimmel CA, Mendola P. Identifying critical windows of exposure for children's health. Environ. Health Perspect. 2000;108(Suppl. 3):451–455. doi: 10.1289/ehp.00108s3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slikker W, Jr, Andersen ME, Bogdanffy MS, Bus JS, Cohen SD, Conolly RB, David RM, Doerrer NG, Dorman DC, Gaylor DW, et al. Dose-dependent transitions in mechanisms of toxicity. Toxicol. Appl. Pharmacol. 2004;201:203–225. doi: 10.1016/j.taap.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Slikker W, Jr, Chang LW. Handbook of Developmental Neurotoxicology. San Diego, CA: Academic Press; 1998. [Google Scholar]
- Stephenson T. How children's responses to drugs differ from adults. Br. J. Clin. Pharmacol. 2005;59:670–673. doi: 10.1111/j.1365-2125.2005.02445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AM. The carcinogenic effects of low-level radiation: a reappraisal of epidemiologists’ methods and observations. Health Phys. 1973;24:223–240. doi: 10.1097/00004032-197302000-00002. [DOI] [PubMed] [Google Scholar]
- Stewart AM, Kneale GW. Radiation dose effects in relation to obstetric x-rays and childhood cancers. Lancet. 1970;1:1185–1188. doi: 10.1016/s0140-6736(70)91782-4. [DOI] [PubMed] [Google Scholar]
- Stewart AM, Webb D, Hewitt D. A survey of childhood malignancies. Br. Med. J. 1958;1:1495–1508. doi: 10.1136/bmj.1.5086.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham LS, Goh BC, Nafziger A, Guo JY, Wang LZ, Soong R, Lee SC. A warfarin-dosing model in Asians that uses single-nucleotide polymorphisms in vitamin K epoxide reductase complex and cytochrome P450 2C9. Clin. Pharmacol. Ther. 2006;80:346–355. doi: 10.1016/j.clpt.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Timchalk C, Nolan RJ, Mendrala AL, Dittenber DA, Brzak KA, Mattsson JL. A physiologically-based pharmacokinetic and pharmacodynamic (PBPK/PD) model for the organophosphate insecticide chlorpyrifos in rats and humans. Toxicol. Sci. 2002;66:34–53. doi: 10.1093/toxsci/66.1.34. [DOI] [PubMed] [Google Scholar]
- Treluyer J-M, Rey E, Sonnier M, Pons G, Cresteil T. Evidence of impaired cisapride metabolism in neonates. Br. J. Clin. Pharmacol. 2001;52:419–425. doi: 10.1046/j.0306-5251.2001.01470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Washington, DC.: US Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment; 2003a. Guidelines for carcinogen risk assessment. EPA/630/R-03/003. Available at: http://www.epa.gov/ncea/raf/cancer2003.htm. Accessed 2008. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Washington, DC.: U.S. Environmental Protection Agency, Risk Assessment Forum; 2003b. Framework for cumulative risk assessment. EPA/630/P-02/001F. Available at: http://cfpub.epa.gov/ncea/raf/recordisplay.cfm?deid=54944. Accessed 2008. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Washington, DC.: U.S. Environmental Protection Agency, Risk Assessment Forum; 2005a. Supplemental guidance for assessing susceptibility from early-life exposure to carcinogens. EPA/630/R-03/003F. Available at: http://www.epa.gov/ttn/atw/childrens_supplement_final.pdf. Accessed 2008. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Washington, DC.: U.S. Environmental Protection Agency, Risk Assessment Forum; 2005b. Guidance on selecting age groups for monitoring and assessing childhood exposures to environmental contaminants. EPA/630/P-03/003F. Available at: http://cfpub2.epa.gov/ncea/cfm/recordisplay.cfm?deid=146583. Accessed 2008. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Washington, DC.: U.S. Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment; 2006a. A framework for assessing health risks of environmental exposures to children. EPA/600/R-05/093F. Available at: http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=158363. Accessed 2008. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Washington, DC.: U.S. Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment; 2006b. Child-specific exposure factors handbook (external review draft). EPA/600/R-06/096A. Available at: http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=56747. Accessed 2008. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Washington, DC.: U.S. Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment; 2008. Physiological parameters database for older adults 1.0 (beta) Available at: http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=188288. Accessed 2008. [Google Scholar]
- U.S. Food and Drug Administration (USFDA) Washington, DC.: U.S. Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research; 2000. E11 clinical investigation of medicinal products in the pediatric population. Available at: http://www.fda.gov/RegulatoryInformation/Guidances/ucm129476.htm. Accessed 2008. [Google Scholar]
- Vingerhoets AJ, Assies J, Goodkin K, Van Heck GL, Bekker MH. Prenatal diethylstilbestrol exposure and self-reported immune-related diseases. Eur. J. Obstet. Gynecol. Reprod. Biol. 1998;77:205–209. doi: 10.1016/s0301-2115(97)00274-1. [DOI] [PubMed] [Google Scholar]
- Wang F, Chang D, Hu FL, Sui H, Han B, Li DD, Zhao YS. DNA repair gene XPD polymorphisms and cancer risk: a meta-analysis based on 56 case-control studies. Cancer Epidemiol. Biomarkers Prev. 2008;17:507–517. doi: 10.1158/1055-9965.EPI-07-2507. [DOI] [PubMed] [Google Scholar]
- Wedlund PJ. The CYP2C19 enzyme polymorphism. Pharmacology. 2000;61:174–183. doi: 10.1159/000028398. [DOI] [PubMed] [Google Scholar]
- Wei Q, Li L, Chen DJ, editors. DNA Repair, Genetic Instability and Cancer. Hackensack, NJ: World Scientific Publishing Co.; 2007. [Google Scholar]
- Weisglas-Kuperus N, Patandin S, Berbers GA, Sas TC, Mulder PG, Sauer PJ, Hooijkaas H. Immunologic effects of background exposure to polychlorinated biphenyls and dioxins in Dutch preschool children. Environ. Health Perspect. 2000;108:1203–1207. doi: 10.1289/ehp.001081203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss CF, Glazko AJ, Weston JK. Chloramphenicol in the newborn infant: a physiological explanation of its toxicity when given in excessive doses. N. Engl. J. Med. 1960;262:787–794. doi: 10.1056/NEJM196004212621601. [DOI] [PubMed] [Google Scholar]
- Willhite CC, Ball GL, McLellan CJ. Derivation of a bisphenol A oral reference dose (RfD) and drinking-water equivalent concentration. J. Toxicol. Environ. Health B Crit. Rev. 2008;11:69–146. doi: 10.1080/10937400701724303. [DOI] [PubMed] [Google Scholar]
- Wilson JMG, Jungner G. Screening for Inborn Errors of Metabolism, Technical Series No. 401. Geneva, Switzerland: World Health Organization; 1968. Principles and practice of screening for disease; p. 163. [Google Scholar]
- Winther JF, Boice JD, Jr, Mulvihill J, Stovall M, Frederiksen K, Tawn EJ, Olsen JH. Chromosomal abnormalities among offspring of childhood-cancer survivors in Denmark: a population-based study. Am. J. Human Genet. 2004;74:1282–1285. doi: 10.1086/421473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood R. Nucleotide excision repair in mammalian cells. J. Biol. Chem. 1997;272:23465–23468. doi: 10.1074/jbc.272.38.23465. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) Geneva, Switzerland: World Health Organization, International Programme on Chemical Safety; 2006. Principles for evaluating health risks in children associated with exposure to chemicals (draft). International Programme on Chemical Safety Environmental Health Criteria document (EHC-237) Available at: http://www.who.int/ipcs/features/ehc/en/index.html. Accessed 2008. [Google Scholar]
- Zaya MJ, Hines RN, Stevens JC. Epirubicin glucuronidation and UGT2B7 developmental expression. Drug Metab. Dispos. 2006;34:2097–2101. doi: 10.1124/dmd.106.011387. [DOI] [PubMed] [Google Scholar]