

Abstract
Aging-associated neurodegenerative diseases significantly influence the quality of life of affected individuals. Genetic approaches, combined with genomic technology, have provided powerful insights into common late-onset diseases, such as age-related macular degeneration (AMD). Here, we discuss current findings on the genetics of AMD to highlight areas of rapid progress and new challenges. We also attempt to integrate available genetic and biochemical data with cellular pathways involved in aging to formulate an integrated model of AMD pathogenesis.
Keywords: protein homeostasis, gene-environment interaction, genetic variation, neurodegeneration, neovascularization, animal models
INTRODUCTION
Many common noninfectious diseases exhibit a more severe clinical presentation in older individuals. These diseases often exhibit complex etiology and can affect different tissues and cell types, with a wide spectrum of clinical outcomes. Prominent aging-associated neurodegenerative diseases are Alzheimer’s disease (AD), Parkinson’s disease (PD), and age-related macular degeneration (AMD), all of which can severely compromise the quality of life and have serious repercussions on both the individual and society at large. These late-onset diseases generally result from the interplay between multiple genetic susceptibility factors and environmental components. Sequencing of the human genome, cataloging of millions of single nucleotide polymorphisms (SNPs) together with the development of a map of common haplo-types, and technological innovations in genotyping are among the major milestones that are facilitating exploration of the genetic basis of common diseases (1, 7, 50). In the field of AMD genetics, these advances have led to the identification of several genetic susceptibility factors and enabled us to start dissecting the relationship between environmental risk factors and the genetic constitution of each individual (66, 118, 148). As a result, new opportunities are emerging for improved understanding of disease pathogenesis that may lead to better management and treatment of AMD. Clinical aspects of AMD are discussed only briefly (for a more in-depth discussion, see Reference 79).
STRUCTURE OF THE HUMAN EYE
To understand the pathobiology of AMD, we briefly describe the structure of the human eye (Figure 1). The perception of light begins in the retina, a part of the central nervous system that is easily accessible to evaluation and relatively amenable to treatment (123). The lens and cornea are optical elements in the eye that focus light on the fovea, a well-defined region of the retina responsible for capturing high-resolution visual information. The fovea is the center of the macula, which is 5–6 mm in diameter, has a dense concentration of photoreceptors, and a ratio of cone to rod photoreceptors that is approximately twice as high as that of the remaining retina. Overall, the human retina has approximately 20 times more rods than cones; rod photoreceptors are highly sensitive to light and allow night vision, whereas cones are responsible for color perception and high visual acuity. Rods and cones are polarized neurons with specialized visual phototransduction proteins that are concentrated in membranous-like structures, the outer segments. These discs have high metabolic demands and undergo continuous renewal in a process controlled by light and circadian rhythms (172). As outer-segment discs are continuously shed at the distal tips of the outer segments and replaced, the retinal pigment epithelium (RPE) is responsible for clearing old shed discs by phagocytosis and supplying both cones and rods with the nutrients and oxygen they need. The RPE also serves as a barrier to the small fenestrated capillaries of the choroid or choriocapillaris, thereby playing a critical role in maintaining photoreceptor health (see Figure 1) (15). A thin stratified extracellular matrix, Bruch’s membrane, separates the RPE from the choriocapillaris.
Figure 1.

(a) Cross-section of the human eye, and schematic of the photoreceptor-retinal pigment epithelium (RPE)-choroidal layers that are affected in AMD. Cone photoreceptors are shown in red, green, or blue. RPE apical processes are intimately associated with photoreceptor outer segments. Two of the rods are shown in the active process of outer segment disc shedding. Melanosomes are shown as dark organelles in RPE cells. (b) Fundus photograph showing the retina of a normal individual. Retinal blood vessels are clearly visible. F, fovea; M, macula; OD, optic disc.
CLINICAL DESCRIPTION OF AMD
AMD is a late-onset, progressive degenerative disease that encompasses a broad spectrum of clinical phenotypes and pathology, primarily affecting the macular region of the human retina. AMD increases in prevalence with advancing age, with a typical onset in the sixth decade. In early stages, AMD causes minimal visual impairment; however, in severe cases it leads to the loss of high-resolution central vision in affected individuals. Numerous definitions and classifications of AMD exist in the literature (13). In this review, we use the Age-Related Eye Disease Study (AREDS) classification of AMD because beneficial preventive therapy decisions can be made based on this classification (4).
An early clinical sign of AMD is the appearance of drusen, which are yellow extracellular deposits of protein and lipid materials beneath the retina (Figure 2). Early AMD is defined by the presence of multiple (>5) small drusen or the presence of at least one intermediate drusen (drusen size of ≥63 μm but <125 μm). Intermediate AMD refers to retina with many intermediate drusen or at least one large drusen (drusen size of ≥125 μm). The large drusen are often accompanied by hyper- or hypopigmentation of the RPE. Advanced AMD, the third category of AMD, includes two different types of late-stage lesions—geographic atrophy (GA) and neovascular (NV) AMD (Figure 2e–h). GA is clinically defined as a discrete area of retinal depigmentation at least 175 μm in diameter with a sharp border and visible choroidal vessels in the absence of neovascularization in the same eye. AMD is a progressive disease resulting from the development of focal loss of retinal photoreceptors and RPE as well as small blood vessels directly beneath the RPE. In NV-AMD, clinical manifestations include serous or hemorrhagic detachment of either the RPE or sensory retina, presence of subretinal fibrous tissue, and eventually widespread RPE atrophy. The onset of vision loss is usually acute with the development of neovascularization under the retina. Neovascular AMD accounts for over 90% of severe central vision loss among patients with AMD (116, 154, 161).
Figure 2.

(a) Fundus photograph of the left eye of a patient with large, “soft” drusen (arrow), indicative of intermediate AMD. The patient has good vision. (b) The optical coherence tomography (OCT) image of the patient in (a) with drusen. This image has the inner retina pointing toward the top with the choroid shown at the bottom of the image, a reverse of our schematic seen in panels (i) and (j). The solid arrow points to corresponding areas of drusen that have caused an elevation of the retinal pigment epithelium, giving it a “bumpy-lumpy” appearance. This section of the OCT shows the center of fovea with the retinal layers shown as a central “foveal depression” (dashed arrow). (c) Fundus photograph of a patient with an elevated RPE detachment, which is the creamy-colored round lesion centered on the macula (outlined by the arrows). The patient still has good vision because the retina has no intraretinal or subretinal fluid. This is another form of advanced AMD that may lead to NV or GA disease. (d) The OCT image of the patient in (c) with elevated RPE detachment (long solid arrow indicates the RPE layer). There is little intraretinal fluid (short solid arrow) and subretinal fluid (dashed arrow) detected. (e) Fundus photograph of the right eye with evidence of advanced AMD, the neovascular form with evidence of subretinal hemorrhage (solid arrow), retinal hard exudates (yellow waxy lesions, dashed arrow), and large drusen. The visual acuity is 20/160. (f) Fluorescein angiogram of patient with neovascular AMD, leakage of the fluorescein dye in the late frames through the abnormal neovascular complex, resulting in hyperfluorescence (white area, solid arrow). The subretinal hemorrhage blocked the dye, resulting in an area of hypofluorescence (dashed arrow). (g) Fundus photograph of patient with the “dry” form of advanced AMD, with GA centered on the macula, a well-defined area of atrophy with the loss of the neuroretina, RPE, and the choriocapillaris. The large vessels within the atrophic area are the remaining vessels of the choroid that can be viewed readily because of the lack of the pigmented layer of the RPE (arrow). Peripheral to the GA are large drusen. The visual acuity is 20/200. (h) The OCT image of the patient in (g) with thinning of the retina and the loss of the RPE and choriocapillaris in the center of the macula (arrow). (i) Schematic of the photoreceptor-RPE-choroid region from an early AMD retina. A large drusen is indicated. (j) Schematic of the photoreceptor-RPE-choroid region from a late AMD retina. Various abnormalities are indicated.
EPIDEMIOLOGY OF AMD
AMD is a leading cause of severe and incurable vision impairment in the United States and other developed countries (31). It accounts for more than 54% of all blindness in the USA, with approximately 1.75 million people of age 40 years or older affected with advanced AMD and another 7.3 million with intermediate AMD (51). Age is the strongest risk factor associated with the development of AMD (3, 112, 140) (Figure 3). The incidence of early AMD, large drusen and pigmentary changes in intermediate AMD, and choroidal neovascularization (CNV) and GA in advanced AMD increase with advancing age (51, 93). The pooled data from two population-based studies reveal an estimated prevalence of advanced AMD of ~0.2% in ages 55 to 64, increasing to 13% in those older than 85 years (140). The number of individuals with AMD is expected to increase worldwide as the population ages and treatments for preventable blindness (such as cataract) become more widely available.
Figure 3.
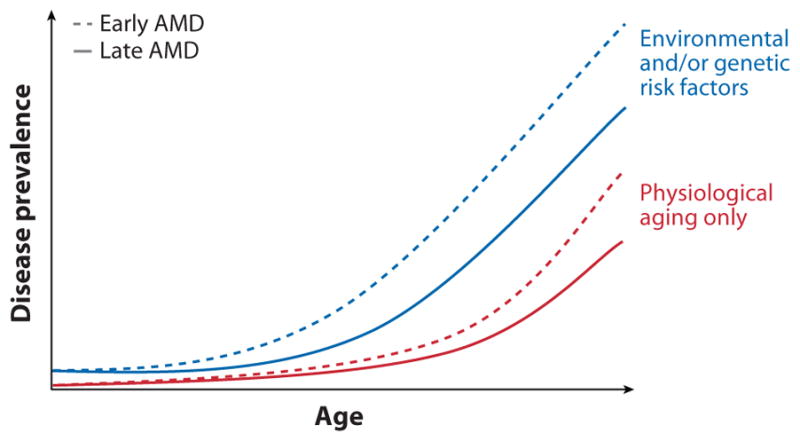
A representation of disease prevalence with age. The incidence of both early and late AMD greatly increases in all individuals at a late age (age 80 and above). We suggest that individuals with genetic susceptibility variants and those who have been exposed to environmental risk factors exhibit the disease at much earlier age (age 60 and above).
Epidemiological studies have revealed differences in the prevalence of advanced AMD among different ethnic groups (51, 3, 141). An analysis of U.S. participants in the Multi-ethnic Study of Atherosclerosis (MESA) showed the prevalence of AMD in persons aged 45 to 85 years to be 2.4% in African-Americans, 4.2% in Hispanics, 4.6% in Chinese-descent individuals, and 5.4% in Caucasians (94).
PATHOLOGY OF AMD
Early pathological changes in AMD include the appearance of basal deposits in Bruch’s membrane, which is made up of the basement membranes of the RPE and the choriocapillaris. These deposits are of two distinct types (59): (a) basal laminar deposits between the RPE and basement membrane, and (b) basal linear deposits, which are composed primarily of membraneous material located external to the RPE basement membrane in Bruch’s membrane and are believed to be more specific to AMD (126). Accumulation of these deposits together with secondary RPE changes may lead to the formation of drusen. Biochemical analysis of drusen has identified glycoconjugates and components found in atherosclerotic plaques including vitronectin, apolipoprotein B and E, alpha-crystallin, complement proteins, and lipids (32, 106, 115).
Drusen can present as small discrete nodules, known as hard drusen, or as large nodules with ill-defined, indistinct boundaries, known as soft drusen (19, 126). Soft drusen are associated with more extensive damage to the retina, RPE, and choroid, and predict more severe clinical outcomes, such as GA and CNV (45). GA shows abnormal RPE with cellular hypotrophy and hypertrophy, hypopigmentation and hyperpigmentation, loss of photoreceptors, Bruch’s membrane thinning, and degeneration of the choriocapillaris (59, 103). Although RPE is the most affected cell layer, the photoreceptor layer is also severely attenuated (87). CNV refers to the growth of new blood vessels from the choroid through Bruch’s membrane and under the RPE (125). These vessels eventually break through the RPE (60), resulting in severe clinical phenotypes including (a) neovascular AMD, where lipid-rich fluid accumulates under the RPE and/or neural retina; (b) hemorrhagic AMD, where blood breaks through the RPE into the subretinal space; and (c) disciform AMD, where fibrous tissue with NV and altered RPE proliferates and replaces neural retina.
Though the initial lesions in both GA and CNV involve RPE and Bruch’s membrane pathology, it is the dysfunction and death of macular photoreceptors that are responsible for impairment of central vision in AMD (78).
Based on epidemiology and pathobiology, prevalence of AMD phenotypes depends on three key risk factors: environmental factors, advanced age, and genetic susceptibility variants (see Figure 3).
ENVIRONMENTAL FACTORS
As in other complex diseases, environmental factors play an important role in AMD development. Smoking is a modifiable and consistently associated environmental factor, typically resulting in about two-fold increased risk of developing advanced AMD (3, 112, 150, 155). A dose effect of enhanced risk of AMD with increasing number of cigarettes smoked has also been reported (86). The mechanism by which smoking affects the retina is unknown, although oxidative insults to the retina have been suggested (43).
The association of cardiovascular disease and its risk factors with AMD have not been consistently reproduced (26, 127, 136, 153). It is plausible that AMD and cardiovascular disease share common risk factors. Results from observational, case control, and population-based studies have also suggested an association of diet with AMD susceptibility. Diets high in antioxidants lutein and zeaxanthin (found mostly in green leafy vegetables) and in omega-3 fatty acids (primarily found in fish) have been linked to a decreased risk of neovascular AMD (48, 134).
Additional environmental factors that may influence AMD pathogenesis include sunlight exposure (33, 35, 85), alcohol use (23, 24, 114), and infection of bacterial pathogens (particularly Chlamydia pneumoniae) (11, 76, 81).
INTEGRATING THE BIOLOGY OF AGING IN STUDIES OF AMD
Aging is a genetically regulated process that is defined by progressive decline of somatic cell functions with age (46, 70). In mammals, aging-associated morphological, molecular, and functional alterations may represent an adaptive response to accumulation of damage (29, 166, 169), followed by cellular dysfunction and degeneration (16, 34, 55, 78). In humans, advanced age is accompanied by increased vulnerability to disease, with progressive and generally irreversible loss of physiological functions; in the eye, these include deficits in visual processing and degenerative changes in retinal neurons (particularly rods and ganglion cells) and the RPE (16, 78). Older individuals commonly have small drusen beneath the retina and cholesterol accumulation in Bruch’s membrane (64, 74). Another sign of aging is the accumulation of lipofuscin, an undegradable fluorescent material; in the RPE, accumulation of lipofuscin may be due in large part to incomplete digestion of shed photoreceptor discs (142). A2E (a by-product of phototransduction, present in lipofuscin) and mitochondrial dysfunction are reported to impair RPE phagocytosis and activate the complement system (156, 178). The pathogenesis of Stargardt’s disease, an early onset disorder whose symptoms resemble those of AMD, is thought to result from the accumulation of A2E in the RPE (110).
Aging is the single strongest risk factor for AMD; hence it is critical to integrate the biology of aging in models of AMD. Adaptive responses of the retina and RPE to aging are reflected in changes in expression of specific genes and cellular pathways (171). These changes likely contribute to the initiation of AMD; therefore, sequence variation in some of these genes may modulate individual specific risk of developing AMD. Aging-associated expression changes in genes associated with mitochondrial function, protein metabolism, and immune response have been identified in several tissues (173) and are consistent with some proposed mechanisms for late-onset neurodegenerative diseases (98, 113, 124, 166). Although an extensive gene expression database exists for the aging mouse (173), knowledge of such changes in retina remains limited. In humans, these studies are hampered by difficulties in obtaining appropriate patient material and inter-individual variability in gene expression profiles of the retina (25). Nevertheless, an initial comparison of young and aging human retinas identified differential expression of a small number of genes involved in stress and immune response, protein and energy metabolism, and inflammation (171). Retinal expression profiles of senescence-accelerated mice highlighted expression changes for a different set of genes (21). Given that gene profiles from the whole retina capture only an average of expression changes across heterogeneous cell populations and that rod photoreceptors are more vulnerable to aging (78), gene expression studies of aging in specific cell types are likely to be more informative. Initial expression analyses of GFP-tagged rod photoreceptors purified from the retinas of mice (5) demonstrate aging-associated changes in genes involved in mitochondrial function, protein degradation, and apoptosis (S. Parapuram & A. Swaroop, unpublished data). These changes may permit rod photoreceptors to adapt to aging and survive. Ultimately, it will be necessary to integrate our knowledge of aging in the human retina and in model organisms with the genetic and environmental risk factors that underlie AMD susceptibility if we are to obtain a comprehensive understanding of early steps in AMD pathogenesis.
GENETIC SUSCEPTIBILITY FACTORS
The contribution of genetics to AMD was carefully documented throughout the 1990s with reports of familial aggregation, concordant phenotypes in twins, and a higher risk of disease in first-degree relatives of affected individuals (69, 89, 91, 132). Twin studies demonstrated that genetic factors play a substantial role in the etiology of AMD, with some estimates suggesting that as much as 71% of the variation in the overall severity of AMD is genetically determined (133). Marked progress in identifying the genetic contributors has been achieved during the past ten years (see also 148).
GENETIC LINKAGE ANALYSIS
Linkage studies of AMD using large families have been difficult because of the late onset of disease, phenotypic variability, and phenocopy effects. Klein and colleagues were the first to report linkage to chromosome 1q25–31 in an apparently autosomal dominant pedigree with dry AMD (92). As with other complex diseases, gene mapping of AMD gradually shifted in focus from studies that include a small number of large families with many affected individuals to studies of much larger numbers of smaller families with affected pairs of siblings and other close relatives (121, 148). These studies of affected relative pairs were designed specifically to facilitate gene identification in complex multifactorial traits with a late age of onset (14, 121). In AMD, several independent studies, and a meta-analysis of multiple genome scans (47), suggested susceptibility loci with large effect at chromosomes 1q31 and 10q26. In addition, these studies provided more modest evidence for linkage on many other chromosomes (2, 77, 104, 137, 160), including a region on chromosome 22 near TIMP3, the gene mutated in Sorsby fundus dystrophy (158) (Figure 4).
Figure 4.

A schematic of human chromosomes showing regions of linkage identified in different studies and association signals (confirmed, and tentative requiring further validation) for AMD susceptibility loci/genes (modified from Reference 148).
Overall, linkage studies have had only limited success in identifying complex disease susceptibility genes; nonetheless, in the case of AMD they ultimately led to the discovery of genetic variants near CFH and ARMS2/HTRA1 as the strongest genetic contributors to AMD susceptibility (see below).
GENETIC ASSOCIATION ANALYSIS
Genetic linkage studies of complex traits can, at best, map disease susceptibility loci to intervals that are several megabases long and generally include hundreds of genes. Narrowing these intervals requires identification of the association between specific polymorphisms and disease. During the past few years, genetic association studies, which typically compare the frequency of specific variants between AMD cases and controls of similar ancestry, have identified several susceptibility loci. These studies can either be targeted to specific genes and candidate regions or attempt to survey common genetic variants across the genome. The latter, termed a genome-wide association study (GWAS) (71, 111), has only recently become feasible, owing to rapid advances in genotyping technology. In AMD, both focused association studies and GWAS have been extremely successful.
GENETIC ASSOCIATION STUDIES OF THE COMPLEMENT PATHWAY GENES
Perhaps the most significant breakthrough in our understanding of the genetics of AMD came in early 2005 with the identification of a strong association between disease and variants in and around the complement factor H (CFH) gene (Figure 5); this gene maps at 1q32, close to a linkage region at 1q25–31 that was identified in several independent studies (see Figure 4). Initial reports of association included candidate gene approach, fine mapping of candidate loci, and the first successful whole-genome association study in humans (40, 63, 65, 95) and were immediately replicated (175). This breakthrough association has now been validated in numerous studies worldwide and has proven significant in several ways.
Figure 5.

Genomic regions at chromosome 1q32 and 10q26 showing the genes and key associated single nucleotide polymorphisms (SNPs) and deletion variants.
First, identification of a strong association between AMD and the CFH region prompted evaluation of genetic association between AMD and several other genes in the complement pathway. These subsequent association studies were extremely successful, and several additional complement genes have now been strongly associated with AMD. These include complement 2 (C2) and/or complement factor B (CFB) (56), complement 3 (C3) (170), and complement factor I (CFI) (44). Together, these association signals account for a large part of the genetic contribution to AMD susceptibility.
Second, identification of a strong association between AMD and CFH prompted fine-mapping studies of the region that illustrate the importance of evaluating a full set of genetic variants in each disease susceptibility locus. These studies point to the issues and challenges that geneticists will encounter in searches for the ultimate “causal” variants at each complex disease susceptibility locus. Initial reports of association with the CFH gene focused on the Y402H coding variant that alters a single amino acid residue in the CFH protein. An early attempt at fine mapping the region showed that >20 variants in and around CFH were even more strongly associated with disease than was the Y402H coding variant, potentially suggesting that regulation of the expression of CFH and neighboring genes, rather than alterations in the coding sequence of CFH, mediates AMD susceptibility (97). This analysis also demonstrated that no single genetic variant could account for the contribution of the CFH locus to disease susceptibility and was soon corroborated by others (e.g., 11, 107). In addition to simple sequence changes, deletions in two genes near CFH, CFHR1 and CFHR3, have been associated with reduced risk of AMD (75, 143) (see Figure 5). Therefore, detailed analyses of every disease susceptibility locus (to discover the full complement of genetic variants it contains and assess their contributions to disease susceptibility) will be required to make accurate predictions about disease risk for each individual.
Third, the association between CFH and AMD provided one of the first examples of how genetic association studies, and in particular GWAS, could accelerate the pace of discovery for complex traits, such as AMD, where progress using more conventional approaches had been relatively modest. In this sense, studies of AMD genetics provided the “proof-of-principle” experiment that has since been reproduced for many other traits and conditions. As geneticists and biologists evaluate the best strategies to follow up the GWAS results and further dissect genetic contributions to complex disease susceptibility, studies of AMD genetics will undoubtedly retain this pioneering role.
GENETIC ASSOCIATION STUDIES OF THE ARMS2/HTRA1 REGION
Outside the complement pathway, the major genetic contributor to AMD susceptibility lies in the ARMS2/HTRA1 region (see Figure 5). The association signal in this region was initially identified as a result of fine-mapping efforts designed to narrow the chromosome 10q26 linkage peak (80, 122). This finding was later confirmed in several targeted studies (107) and in a GWAS (37). At least three potential candidate genes reside in the region of association; however, the identity of the gene that affects disease susceptibility is subject of controversy. Whereas two reports implicated HTRA1 as a target of AMD susceptibility (37, 167), recent studies support the original claim (122) of ARMS2 (previously LOC387715) as a stronger candidate (52, 82). The ARMS2 gene sequence is detected in primates and likely encodes a mitochondrial protein (82). Equally significant, the AMD-associated deletion-insertion polymorphism in ARMS2 is shown to mediate mRNA turnover (52). Genetic studies also suggest modification of the susceptibility effect of ARMS2 by smoking (128), consistent with its involvement in mitochondrial function (66, 82). It is, nonetheless, possible that susceptibility variants within the ARMS2 gene affect the activity of HTRA1 promoter, which is only a few kilobases from ARMS2, so that both genes ARMS2 and HTRA1 influence AMD susceptibility. Biological and functional studies of ARMS2 have been difficult because of the lack of a homologous gene in common model organisms (such as mouse or zebra fish). Additional investigations are necessary to define the role of ARMS2 and associated causal variants in AMD pathogenesis.
CANDIDATE GENE ASSOCIATION STUDIES
A multitude of candidate gene association studies have been carried out for AMD. Their findings, however, have not been replicated as widely or as strongly as those summarized above and should, for now, be treated as tentative. Typically, these genes have been selected based on a priori knowledge of the biological pathways involved in disease or on investigator prejudices about the pathways likely to contribute to disease susceptibility (7, 149). We caution that, in the absence of large, definitive, confirmatory studies, the possibility of publication bias cannot be discounted.
Apolipoprotein E (APOE) was an early target of candidate gene studies because of its presence in drusen, its key role in transport of lipids and cholesterol, and established association in Alzheimer’s disease. Individually, these studies have been relatively small, and evidence for association has been modest, although together the published studies provide stronger evidence of an association between APOE and AMD (10, 129, 177).
Other candidate gene studies focused on genes implicated in macular dystrophies with Mendelian inheritance and phenotypic similarities to AMD (146). Genes in this category include those implicated in Stargardt’s disease (caused by mutations in ABCA4), Sorsby fundus dystrophy (TIMP3), Best’s disease (VMD2), Malattia Leventinese (EFEMP1 or Fibulin-3), Stargardt-like dominant macular dystrophy (ELOVL4), butterfly dystrophy and bull’s eye maculopathy (RDS). A gene (C1QTNF5) mutated in patients with an autosomal dominant late-onset retinal/macular degeneration with sub-RPE deposits similar to AMD has also been examined (68). Except for two ABCA4 alleles (6), no variant in any other inherited macular dystrophy gene appears to show a significant association with AMD (148).
A natural progression of studies that examine genes previously implicated in Mendelian syndromes is the assessment of mutations in genes known to have similar structure or function. For example, the findings of Hemi-centin 1/Fibulin 6 variant in autosomal dominant dry AMD (131) and a mutation in EFEMP1/Fibulin-3 identified in Malattia Leventinese and Doyne honeycomb retinal dystrophy led to the screening of five fibulin genes for mutations associated with AMD; this screeening revealed a variant in fibulin-5 in AMD patients (145).
Given our relatively limited understanding of the biological pathways underlying AMD susceptibility, a wide variety of other genes have been tested for association. Consistent with the role of mitochondria in neurodegenerative disease, a specific mitochondrial DNA polymorphism, A4917G, is reported to be associated with AMD (20). Toll-like receptor 4 (TLR4) was considered an attractive candidate due to its key role in innate immunity, potential function in phagocytosis, and location to a region that exhibits some evidence of linkage to AMD; an association between the D299G TLR4 variant and AMD was initially reported (176) but has not been validated (36, 39). More recently, an association between AMD and another Toll-like receptor 3 (TLR3) was reported (168), but this association was not replicated in other large cohorts (41). A polymorphism in the 5′-upstream region of ERCC6, a gene implicated in DNA repair caused by oxidative stress and/or aging, has been associated with AMD susceptibility and was even suggested to exhibit epistatic interaction with CFH variants (152). Because of a potential role of retinal microglia in AMD, variants in the chemokine receptor gene CX3CR1 were examined for association with AMD; homozygosity of the M280 allele showed defective cell migration and appeared to be associated with higher AMD susceptibility (30). Multiple associations between AMD and HLA and cytokine genes have also been reported (57, 58). Screening of potential functional candidate genes identified association with an in-tronic variant within the SERPING1 gene (42); however, other studies failed to replicate these findings (119). These are all attractive candidates based on our current understanding of pathogenic mechanisms underlying AMD, but—as with other genetic association signals–the original signals require validation in large and independent samples (71, 111).
There are now several examples where variants in loci implicated in Mendelian disorders also contribute to susceptibility to more common forms of disease; these include discoveries related to the genetics of blood lipid levels and coronary artery disease (83, 162), genetics of obesity (100, 163), and the genetics of height (96, 159). In all these instances, common variants that contribute to trait variation in loci known to harbor Mendelian variants were missed in several earlier studies. In particular, genetic association studies apparently failed to notice positive signals in these candidate loci because they (a) defined the locus too narrowly (e.g., by focusing only on coding regions and a short flanking promoter sequence, although it now appears that regulatory variants can be located 10s or 100s of kb away from the gene); (b) relied on relatively small sample sizes (GWASs typically examine thousands of individuals and are often better powered to detect modest association signals than previous studies); or (c) did not comprehensively evaluate genetic variants in the candidate locus. When large and well-powered studies that examine all common variants in each of these candidate genes are carried out, we expect that some of the candidate loci implicated in disorders related to AMD may eventually be shown to also play a role in susceptibility to AMD.
PATHOGENIC MECHANISMS: CELLULAR PROCESSES AND PRIMARY AFFECTED CELL TYPES
The preceding discussion points to the complexities associated with delineating the role of genetic variants in mechanisms underlying the pathogenesis of AMD. This is not unexpected as AMD presents itself clinically in a variety of forms and affects many distinct cellular processes. To further our understanding of disease, all findings related to aging biology, genetics, and environmental risk factors must eventually be integrated into a unifying mechanistic model of AMD. A significant overlap in functional pathways highlighted by these disparate research strategies is gradually emerging.
Let us discuss the biological impact of advanced age, which is the primary risk factor for AMD. Numerous studies have demonstrated a central role of mitochondria and energy homeostasis in the pathogenesis of aging-associated neurodegenerative diseases (98, 157, 166). As photoreceptors have high oxygen consumption and energy demands, one can hypothesize increased susceptibility to DNA damage and production of reactive oxygen species in the aging mitochondria of these cells (61, 99, 165). Photoreceptor outer-segment renewal is suggested to serve as a means to eliminate these reactive oxygen species and other damaging molecules, minimizing resulting DNA damage (164). A deficiency in antioxidant processes or increased oxidative stress could therefore make the photoreceptors and consequently RPE vulnerable to disease. Protein homeostasis is another important aspect that requires serious consideration (113). Cellular function and adaptation requires appropriate folding of proteins and elimination of damaged proteins by the combined activities of chaperone networks and protein degradation systems; aging-associated decline in these pathways can lead to protein aggregates that can compromise biochemical functions and cellular health (113, 124). Notably, protein aggregates (lipofuscin and drusen) are hallmarks of retinal aging as well as of AMD. Gene profiles of aging retina and photoreceptors (described above) provide strong support for significant dysfunction in both energy and protein homeostasis.
AN INTEGRATED MODEL OF AMD PATHOGENESIS
Based on extensive biochemical, pathobiological, and genetic studies implicating oxidative stress and inflammation (12, 64, 165, 174), compromised energy and protein metabolism in aging retinal photoreceptors and RPE likely constitute one of the early steps in AMD pathogenesis (Figure 6). The resulting oxidative stress and protein misfolding are followed by lipid peroxidation and formation of protein aggregates, which may cause damage to outer-segment membrane biogenesis and RPE phagocytosis. These initial aging-associated defects are further compounded by genetic susceptibility and environmental factors. We speculate that the ARMS2 protein plays a specific role in primate life span-dependent mitochondrial adaptation. Similarly, smoking can further exacerbate the oxidative stress and add potentially toxic molecules. Whereas the primary triggers of AMD may originate in the photoreceptors because of their high membrane and protein turnover and exposure to light and oxygen, we believe the RPE quickly becomes involved because of its close relationship with photoreceptors. Even a slight decrease in the rate of clearing toxic protein and lipid debris could lead to their accumulation and even aggregation. This, in turn, could cause RPE damage and release of chemokines and cytokines (139). Decreased proteasome function during retinal aging (101), studies on A2E and Abca4-knockout mice (110, 156, 178), and changes in the RPE mitochondrial proteome (117) support this hypothesis. In addition, the CEP-mouse model demonstrates the role of oxidative damage in inducing inflammation and eventually GA-like lesions (72).
Figure 6.

An integrated model of AMD pathogenesis. Aging is the primary driver of early cellular defects in photoreceptors and retinal pigment epithelium (RPE); these changes are exacerbated (or inhibited) by specific genetics variants. Complement and immune response pathways play critical roles in the onset and severity of disease.
Genetic studies have established the roles of chronic inflammation and complement pathway in AMD pathology. It has been proposed that AMD is a systemic disease with specific manifestations in the macula of older individuals (118, 130). However, we believe that such macula-specific immune responses are unlikely without specific initiators of the disease process. We suggest that differential immune response to aging-associated RPE damage (as elaborated in the preceding paragraph) leads to specific clinical manifestations. Common and specific variants in complement and inflammatory response genes are therefore likely to play critical roles in generating an array of AMD phenotypes.
In attempting to integrate current knowledge on AMD, we have found a “multiple-hit” model of disease susceptibility to provide a convenient intellectual framework. Each hit represents the addition of a genetic, environmental, or aging-related risk factor that increases disease susceptibility for each individual. A clinically relevant phenotype is identified only when a threshold effect is reached after combining the susceptibility factors affecting each individual with stochastic contributions to risk (see Figures 3 and 6). Protective genetic haplo-types can reduce their impact on disease risk.
Our multiple-hit model can be summarized as follows:
Initiation–genetic susceptibility: Variants in genes associated with a late-onset disease (such as AMD) are present since birth, implying that these are not sufficient for disease causation. We suggest that the genetic variants alter specific cellular function(s) or pathway(s) in different cell types (such as photoreceptors and RPE) at early stages of life but that the functional consequences are not severe enough to produce a disease phenotype.
Modification–aging-associated changes: Multiple cellular pathways are concurrently affected during the normal aging process. These changes may initiate the disease process and/or exacerbate the impact of genetic susceptibility variants. Some of the changes (such as those involving mitochondrial function) may be overlapping and synergistic, whereas others may simply be additive. Genetic variants and aging together may lead to milder disease phenotypes and at a relatively late stage.
Clinical manifestation–impact of environment: Environmental factors, including smoking, high-fat and antioxidant-deficient diet, chronic infection and/or inflammation, may provide the final trigger(s) for disease causation. The adaptive cellular mechanisms already weakened by genetic variants and aging may finally break down, leading to severe disease phenotypes and advanced stages of AMD.
In this model, cumulative genetic load (i.e., net effect of different susceptibility or protective alleles) and exposure to environmental factors unique to each individual will produce variable age of onset and a range of AMD phenotypes. We also emphasize that this model, although conceptually convenient, obviously might not reflect the full complexity of susceptibility to AMD.
AMD AND OTHER LATE-ONSET NEURODEGENERATIVE DISEASES
Alzheimer’s (AD) and Parkinson’s (PD) are late-onset neurodegenerative diseases (17) that share similarities with AMD. An association of impaired cognitive function with advanced AMD was reported in AREDS (27). Additionally, a prospective population-based study identified an increased risk of developing AD in individuals with advanced AMD (88). Inheritance of the e4 allele of APOE is the only well-established genetic risk factor for sporadic AD (147). Mitochondrial dysfunction and oxidative stress, including mitochondrial DNA damage, have been implicated in all three diseases—AD, PD, and AMD (12, 17, 98, 165). The pathological hallmarks of AD include the presence of senile plaques, generated by accumulation of the amyloid beta (Aβ) peptide (138), whereas those of PD include Lewy bodies, which are cytoplasmic aggregates of fibrillar misfolded proteins (17). Toxic amyloid oligomers have been demonstrated in drusen, basal deposits and RPE of human donor AMD eyes but not in control age-matched eyes without drusen (102). Other components of amyloid deposits and drusen include complement and immune modulators, suggesting a potential role for inflammation in both AMD and AD (9).
ANIMAL MODELS
AMD primarily affects the macula, a specialized region identified in the retina of primates, but not in other mammals or vertebrates. Notably, the human ARMS2 gene appears to have a highly conserved homolog only in non-human primates (49). Because of inherent difficulties associated with using monkeys, mouse models of AMD are being developed to dissect the underlying mechanisms and evaluate treatment paradigms (108). Despite their relatively short life span and absence of a fovea, mouse models offer the ability to dissect distinct characteristics of a complex human disease and investigate contributions of individual components to the complete phenotype. In our opinion, an ideal mouse model would incorporate all three aspects of AMD in humans: genetic susceptibility, advanced age, and environmental factors. No mouse model perfectly mimics AMD in all three respects; in this review we focus on a few selected mouse models that appear robust and reproducible for experimental manipulation [for more details, see other reviews (108, 120)].
The Sod1−/− mice lacking the Cu/Zn-superoxide dismutase 1 gene, a key regulator of oxidative stress in the cytoplasm, exhibit age-dependent development of drusen, sub-RPE deposits, and CNV (67). These deposits are immunopositive for many of the markers present in human drusen, supporting a role for oxidative stress in the pathogenesis of AMD. However, these mice also develop a progressive retinal degeneration, which is not associated with AMD.
The APOE4 mouse carries a targeted replacement of the mouse gene with the human allele. APOE4 mice aged over one year and fed a high-fat diet enriched with cholesterol (HFC) for 8 weeks (APOE4-HFC mice) develop pathological features characteristic of both dry and wet human AMD (105). Pathologies observed include diffuse lipid- and protein-rich drusen-like sub-RPE deposits that are immunopositive for complement and inflammatory markers (Figure 7), thickening of Bruch’s membrane, patchy regions of RPE atrophy, and CNV. This phenotype presents in a temporal, incompletely penetrant and noninvasive manner that is analogous to human AMD progression (105). Visual deficits detected in electroretinograms are also associated with the development of the AMD phenotype in these mice.
Figure 7.

Activated complement components in sub-RPE (retinal pigment epithelium) deposits. (a) Immuno-localization of C3 fragments [C3b/iC3b/C3c (red)] and apoE (green) in basal, sub-RPE deposits (arrowheads) and Bruch’s membrane of 75-week-old APOE4-HFC mouse eye. C3 fragments are found in small, discrete patches that appear to be associated with individual RPE cells (blue is nuclear DAPI stain). (b) Immuno-localization of C5b-9 (red) in human AMD donor eye (cyan is lipofuscin autofluorescence from the RPE). C5b-9 immunoreactivity is associated with similar sub-RPE deposits (arrowheads). OS, outer segments. Images courtesy of J-D. Ding, Duke University (a) and L. Johnson, University of California, Santa Barbara (b).
Mouse models of targeted knockout of chemokines or their receptors appear to exhibit some characteristics of AMD. Aged mice deficient in Ccl-2 (monocyte chemoattractant protein-1) or its cognate receptor Ccr2 (C-C chemokine receptor-2), which are involved in recruiting macrophages to loci of complement opsonization, sporadically present drusen accumulation and lipofuscin deposition in swollen and vacuolated RPE cells (8). In CX3C chemokine receptor (CX3CR1) knockout mouse, subretinal microglia accumulate with age, with albino background, and in response to laser injury (30). Some of these microglia are bloated with ingested lipid and account for the drusen-like appearance on fundus images. A double knockout mouse, generated by crossing Ccl2−/− and Cx3cr1−/− mice, develops a fairly severe phenotype as early as 6 weeks and includes photoreceptor degeneration, RPE damage, basal deposits, and in some animals even CNV (151).
Because of the established role of the complement pathway, attempts are under way to make transgenic mouse strains expressing the human allotypic variants of CFH associated with AMD. Low protein sequence similarity (70%) between the human and mouse CFH proteins may complicate any resulting phenotypes. The Cfh−/− mice exhibit an age-related decrease in visual acuity and impaired photoreceptor function with evidence of C3 deposition and ultrastructural changes in the retina, but less basal membrane thickening and less sub-RPE drusen-like material compared with age-matched wild-type controls (28). Another approach to test the effect of complement activation on AMD pathogenesis in vivo may be by dysregulating complement inhibition in a mouse model through the use of another regulator of the alternative complement pathway.
A nontransgenic mouse model deserves special mention as it is based on oxidative stress and elicitation of a systemic immune response to carboxyethylpyrrole (CEP), an ocular antigen found in eye tissue and plasma from AMD patients (73). As they age, these mice accumulate sub-RPE deposits that are immunopositive for complement components (C3d) in Bruch’s membrane. Another mouse model carrying the R345W mutation in the EFEMP1 gene (responsible for Malattia Leventinese) closely recapitulates the early pathophysiology of human AMD patients (53, 109); this includes small, isolated sub-RPE deposits that increase in size and number, eventually becoming continuous sheets. In older mice, membranous debris is observed within the sub-RPE deposits and within Bruch’s membrane.
Although we do not seem to have an ideal model that recapitulates the whole spectrum of AMD disease phenotype, many new mouse models are emerging to test molecular mechanisms and examine novel targets for therapies. For example, anti-Aβ antibodies are being evaluated to prevent retinal damage in APOE4-HFC mice (38). Although some might argue that the slow progression of disease in the advanced age-dependent mouse models (e.g., EFEMP1ki/ki, CEP, or APOE4) prohibitively increases the cost of research, we believe that aging is an important risk factor in AMD and should be a key component of any model of complex multifactorial aspects of AMD pathology.
MANAGEMENT OF AMD
Dietary Supplements
Because few treatment options exist, prevention is the first approach to reduce vision loss in AMD. Control of modifiable risks factors such as smoking, hypertension, and elevated BMI may reduce the risk of developing AMD by as much as half. AREDS, a multicenter randomized and controlled clinical trial of oral supplements, demonstrated that high doses of antioxidant vitamin C, vitamin E, beta-carotene, zinc, and copper are effective in slowing the progression of advanced stages of AMD by 25% (4). This formulation, if given for the next 5 years to all those who are at risk in the United States, has the potential to prevent 300,000 individuals from developing advanced AMD. Dietary supplements are being further evaluated in a new clinical trial, AREDS2, in which lutein, zeaxanthin, and/or omega-3 fatty acids (1 gm) will be evaluated for the prevention of advanced AMD (http://www.areds2.org).
Treatment of Neovascular AMD
For patients who have already developed neo-vascular AMD, early detection is associated with a better prognosis. The treatment of neo-vascular AMD has significantly improved with the development of inhibitors for vascular endothelial growth factor (VEGF), which acts as both a neuroprotective and angiogenic factor (18). In June 2006, the FDA approved ranibizumab (Lucentis), a fragment of a monoclonal antibody that binds to and inhibits all forms of VEGF-A, for the treatment of neo-vascular AMD (144). This drug, when injected intravitreally on a monthly basis, prevented vision loss and was the first AMD treatment associated with improvements in vision for many patients. Differences in visual acuity score in the treated and control groups were considerable, with a mean increase of 7 letters in the treated group compared with a decrease of 10 letters in the placebo, sham-injected group (P < 0.001) (144). A randomized trial comparing bevacizumab (a closely related antibody) with ranibizumab will directly evaluate the potential effects of reduced treatment frequency to alleviate the burden of monthly intravitreal injections (http://www.med.upenn.edu/cpob/studies/CATT.shtml).
THE FUTURE OF AMD TREATMENT
We expect that in the near future, genetic testing will complement clinical exams, to identify patients at risk of progressing to the advanced forms of AMD. Currently, by using a simple scale for severity of AMD, the presence of certain ocular lesions can be utilized to make useful predictions about disease progression. For example, persons with bilateral large drusen and pigmentary changes have a 50% risk of progressing to advanced AMD in one eye over the next 5 years. Additional evaluation of the genetic mutations may provide increased precision in these predictions. A study of AREDS participants demonstrated that the presence of CFH polymorphisms and ARMS2-A69S independently contributed to progression to advanced AMD. The presence of these polymorphisms with the AREDS-type dietary supplements, smoking, and elevated body mass index (25 or greater) increased the susceptibility to advanced AMD (135).
Therapies of the future may use genetic information to modulate and target individual therapies. A preliminary analysis of interaction between CFH and ARMS2 susceptibility variants with the AREDS-type dietary supplements (90) suggests that oral supplementation with zinc was especially helpful for individuals with low-risk disease alleles at CFH.
ANTICIPATED PROGRESS IN GENETIC STUDIES
The pace of discovery in genetic studies of AMD has been extremely rapid, and we expect major advances within the next few years. GWAS including thousands of AMD cases and controls are now under way and will enable us to further examine the genome in an unbiased manner. We believe these studies will implicate several additional loci in disease susceptibility and clarify the role of variants examined in previous candidate gene studies. In each identified region/gene/locus, we expect that a detailed analysis of genetic variation, aided by advances in sequencing technologies, will enable us to thoroughly assess their contributions to disease susceptibility. In parallel, because of significant differences in disease prevalence and phenotypes, we believe it will be critical to study genetic contributions to AMD in different ethnic groups. Much of the AMD genetics research has focused on Caucasians; nonetheless, genetic analyses of relatively small cohorts of other ethnic groups have begun to reveal interesting insights (22, 37, 54, 84). Examination of individuals of different ethnicities (by resequencing the associated genomic regions) will be particularly helpful in attempts to identify causal alleles at each AMD susceptibility locus, since linkage disequilibrium will limit the resolution of genetic studies that focus on a single population.
We believe that changes in the management of AMD (as well as other late-onset degenerative diseases) will not occur in a vacuum. With the rapid decrease in costs of genome sequencing and genotyping and a concomitant increase in our understanding of the genetic basis of complex and common disease, we predict that within the next decade genetic information will be used to tailor individual diagnosis and treatment plans and become an integral part of the personalized medical care.
Acknowledgments
We sincerely apologize to our colleagues whose work (particularly on the epidemiology and pathology of AMD, and on CFH and ARMS2/HTRA1) could not be cited because of page limitations. We are grateful to John Heckenlively, Sheldon Miller, Dwight Stambolian, Robert Fariss, Nicholas Katsanis, Fredrick Ferris, Robert Nussenblatt, Wei Chen, Kari Branham, Atsuhiro Kanda, Rivka Rachel and Mohammad Othman for discussions, comments, and assistance. We are grateful to Tiziana Cogliati and Lydia Kibluk for excellent assistance with illustrations. Our research is supported by the intramural program of the National Eye Institute and by grants from the National Institutes of Health, The Foundation Fighting Blindness, Macula Vision Research Foundation, and Research to Prevent Blindness.
Glossary
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- AMD
age-related macular degeneration
- RPE
retinal pigment epithelium
- AREDS
Age-Related Eye Disease Study
- GA
geographic atrophy
- NV
neovascular
- CNV
choroidal neovascularization
- GWAS
genome-wide association study
- HFC
high-fat diet enriched with cholesterol
- VEGF
vascular endothelial growth factor
Footnotes
DISCLOSURE STATEMENT
G.A. is co-inventor on a University of Michigan patent covering the use of genetic markers for diagnosis of AMD.
LITERATURE CITED
- 1.Wellcome Trust Case Control Consort. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abecasis GR, Yashar BM, Zhao Y, Ghiasvand NM, Zareparsi S, et al. Age-related macular degeneration: a high-resolution genome scan for susceptibility loci in a population enriched for late-stage disease. Am J Hum Genet. 2004;74:482–94. doi: 10.1086/382786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology. 2000;107:2224–32. doi: 10.1016/s0161-6420(00)00409-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–36. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, et al. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc Natl Acad Sci USA. 2006;103:3890–95. doi: 10.1073/pnas.0508214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000;67:487–91. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–88. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–97. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 9.Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78:243–56. doi: 10.1016/j.exer.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Baird PN, Richardson AJ, Robman LD, Dimitrov PN, Tikellis G, et al. Apolipoprotein (APOE) gene is associated with progression of age-related macular degeneration (AMD) Hum Mutat. 2006;27:337–42. doi: 10.1002/humu.20288. [DOI] [PubMed] [Google Scholar]
- 11.Baird PN, Robman LD, Richardson AJ, Dimitrov PN, Tikellis G, et al. Gene-environment interaction in progression of AMD: the CFH gene, smoking and exposure to chronic infection. Hum Mol Genet. 2008;17:1299–305. doi: 10.1093/hmg/ddn018. [DOI] [PubMed] [Google Scholar]
- 12.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–34. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 13.Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39:367–74. doi: 10.1016/s0039-6257(05)80092-x. [DOI] [PubMed] [Google Scholar]
- 14.Bishop DT, Williamson JA. The power of identity-by-state methods for linkage analysis. Am J Hum Genet. 1990;46:254–65. [PMC free article] [PubMed] [Google Scholar]
- 15.Bok D. Retinal photoreceptor-pigment epithelium interactions. Friedenwald lecture. Investig Ophthalmol Vis Sci. 1985;26:1659–94. [PubMed] [Google Scholar]
- 16.Bonnel S, Mohand-Said S, Sahel JA. The aging of the retina. Exp Gerontol. 2003;38:825–31. doi: 10.1016/s0531-5565(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 17.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 18.Breen EC. VEGF in biological control. J Cell Biochem. 2007;102:1358–67. doi: 10.1002/jcb.21579. [DOI] [PubMed] [Google Scholar]
- 19.Bressler NM, Silva JC, Bressler SB, Fine SL, Green WR. Clinicopathologic correlation of drusen and retinal pigment epithelial abnormalities in age-related macular degeneration. Retina. 1994;14:130–42. [PubMed] [Google Scholar]
- 20.Canter JA, Olson LM, Spencer K, Schnetz-Boutaud N, Anderson B, et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE. 2008;3:e2091. doi: 10.1371/journal.pone.0002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter TA, Greenhall JA, Yoshida S, Fuchs S, Helton R, et al. Mechanisms of aging in senescence-accelerated mice. Genome Biol. 2005;6:R48. doi: 10.1186/gb-2005-6-6-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen LJ, Liu DT, Tam PO, Chan WM, Liu K, et al. Association of complement factor H polymorphisms with exudative age-related macular degeneration. Mol Vis. 2006;12:1536–42. [PubMed] [Google Scholar]
- 23.Cho E, Hankinson SE, Willett WC, Stampfer MJ, Spiegelman D, et al. Prospective study of alcohol consumption and the risk of age-related macular degeneration. Arch Ophthalmol. 2000;118:681–88. doi: 10.1001/archopht.118.5.681. [DOI] [PubMed] [Google Scholar]
- 24.Chong EW, Kreis AJ, Wong TY, Simpson JA, Guymer RH. Alcohol consumption and the risk of age-related macular degeneration: a systematic review and meta-analysis. Am J Ophthalmol. 2008;145:707–15. doi: 10.1016/j.ajo.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Chowers I, Liu D, Farkas RH, Gunatilaka TL, Hackam AS, et al. Gene expression variation in the adult human retina. Hum Mol Genet. 2003;12:2881–93. doi: 10.1093/hmg/ddg326. [DOI] [PubMed] [Google Scholar]
- 26.Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL., 3rd Risk factors for the incidence of advanced age-related macular degeneration in the Age-Related Eye Disease Study (AREDS): AREDS report no. 19. Ophthalmology. 2005;112:533–39. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clemons TE, Rankin MW, McBee WL. Cognitive impairment in the Age-Related Eye Disease Study: AREDS report no. 16. Arch Ophthalmol. 2006;124:537–43. doi: 10.1001/archopht.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coffey PJ, Gias C, McDermott CJ, Lundh P, Pickering MC, et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci USA. 2007;104:16651–56. doi: 10.1073/pnas.0705079104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008;9:759–67. doi: 10.1038/nrn2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Investig. 2007;117:2920–28. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Congdon N, O’Colmain B, Klaver CC, Klein R, Munoz B, et al. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–85. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- 32.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–87. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cruickshanks KJ, Klein R, Klein BE, Nondahl DM. Sunlight and the 5-year incidence of early age-related maculopathy: the Beaver Dam Eye Study. Arch Ophthalmol. 2001;119:246–50. [PubMed] [Google Scholar]
- 34.Curcio CA, Owsley C, Jackson GR. Spare the rods, save the cones in aging and age-related maculopathy. Invest Ophthalmol Vis Sci. 2000;41:2015–18. [PubMed] [Google Scholar]
- 35.Delcourt C, Carriere I, Ponton-Sanchez A, Fourrey S, Lacroux A, Papoz L. Light exposure and the risk of age-related macular degeneration: the Pathologies Oculaires Liées a l’Age (POLA) study. Arch Ophthalmol. 2001;119:1463–68. doi: 10.1001/archopht.119.10.1463. [DOI] [PubMed] [Google Scholar]
- 36.Despriet DD, Bergen AA, Merriam JE, Zernant J, Barile GR, et al. Comprehensive analysis of the candidate genes CCL2, CCR2, and TLR4 in age-related macular degeneration. Investig Ophthalmol Vis Sci. 2008;49:364–71. doi: 10.1167/iovs.07-0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–92. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 38.Ding JD, Lin J, Mace BE, Herrmann R, Sullivan P, Bowes Rickman C. Targeting age-related macular degeneration with Alzheimer’s disease based immunotherapies: anti-amyloid-beta antibody attenuates pathologies in an age-related macular degeneration mouse model. Vision Res. 2008;48:339–45. doi: 10.1016/j.visres.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edwards AO, Chen D, Fridley BL, James KM, Wu Y, et al. Toll-like receptor polymorphisms and age-related macular degeneration. Investig Ophthalmol Vis Sci. 2008;49:1652–59. doi: 10.1167/iovs.07-1378. [DOI] [PubMed] [Google Scholar]
- 40.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–24. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 41.Edwards AO, Swaroop A, Seddon JM. Geographic atrophy in age-related macular degeneration and TLR3. N Engl J Med. 2009;360:2254–5. [PubMed] [Google Scholar]
- 42.Ennis S, Jomary C, Mullins R, Cree A, Chen X, et al. Association between the SERPING1 gene and age-related macular degeneration: a two-stage case-control study. Lancet. 2008;372:1828–34. doi: 10.1016/S0140-6736(08)61348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Investig Ophthalmol Vis Sci. 2006;47:729–37. doi: 10.1167/iovs.05-0719. [DOI] [PubMed] [Google Scholar]
- 44.Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100–4. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferris FL, Davis MD, Clemons TE, Lee LY, Chew EY, et al. A simplified severity scale for age-related macular degeneration: AREDS Report No. 18. Arch Ophthalmol. 2005;123:1570–74. doi: 10.1001/archopht.123.11.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finch CE, Ruvkun G. The genetics of aging. Annu Rev Genomics Hum Genet. 2001;2:435–62. doi: 10.1146/annurev.genom.2.1.435. [DOI] [PubMed] [Google Scholar]
- 47.Fisher SA, Abecasis GR, Yashar BM, Zareparsi S, Swaroop A, et al. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet. 2005;14:2257–64. doi: 10.1093/hmg/ddi230. [DOI] [PubMed] [Google Scholar]
- 48.Flood V, Smith W, Wang JJ, Manzi F, Webb K, Mitchell P. Dietary antioxidant intake and incidence of early age-related maculopathy: the Blue Mountains Eye Study. Ophthalmology. 2002;109:2272–78. doi: 10.1016/s0161-6420(02)01263-0. [DOI] [PubMed] [Google Scholar]
- 49.Francis PJ, Appukuttan B, Simmons E, Landauer N, Stoddard J, et al. Rhesus monkeys and humans share common susceptibility genes for age-related macular disease. Hum Mol Genet. 2008;17:2673–80. doi: 10.1093/hmg/ddn167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 52.Fritsche LG, Loenhardt T, Janssen A, Fisher SA, Rivera A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40:892–96. doi: 10.1038/ng.170. [DOI] [PubMed] [Google Scholar]
- 53.Fu L, Garland D, Yang Z, Shukla D, Rajendran A, et al. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet. 2007;16:2411–22. doi: 10.1093/hmg/ddm198. [DOI] [PubMed] [Google Scholar]
- 54.Fuse N, Miyazawa A, Mengkegale M, Yoshida M, Wakusawa R, et al. Polymorphisms in Complement Factor H and Hemicentin-1 genes in a Japanese population with dry-type age-related macular degeneration. Am J Ophthalmol. 2006;142:1074–76. doi: 10.1016/j.ajo.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 55.Gao H, Hollyfield JG. Aging of the human retina. Differential loss of neurons and retinal pigment epithelial cells. Investig Ophthalmol Vis Sci. 1992;33:1–17. [PubMed] [Google Scholar]
- 56.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goverdhan SV, Ennis S, Hannan SR, Madhusudhana KC, Cree AJ, et al. Interleukin-8 promoter polymorphism-251A/T is a risk factor for age-related macular degeneration. Br J Ophthalmol. 2008;92:537–40. doi: 10.1136/bjo.2007.123190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goverdhan SV, Howell MW, Mullins RF, Osmond C, Hodgkins PR, et al. Association of HLA class I and class II polymorphisms with age-related macular degeneration. Investig Ophthalmol Vis Sci. 2005;46:1726–34. doi: 10.1167/iovs.04-0928. [DOI] [PubMed] [Google Scholar]
- 59.Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27. [PubMed] [Google Scholar]
- 60.Grossniklaus HE, Green WR. Choroidal neovascularization. Am J Ophthalmol. 2004;137:496–503. doi: 10.1016/j.ajo.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 61.Guarente L. Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–76. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gupta SK, Murthy GV, Morrison N, Price GM, Dherani M, et al. Prevalence of early and late age-related macular degeneration in a rural population in northern India: the INDEYE feasibility study. Investig Ophthalmol Vis Sci. 2007;48:1007–11. doi: 10.1167/iovs.06-0712. [DOI] [PubMed] [Google Scholar]
- 63.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–32. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 65.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 66.Haines JL, Spencer KM, Pericak-Vance MA. Bringing the genetics of macular degeneration into focus. Proc Natl Acad Sci USA. 2007;104:16725–26. doi: 10.1073/pnas.0708151104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hashizume K, Hirasawa M, Imamura Y, Noda S, Shimizu T, et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am J Pathol. 2008;172:1325–31. doi: 10.2353/ajpath.2008.070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hayward C, Shu X, Cideciyan AV, Lennon A, Barran P, et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: a genetic model for age-related macular degeneration. Hum Mol Genet. 2003;12:2657–67. doi: 10.1093/hmg/ddg289. [DOI] [PubMed] [Google Scholar]
- 69.Heiba IM, Elston RC, Klein BE, Klein R. Sibling correlations and segregation analysis of age-related maculopathy: the Beaver Dam Eye Study. Genet Epidemiol. 1994;11:51–67. doi: 10.1002/gepi.1370110106. [DOI] [PubMed] [Google Scholar]
- 70.Hekimi S, Guarente L. Genetics and the specificity of the aging process. Science. 2003;299:1351–54. doi: 10.1126/science.1082358. [DOI] [PubMed] [Google Scholar]
- 71.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 72.Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–98. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hollyfield JG, Bonilha VL, Rayborn ME, Yang XP, Shadrach KG, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–98. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang JD, Curcio CA, Johnson M. Morphometric analysis of lipoprotein-like particle accumulation in aging human macular Bruch’s membrane. Investig Ophthalmol Vis Sci. 2008;49:2721–27. doi: 10.1167/iovs.07-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–77. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 76.Ishida O, Oku H, Ikeda T, Nishimura M, Kawagoe K, Nakamura K. Is Chlamydia pneumoniae infection a risk factor for age-related macular degeneration? Br J Ophthalmol. 2003;87:523–24. doi: 10.1136/bjo.87.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iyengar SK, Song D, Klein BE, Klein R, Schick JH, et al. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am J Hum Genet. 2004;74:20–39. doi: 10.1086/380912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jackson GR, Owsley C, Curcio CA. Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res Rev. 2002;1:381–96. doi: 10.1016/s1568-1637(02)00007-7. [DOI] [PubMed] [Google Scholar]
- 79.Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606–17. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 80.Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77:389–407. doi: 10.1086/444437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kalayoglu MV, Galvan C, Mahdi OS, Byrne GI, Mansour S. Serological association between Chlamydia pneumoniae infection and age-related macular degeneration. Arch Ophthalmol. 2003;121:478–82. doi: 10.1001/archopht.121.4.478. [DOI] [PubMed] [Google Scholar]
- 82.Kanda A, Chen W, Othman M, Branham KE, Brooks M, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci USA. 2007;104:16227–32. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaur I, Katta S, Hussain A, Hussain N, Mathai A, et al. Variants in the 10q26 gene cluster (LOC387715 and HTRA1) exhibit enhanced risk of age-related macular degeneration along with CFH in Indian patients. Investig Ophthalmol Vis Sci. 2008;49:1771–76. doi: 10.1167/iovs.07-0560. [DOI] [PubMed] [Google Scholar]
- 85.Khan JC, Shahid H, Thurlby DA, Bradley M, Clayton DG, et al. Age-related macular degeneration and sun exposure, iris colour, and skin sensitivity to sunlight. Br J Ophthalmol. 2006;90:29–32. doi: 10.1136/bjo.2005.073825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khan JC, Thurlby DA, Shahid H, Clayton DG, Yates JR, et al. Smoking and age-related macular degeneration: The number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br J Ophthalmol. 2006;90:75–80. doi: 10.1136/bjo.2005.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim SY, Sadda S, Pearlman J, Humayun MS, de Juan E, Jr, et al. Morphometric analysis of the macula in eyes with disciform age-related macular degeneration. Retina. 2002;22:471–77. doi: 10.1097/00006982-200208000-00012. [DOI] [PubMed] [Google Scholar]
- 88.Klaver CC, Ott A, Hofman A, Assink JJ, Breteler MM, de Jong PT. Is age-related maculopathy associated with Alzheimer’s disease? The Rotterdam Study. Am J Epidemiol. 1999;150:963–68. doi: 10.1093/oxfordjournals.aje.a010105. [DOI] [PubMed] [Google Scholar]
- 89.Klaver CC, Wolfs RC, Assink JJ, van Duijn CM, Hofman A, de Jong PT. Genetic risk of age-related maculopathy. Population-based familial aggregation study. Arch Ophthalmol. 1998;116:1646–51. doi: 10.1001/archopht.116.12.1646. [DOI] [PubMed] [Google Scholar]
- 90.Klein ML, Francis PJ, Rosner B, Reynolds R, Hamon SC, et al. CFH and LOC387715/ARMS2 genotypes and treatment with antioxidants and zinc for age-related macular degeneration. Ophthalmology. 2008;115:1019–25. doi: 10.1016/j.ophtha.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 91.Klein ML, Mauldin WM, Stoumbos VD. Heredity and age-related macular degeneration. Observations in monozygotic twins. Arch Ophthalmol. 1994;112:932–37. doi: 10.1001/archopht.1994.01090190080025. [DOI] [PubMed] [Google Scholar]
- 92.Klein ML, Schultz DW, Edwards A, Matise TC, Rust K, et al. Age-related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Arch Ophthalmol. 1998;116:1082–88. doi: 10.1001/archopht.116.8.1082. [DOI] [PubMed] [Google Scholar]
- 93.Klein R, Klein BE, Knudtson MD, Meuer SM, Swift M, Gangnon RE. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007;114:253–62. doi: 10.1016/j.ophtha.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 94.Klein R, Klein BE, Knudtson MD, Wong TY, Cotch MF, et al. Prevalence of age-related macular degeneration in 4 racial/ethnic groups in the multi-ethnic study of atherosclerosis. Ophthalmology. 2006;113:373–80. doi: 10.1016/j.ophtha.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 95.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–89. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet. 2008;40:584–91. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li M, Atmaca-Sonmez P, Othman M, Branham KE, Khanna R, et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet. 2006;38:1049–54. doi: 10.1038/ng1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 99.Loeb LA, Wallace DC, Martin GM. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc Natl Acad Sci USA. 2005;102:18769–70. doi: 10.1073/pnas.0509776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–75. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Louie JL, Kapphahn RJ, Ferrington DA. Proteasome function and protein oxidation in the aged retina. Exp Eye Res. 2002;75:271–84. [PubMed] [Google Scholar]
- 102.Luibl V, Isas JM, Kayed R, Glabe CG, Langen R, Chen J. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J Clin Investig. 2006;116:378–85. doi: 10.1172/JCI25843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lutty G, Grunwald J, Majji AB, Uyama M, Yoneya S. Changes in choriocapillaris and retinal pigment epithelium in age-related macular degeneration. Mol Vis. 1999;5:35. [PubMed] [Google Scholar]
- 104.Majewski J, Schultz DW, Weleber RG, Schain MB, Edwards AO, et al. Age-related macular degeneration—a genome scan in extended families. Am J Hum Genet. 2003;73:540–50. doi: 10.1086/377701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Malek G, Johnson LV, Mace BE, Saloupis P, Schmechel DE, et al. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci USA. 2005;102:11900–5. doi: 10.1073/pnas.0503015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Malek G, Li CM, Guidry C, Medeiros NE, Curcio CA. Apolipoprotein B in cholesterol-containing drusen and basal deposits of human eyes with age-related maculopathy. Am J Pathol. 2003;162:413–25. doi: 10.1016/S0002-9440(10)63836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Maller J, George S, Purcell S, Fagerness J, Altshuler D, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–59. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 108.Marmorstein AD, Marmorstein LY. The challenge of modeling macular degeneration in mice. Trends Genet. 2007;23:225–31. doi: 10.1016/j.tig.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 109.Marmorstein LY, McLaughlin PJ, Peachey NS, Sasaki T, Marmorstein AD. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum Mol Genet. 2007;16:2423–32. doi: 10.1093/hmg/ddm199. [DOI] [PubMed] [Google Scholar]
- 110.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154–59. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 112.McCarty CA, Mukesh BN, Fu CL, Mitchell P, Wang JJ, Taylor HR. Risk factors for age-related maculopathy: the Visual Impairment Project. Arch Ophthalmol. 2001;119:1455–62. doi: 10.1001/archopht.119.10.1455. [DOI] [PubMed] [Google Scholar]
- 113.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–38. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Moss SE, Klein R, Klein BE, Jensen SC, Meuer SM. Alcohol consumption and the 5-year incidence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 1998;105:789–94. doi: 10.1016/S0161-6420(98)95016-3. [DOI] [PubMed] [Google Scholar]
- 115.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–46. [PubMed] [Google Scholar]
- 116.Munoz B, West SK, Rubin GS, Schein OD, Quigley HA, et al. Causes of blindness and visual impairment in a population of older Americans: The Salisbury Eye Evaluation Study. Arch Ophthalmol. 2000;118:819–25. doi: 10.1001/archopht.118.6.819. [DOI] [PubMed] [Google Scholar]
- 117.Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig Ophthalmol Vis Sci. 2008;49:2848–55. doi: 10.1167/iovs.07-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nussenblatt RB, Ferris F., 3rd Age-related macular degeneration and the immune response: implications for therapy. Am J Ophthalmol. 2007;144:618–26. doi: 10.1016/j.ajo.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Park KH, Ryu E, Tosakulwong N, Wu Y, Edwards AO. Common variation in the SERPING1 gene is not associated with age-related macular degeneration in two independent groups of subjects. Mol Vis. 2009;15:200–7. [PMC free article] [PubMed] [Google Scholar]
- 120.Rattner A, Nathans J. Macular degeneration: recent advances and therapeutic opportunities. Nat Rev Neurosci. 2006;7:860–72. doi: 10.1038/nrn2007. [DOI] [PubMed] [Google Scholar]
- 121.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–17. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 122.Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14:3227–36. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- 123.Rodieck RW. The First Steps in Seeing. Sinauer; Sunderland, MA: 1998. [Google Scholar]
- 124.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–86. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 125.Sarks JP, Sarks SH, Killingsworth MC. Morphology of early choroidal neovascularisation in age-related macular degeneration: correlation with activity. Eye. 1997;11(Part 4):515–22. doi: 10.1038/eye.1997.137. [DOI] [PubMed] [Google Scholar]
- 126.Sarks S, Cherepanoff S, Killingsworth M, Sarks J. Relationship of Basal laminar deposit and membranous debris to the clinical presentation of early age-related macular degeneration. Investig Ophthalmol Vis Sci. 2007;48:968–77. doi: 10.1167/iovs.06-0443. [DOI] [PubMed] [Google Scholar]
- 127.Schaumberg DA, Christen WG, Buring JE, Glynn RJ, Rifai N, Ridker PM. High-sensitivity C-reactive protein, other markers of inflammation, and the incidence of macular degeneration in women. Arch Ophthalmol. 2007;125:300–5. doi: 10.1001/archopht.125.3.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schmidt S, Hauser MA, Scott WK, Postel EA, Agarwal A, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006;78:852–64. doi: 10.1086/503822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schmidt S, Klaver C, Saunders A, Postel E, De La Paz M, et al. A pooled case-control study of the apolipoprotein E (APOE) gene in age-related maculopathy. Ophthalmic Genet. 2002;23:209–23. doi: 10.1076/opge.23.4.209.13883. [DOI] [PubMed] [Google Scholar]
- 130.Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schultz DW, Klein ML, Humpert AJ, Luzier CW, Persun V, et al. Analysis of the ARMD1 locus: evidence that a mutation in HEMICENTIN-1 is associated with age-related macular degeneration in a large family. Hum Mol Genet. 2003;12:3315–23. doi: 10.1093/hmg/ddg348. [DOI] [PubMed] [Google Scholar]
- 132.Seddon JM, Ajani UA, Mitchell BD. Familial aggregation of age-related maculopathy. Am J Ophthalmol. 1997;123:199–206. doi: 10.1016/s0002-9394(14)71036-0. [DOI] [PubMed] [Google Scholar]
- 133.Seddon JM, Cote J, Page WF, Aggen SH, Neale MC. The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch Ophthalmol. 2005;123:321–27. doi: 10.1001/archopht.123.3.321. [DOI] [PubMed] [Google Scholar]
- 134.Seddon JM, Cote J, Rosner B. Progression of age-related macular degeneration: association with dietary fat, transunsaturated fat, nuts, and fish intake. Arch Ophthalmol. 2003;121:1728–37. doi: 10.1001/archopht.121.12.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Seddon JM, Francis PJ, George S, Schultz DW, Rosner B, Klein ML. Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA. 2007;297:1793–800. doi: 10.1001/jama.297.16.1793. [DOI] [PubMed] [Google Scholar]
- 136.Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol. 2005;123:774–82. doi: 10.1001/archopht.123.6.774. [DOI] [PubMed] [Google Scholar]
- 137.Seddon JM, Santangelo SL, Book K, Chong S, Cote J. A genomewide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am J Hum Genet. 2003;73:780–90. doi: 10.1086/378505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 139.Shi G, Maminishkis A, Banzon T, Jalickee S, Li R, et al. Control of chemokine gradients by the retinal pigment epithelium. Investig Ophthalmol Vis Sci. 2008;49:4620–30. doi: 10.1167/iovs.08-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Smith W, Assink J, Klein R, Mitchell P, Klaver CC, et al. Risk factors for age-related macular degeneration: pooled findings from three continents. Ophthalmology. 2001;108:697–704. doi: 10.1016/s0161-6420(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 141.Sommer A, Tielsch JM, Katz J, Quigley HA, Gottsch JD, et al. Racial differences in the cause-specific prevalence of blindness in east Baltimore. N Engl J Med. 1991;325:1412–17. doi: 10.1056/NEJM199111143252004. [DOI] [PubMed] [Google Scholar]
- 142.Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, et al. A2E, a byproduct of the visual cycle. Vision Res. 2003;43:2983–90. doi: 10.1016/s0042-6989(03)00475-9. [DOI] [PubMed] [Google Scholar]
- 143.Spencer KL, Hauser MA, Olson LM, Schmidt S, Scott WK, et al. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum Mol Genet. 2008;17:971–77. doi: 10.1093/hmg/ddm369. [DOI] [PubMed] [Google Scholar]
- 144.Steinbrook R. The price of sight–ranibizumab, bevacizumab, and the treatment of macular degeneration. N Engl J Med. 2006;355:1409–12. doi: 10.1056/NEJMp068185. [DOI] [PubMed] [Google Scholar]
- 145.Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–53. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- 146.Stone EM, Sheffield VC, Hageman GS. Molecular genetics of age-related macular degeneration. Hum Mol Genet. 2001;10:2285–92. doi: 10.1093/hmg/10.20.2285. [DOI] [PubMed] [Google Scholar]
- 147.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Swaroop A, Branham KE, Chen W, Abecasis G. Genetic susceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits. Hum Mol Genet. 2007;16(Spec 2):R174–82. doi: 10.1093/hmg/ddm212. [DOI] [PubMed] [Google Scholar]
- 149.Tabor HK, Risch NJ, Myers RM. Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet. 2002;3:391–97. doi: 10.1038/nrg796. [DOI] [PubMed] [Google Scholar]
- 150.Tomany SC, Wang JJ, Van Leeuwen R, Klein R, Mitchell P, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111:1280–87. doi: 10.1016/j.ophtha.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 151.Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, et al. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Investig Ophthalmol Vis Sci. 2007;48:3827–36. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tuo J, Ning B, Bojanowski CM, Lin ZN, Ross RJ, et al. Synergic effect of polymorphisms in ERCC6 5′ flanking region and complement factor H on age-related macular degeneration predisposition. Proc Natl Acad Sci USA. 2006;103:9256–61. doi: 10.1073/pnas.0603485103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vine AK, Stader J, Branham K, Musch DC, Swaroop A. Biomarkers of cardiovascular disease as risk factors for age-related macular degeneration. Ophthalmology. 2005;112:2076–80. doi: 10.1016/j.ophtha.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 154.Vingerling JR, Dielemans I, Hofman A, Grobbee DE, Hijmering M, et al. The prevalence of age-related maculopathy in the Rotterdam Study. Ophthalmology. 1995;102:205–10. doi: 10.1016/s0161-6420(95)31034-2. [DOI] [PubMed] [Google Scholar]
- 155.Vingerling JR, Hofman A, Grobbee DE, de Jong PT. Age-related macular degeneration and smoking. The Rotterdam Study. Arch Ophthalmol. 1996;114:1193–96. doi: 10.1001/archopht.1996.01100140393005. [DOI] [PubMed] [Google Scholar]
- 156.Vives-Bauza C, Anand M, Shirazi AK, Magrane J, Gao J, et al. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem. 2008;283:24770–80. doi: 10.1074/jbc.M800706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8:352–56. doi: 10.1038/ng1294-352. [DOI] [PubMed] [Google Scholar]
- 159.Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–83. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Weeks DE, Conley YP, Tsai HJ, Mah TS, Schmidt S, et al. Age-related maculopathy: a genomewide scan with continued evidence of susceptibility loci within the 1q31, 10q26, and 17q25 regions. Am J Hum Genet. 2004;75:174–89. doi: 10.1086/422476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Weih LM, VanNewkirk MR, McCarty CA, Taylor HR. Age-specific causes of bilateral visual impairment. Arch Ophthalmol. 2000;118:264–69. doi: 10.1001/archopht.118.2.264. [DOI] [PubMed] [Google Scholar]
- 162.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–69. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Winkler BS. An hypothesis to account for the renewal of outer segments in rod and cone photoreceptor cells: renewal as a surrogate antioxidant. Investig Ophthalmol Vis Sci. 2008;49:3259–61. doi: 10.1167/iovs.08-1785. [DOI] [PubMed] [Google Scholar]
- 165.Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32. [PMC free article] [PubMed] [Google Scholar]
- 166.Wright AF, Jacobson SG, Cideciyan AV, Roman AJ, Shu X, et al. Lifespan and mitochondrial control of neurodegeneration. Nat Genet. 2004;36:1153–58. doi: 10.1038/ng1448. [DOI] [PubMed] [Google Scholar]
- 167.Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992–93. doi: 10.1126/science.1133811. [DOI] [PubMed] [Google Scholar]
- 168.Yang Z, Stratton C, Francis PJ, Kleinman ME, Tan PL, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008;359:1456–63. doi: 10.1056/NEJMoa0802437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- 170.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–61. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 171.Yoshida S, Yashar BM, Hiriyanna S, Swaroop A. Microarray analysis of gene expression in the aging human retina. Investig Ophthalmol Vis Sci. 2002;43:2554–60. [PubMed] [Google Scholar]
- 172.Young RW. Visual cells, daily rhythms, and vision research. Vision Res. 1978;18:573–78. doi: 10.1016/0042-6989(78)90205-5. [DOI] [PubMed] [Google Scholar]
- 173.Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, et al. AGEMAP: a gene expression database for aging in mice. PLoS Genet. 2007;3:e201. doi: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122:598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- 175.Zareparsi S, Branham KE, Li M, Shah S, Klein RJ, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet. 2005;77:149–53. doi: 10.1086/431426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zareparsi S, Buraczynska M, Branham KE, Shah S, Eng D, et al. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Hum Mol Genet. 2005;14:1449–55. doi: 10.1093/hmg/ddi154. [DOI] [PubMed] [Google Scholar]
- 177.Zareparsi S, Reddick AC, Branham KE, Moore KB, Jessup L, et al. Association of apolipoprotein E alleles with susceptibility to age-related macular degeneration in a large cohort from a single center. Investig Ophthalmol Vis Sci. 2004;45:1306–10. doi: 10.1167/iovs.03-1253. [DOI] [PubMed] [Google Scholar]
- 178.Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2006;103:16182–87. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]