

Abstract
Background
Waved with open eyelids 2 (woe2) is a novel autosomal recessive mouse mutation that arose spontaneously in our animal facility. Upon initial evaluation, mutant mice exhibited eyelids open at birth (EOB) and wavy fur phenotypes. The goals of this study were to phenotypically characterize the woe2 mice and to identify the gene harboring the mutation responsible for the woe2 phenotype.
Results
Histological analysis of woe2 embryos identified the failure of embryonic eyelid closure. Clinical and histological analysis of woe2 adult eyes identified severe corneal opacities, abnormalities of the anterior segment of the eye, and the absence of meibomian glands. Abnormalities in the fur texture and the absence of meibomian glands prompted us to evaluate other epidermal appendages: skin, teeth, and nails--as well as lacrimal, mammary, salivary, sebaceous and sweat glands. No obvious morphological differences between WT and woe2 mice were identified in these tissues. However, the analysis of woe2 identified cardiac abnormalities. Positional cloning of the woe2 locus identified a 1308 bp deletion in the Ppp1r13l gene. The deletion resulted in an aberrant Ppp1r13lΔexon9-11 transcript that lacks exons 9, 10 and 11 resulting in a premature stop and a loss of 223 amino acids from the C-terminal end of the putative mutant PPP1R13L protein. Immunohistological analysis during eye development identified expression of PPP1R13L in the palpebral epidermis, palpebral and bulbar conjunctiva, corneal epithelium and meibomian glands.
Conclusions
The woe2 mouse harbors a novel deletion within the Ppp1r13l gene, likely resulting in a complete loss of PPP1R13L function. Results from this study provide evidence that PPP1R13L has an essential role in embryonic eyelid closure as well in development of meibomian glands and the anterior segment of the eye. The woe2 mice are a useful model for investigation of the role of PPP1R13L, especially during ocular and eyelid development.
Keywords: EOB, Eyelid, Embryonic, Meibomian, Mapping, Microsatellite, Locus, Mutation, Deletion
Background
As a part of mammalian ocular development the eyelids form, grow over the cornea, meet, and temporarily fuse [1]. The closed eyelids serve as a protective barrier preventing premature exposure of developing ocular structures to the environment. Once conjunctival and corneal epithelia reach their maturity, the eyelid junction begins to break down and the eyelids reopen [2]. Whether different mammalian species are born with the eyelids open or closed is determined by the stage of the ocular development specific for the species at the time of birth [1]. In mice, eyelid formation is initiated at embryonic day 11.5 (E11.5) by invagination of the dorsal and ventral periocular ectoderm resulting in the formation of the primitive eyelid [1,3]. By E15.5, the leading edges of the eyelid form at the tip of the primitive eyelids and start to migrate towards each other. By E16.5, the eyelids meet and form a junction and by E17 embryonic eyelid closure is fully completed [1,3]. Mice are born with their eyelids closed and they remain closed until postnatal days 10 to 12 (P10-12) at which time they reopen [2]. When eyelids fail to close properly before birth, the result is often severe corneal inflammation, defects in the anterior segment and blindness [3,4]. Forward and reverse genetic approaches have identified genes essential for embryonic eyelid closure that, when mutated, lead to the “eyelids-open-at-birth” (EOB) phenotype. Given that mice are born with their eyelids closed, the EOB phenotype is easily identified by observation of the eyelids of newborn pups. EOB phenotypes have been identified to be associated with many genotypes; in fact, Mouse Genome Informatics (MGI) (http://www.informatics.jax.org/) lists 138 genotypes associated with the EOB phenotype indicating numerous genes and molecular pathways associated with this process.
A subgroup of mutant mice with the EOB phenotype also exhibit wavy fur. Molecular and genetic analyses showed that the majority of the mice with EOB and wavy fur phenotypes have defects in epidermal growth factor receptor (EGFR) signaling. Specifically, mice with mutations in the transforming growth factor alpha (Tgfa) [5,6], a disintegrin and metallopeptidase domain 17 (Adam17) [7-9] and Egfr which encodes the EGFR receptor [10-14] all exhibit EOB and wavy fur phenotypes. EGFR signaling plays an essential role in regulating the eyelid leading edge migration through activation of the EGFR-ERK signaling cascade [15]. Interestingly, another mouse mutant termed waved 3 (wa3) also exhibits the EOB and wavy fur phenotypes similar to those observed in mice with EGFR signaling defects [16]. The wa3 mice have a mutation in Ppp1r13l [16], a gene that belongs to the Apoptosis Stimulating Proteins of p53 (ASPP) family of proteins. Although PPP1R13L is a highly conserved protein from C. elegans to human [17] the role of PPP1R13L remains poorly understood. It has been shown that PPP1R13L acts as a regulator of p53-mediated apoptosis [17] and as a regulator of the NF-кB subunit p65-RelA gene expression [18]. Recently, it was also shown that PPP1R13L, via its regulation of p63, is a key regulator of epithelial homeostasis [19] and epithelial stratification [20].
Here we report a novel autosomal recessive mouse mutation that arose spontaneously in our mouse colony. Initial observations showed that the mutant mice exhibit EOB and wavy fur phenotypes. The identified phenotypes observed in the mutant mice resemble those in waved with open eyelids (woe) mice carrying a hypermorphic mutation in Adam17, previously studied in our lab [8]. Thus, we termed the newly identified mutant mice waved with open eyelids 2 (woe2). A detailed examination of woe2 phenotypes revealed that a defect in embryonic eyelid closure is responsible for the woe2 EOB phenotype observed at birth. Additional ocular phenotypes in woe2 mice include severe corneal opacities, defects in the structures of the anterior segment of the eye, and the absence of the meibomian glands. In addition to ocular and wavy fur phenotypes, woe2 mice also exhibited severe cardiac defects. Genetic analysis showed that the woe2 phenotypes are due to a 1308 bp deletion in the Pppr13l gene. The identified deletion results in aberrantly spliced Ppp1r13l transcript and a putative truncated PPP1R13L protein lacking C-terminal functional domains. These findings uncover previously unidentified roles for PPP1R13L during eyelid and ocular development.
Methods
Mice
The woe2 mutation arose spontaneously on a mixed C57BL/6X129/SvJ background. The woe2 locus was maintained by brother-sister breedings. The C3A.BLiA-Pde6b+/J strain, which is a Pde6b(+) strain of C3H/HeJ (http://jaxmice.jax.org/strain/001912.html), and C57BL/6J were obtained from the Jackson Laboratory (Bar Harbor, ME). All strains exhibited normal breeding patterns and litter sizes. The treatment and use of all animals in this study was compliant with all protocols and provisions approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical College of Wisconsin.
Clinical evaluation, histology and electron microscopy
For clinical analysis, mouse eyes were examined with a Topcon SL-D8Z slit lamp biomicroscope, following mydriasis with 1% Atropine Sulfate (Bausch & Lomb). The eyes were imaged with a Nikon SLR-based Photo Slit Lamp imaging system as previously described [21]. For tissue analysis, E0.5 was defined as the morning of the day that a vaginal plug was first observed in a female. Embryonic and postnatal tissues were collected and fixed in either Zinc-formalin, Davidson’s solution, or 4% paraformaldehyde, then embedded in paraffin and sectioned to 4 μm thickness and stained with H&E using standard procedures as previously described [8]. For scanning electron microscopy (SEM), E15.5 and E16.5 wild-type and woe2 embryo heads were collected, fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, rinsed in buffer and dehydrated in ethanol. The samples were then critical-point dried in a Bal-tec CPD050, gold sputter coated in a Denton Desk II and viewed in a FEI XL30 SEM.
Immunohistochemistry
Antigen retrieval was performed in 1x citrate Buffer (Invitrogen) warmed to 95°C for 20 minutes. Sections were allowed to cool to room temperature and subsequently blocked in 10% normal goat serum with 1% bovine serum albumin in PBS for one hour. Slides were incubated at 4°C for one hour with monoclonal anti- PPP1R13L primary antibody (Sigma-Aldrich) at 1:200. Slides were then washed 3x in PBST and incubated for 20 minutes with goatαmouse Alexa633 (Invitrogen) secondary antibody at 1:1000. Slides were washed 3x in PBST and mounted with Vectashield mounting medium containing DAPI (Vector Labs).
Linkage mapping
The woe2 homozygote mice were outcrossed to C3A.BliA-Pde6β+/J; the resulting F1 progeny were backcrossed which generated 164 F2 progeny. These progeny were evaluated at three weeks of age for the presence of the wavy fur and were clinically evaluated for ocular abnormalities. Following phenotyping, the F2 progeny were euthanized and tissues were collected. Genomic DNA was purified from collected tissues as previously described [22]. Initially, 25 F2 progeny were genotyped as previously described [22] with polymorphic microsatellites from Chr. 6: D6Mit188, D6Mit320, D6Mit323; Chr. 7: D7Mit340, D7Mit160, D7Mit148; Chr. 11: D11Mit19, D11Mit229, D11Mit294; Chr. 12: D12Mit12, D12Mit147, D12Mit52. For fine mapping on chromosome 7 all 164 progeny were genotyped with D7Mit1, D7Mit22, D7Mit224, D7Mit340, D7Mit160, and D7Mit148. Linkage was determined using Map Manager QTX software (http://www.mapmanager.org).
Sequence analysis of Ppp1r13l
For exonic and intron/exon junction genomic sequence analysis, primers were designed to anneal 50 bp upstream and downstream from intron/exon junctions. PCR products were amplified and sequenced as previously described [22]. For cDNA analysis, RT-PCR was performed using total RNA from wild-type, woe2/+, and woe2/woe2 tissues, utilizing hearts, spleens, or eyes as previously described [22]. The list of primer sequences is summarized in Additional file 1: Table S1. Long range PCR was conducted using the GeneAmp XL PCR kit (Roche, Branchburg, NJ) with primers (5’ CTGTCCACAATTACAGAGGGATCTGAA G3’) and (5’ CGGAAAGACAGCTCATCCCCAAACTCT 3’) as previously described [22]. DNAStar software was utilized for comparative analysis of the sequences.
Results
The woe2 phenotypes
At birth, woe2 pups exhibit the fully penetrant, bilateral EOB phenotype (Figure 1B). Histological analysis of woe2 eyes at P0.5 confirmed open eyelids at birth and also identified hyperkeratosis of the eyelids (Figure 1D). In addition to the eyelid defects, stromal keratitis, the absence of the conjunctival sacs, anterior synechiae, and hyperplastic corneas were also noted (Figure 1D). To determine if the EOB phenotype in woe2 newborn pups was due to a defect in embryonic eyelid closure or due to premature eyelid opening at birth, we histologically evaluated E18.5 embryonic WT and woe2 eyes. In WT mice, eyelid closure was completed between the two eyelids (Figure 1E), while in contrast, woe2 mice exhibited open eyelids and exposed corneas (Figure 1F). This finding indicated that the EOB observed in the woe2 newborn pups was due to the failure of embryonic eyelid closure.
Figure 1.
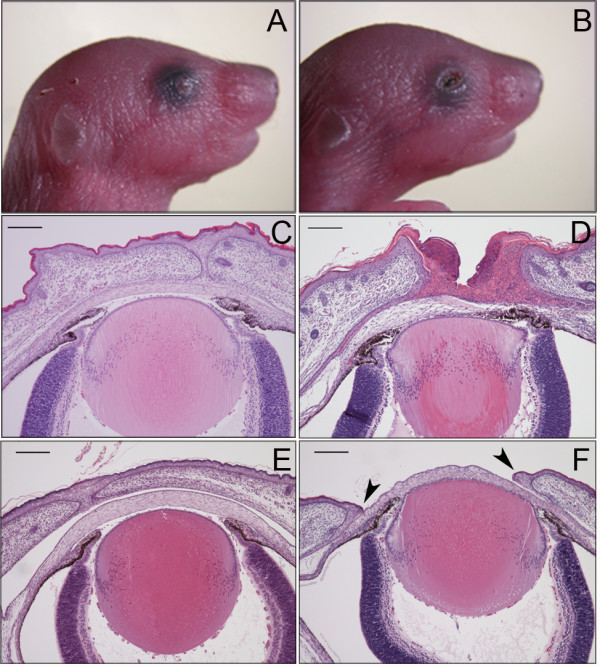
The EOB phenotype in woe2 mice. All woe2 neonates were born with bilateral open eyelids (B) in contrast to the WT neonates that have closed eyelids (A). Histological analysis of P0.5 woe2 mice confirmed the open eyelid phenotype and also revealed hyperkeratosis of the eyelids, stromal keratitis, absence of the conjunctival sacs, anterior synechiae, and hyperplastic corneas (D) features that were absent in the WT age matched controls (C). Further histological analysis of woe2 mice at E18.5 (F) showed failure of the embryonic eyelid closure (arrows) whereas WT controls exhibited embryonic eyelid closure (E).
As woe2 mice progressed through development, their eyes appeared smaller in size when compared to WT controls. To further investigate the woe2 eye phenotype, we clinically evaluated P28 eyes. The analysis revealed opaque and vascularized corneas (Figure 2B) which precluded further clinical investigation. Clinical eye evaluation also identified that in about 25% of P28 woe2 mice, unilateral or bilateral eyelid closure was present (Figure 2C). Histological analysis of P28 woe2 eyes confirmed severely disrupted corneas and further identified anterior segment dysgenesis with extensive anterior synechiae, (Figure 2E) while no obvious morphological abnormalities of the lens and the retina were noted (not shown). Histological analysis of the woe2 eyes which appeared closed (Figure 2C) revealed severely microphthalmic eyes as a result of aphakia (Figure 2F). In addition, highly disorganized corneal, iris and retinal structures were observed (Figure 2F). Histological analysis of P28 eye globes also revealed the absence of meibomian glands in the P28 woe2 eyelids (Figure 2H).
Figure 2.

The woe2 eye phenotype at P28. Clinical evaluation of woe2 eyes identified microphthalmia with corneal opacities and neovascularization (B) when compared with WT mice (A). In about 25% of woe2 animals, unilateral or bilateral eyelid closure was noted (C). Histological analysis of woe2 eyes (E) identified anterior segment abnormalities with extensive anterior synechia (arrow), corneal hypertrophy (arrowhead) and stromal vascularization. Histological analysis of closed woe2 eyes (C) revealed aphakia with highly disorganized cornea, iris and retina (F). Histological analysis of adult woe2 eyelids (H) identified absence of meibomian glands (arrow) whereas meibomian glands were present in age-matched WT eyelids (G) (asterisk). Scale bar in D-H = 100 μm.
In addition to the ocular phenotypes, woe2 mice start to exhibit wavy fur around P14 that remains wavy in texture throughout the life of the animal (Figure 3A). Abnormalities in the fur texture and the absence of meibomian glands identified in woe2 eyelids (Figure 2H) prompted us to evaluate other epidermal appendages in woe2. Clinical and histological analysis of the skin, teeth, and nails as well as the lacrimal, mammary, salivary, sebaceous and sweat glands did not identify any obvious morphological differences between WT and woe2 mice (not shown). However, the adult woe2 mice exhibited cardiac abnormalities characterized with opaque white plaques covering approximately 2-15% of the exposed surface (Figure 3B). The mineralized foci tended to occur in the myocardium adjacent to the epicardium, although similar lesions occurred in the interventricular septum as well (not shown). On average, woe2 mice lived until around 9 months of age indicating shortened life spans when compared to age-matched C57BL/6 J mice. The initial breeding pattern revealed that the woe2 phenotypes were inherited as a fully penetrant single autosomal recessive locus.
Figure 3.
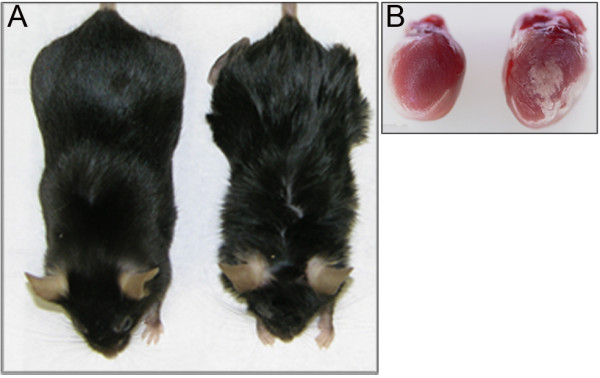
Non-ocular woe2 phenotypes at P28. The fur (A) in woe2 mice (right) showed waviness when compared to the smooth coat in WT mice (left). Hearts (B) of woe2 mice showed mineralized foci and opaque white plaques (right) that were absent in age-matched wild-type controls (left).
Positional cloning of woe2
The observed phenotypes of woe2 mice resembled phenotypes of mice with mutations in Tgfa, Egfr, Adam17 and Ppp1r13l genes [5-11,14,16]. Therefore, we hypothesized that woe2 may be a new mutant allele in one of these four genes. To test that hypothesis, we utilized a genetic approach. Tgfa, Ppp1r13l, Egfr, and Adam17 genes map to chromosomes 6, 7, 11 and 12 respectively (http://www.informatics.jax.org/). Therefore, we selected microsatellite markers from chromosomes 6, 7, 11 and 12 and tested segregation of the woe2 locus with these microsatellites. No linkage was observed between the woe2 locus and microsatellite markers on chromosomes 6, 11 and 12, but linkage of the woe2 locus to chromosome 7 was established with LOD > 10. To refine the genetic position of the woe2 locus we selected additional microsatellite markers from chromosome 7. The analysis mapped the woe2 locus to a 6.8 cM region between D7Mit340 and D7Mit1 (Figure 4A). The established woe2 critical region contained the Ppp1r13l gene. Therefore, we proceeded to evaluate Ppp1r13l as a candidate gene.
Figure 4.

The genetic analysis of the woe2 locus. The woe2 locus was mapped to mouse chromosome 7 (A) between D7Mit340 and D7Mit1 establishing Ppp1r13l as a candidate gene. Each column represents the haplotype found in progeny of woe2 mutants (C57BLX129/SvJ background) when backcrossed to the C3A.BliAPde6+/J strain. Black boxes represent the C3ABliA-Pde6+/J allele and white boxes represent the C57BL6X129/SvJ allele. The number of offspring inheriting each haplotype is listed at the bottom of each column. Sequence analysis of genomic DNA from woe2 tissues (B) identified a 1308 bp deletion encompassing the genomic region from Ppp1r13l intron 8 to intron 11. RT-PCR analysis from WT tissues (C) identified a single Ppp1r13l transcript that matched the Ppp1r13l reference sequence (NM_001010836). RT-PCR analysis from woe2 tissues (D) identified a single aberrantly spliced Ppp1r13l transcript with missing exons 9, 10 and 11. (E) depicts a schematic of wild type PPP1R13L protein (NP_001010836) (top) conserved domains: Pro-rich domain (54–598), ANK1 domain (655–684), ANK2 (688–717), and SH3 (754–816) domain. The woe2 putative PPP1R13Lp.E602TX7 protein (bottom figure) shows, following Met601, an addition of 7 novel amino acids (TWNKAWD) depicted by a gray box, a premature stop and a loss of 223 amino acids from the C-terminus encompassing ANK1, ANK2 and SH3 domains. Numbers on the top of each figure in (E) represent the amino acid residues.
Genomic sequence analysis of Ppp1r13l exons 1–8 and exon 12 including intron/exon junctions did not identify any difference between the Ppp1r13l reference sequence (NC_000073.5) and sequences from woe2. Although conventional PCR produced PCR products for exons 9, 10, and 11 when using genomic DNA from WT animals, it did not produce any PCR products when using genomic DNA from woe2 animals. This suggested a possible genomic rearrangement across the Ppp1r13l exon 9-11 region in the woe2 animals. Long range-PCR from exon 8 to exon 12 and subsequent sequencing revealed a 1308 bp deletion in woe2 (Figure 4B). The deletion break points were identified in Ppp1r13l intron 8 at −271 bp upstream from the 5' end of exon 9 and in intron 11 at −1203 bp upstream from the 5' end of exon 12 (Figure 4B).
To determine the molecular consequences of the 1308 bp deletion on the splicing of the Ppp1r13l transcript in the woe2 tissues, we proceeded to evaluate Ppp1r13l cDNA. RT-PCR analysis from woe2 tissues identified a single Ppp1r13l transcript that exhibited aberrant splicing from exon 8 to exon 12 with exons 9, 10 and 11 missing (Figure 4D). RT-PCR analysis of Ppp1r13l from WT tissues identified a transcript that matched the Ppp1r13l reference sequence (NM_001010836.3) (Figure 4C). The aberrant Ppp1r13lΔexon9-11 transcript identified in woe2 encodes a putative PPP1R13L mutant protein with a frame shift following M601, an addition of 7 amino acids (TWNKAWD), and a premature stop. The putative mutant PPP1R13L lacks 223 amino acids from the C-terminal end (Figure 4E).
The woe2 EOB phenotype
To further investigate the embryonic eyelid closure defect that lead to the EOB phenotype in the woe2 mice, we evaluated the embryonic eyelids with scanning electron microscopy (SEM) just prior to the embryonic eyelid closure. At E15.5, the leading edges were present in both WT and woe2 upper (Figures 5A) and lower eyelids (Figure 5B). Higher magnification views showed presence of rounded periderm cells in both the WT and woe2 eyelid margins (Figures 5A', 5A", 5B' and 5B"). At E16.5, in WT mice, the eyelid closure was completed (Figure 5C) and flattened cells were present at the eyelid junction (Figures 5C' and 5C"). By contrast, the woe2 eyelids, at E16.5, remained wide open (Figure 5D). Rounded peridemal cells were infrequently seen at this stage at the leading edge of woe2 (Figure 5D' and 5D").
Figure 5.

Scanning electron micrographs showing impaired embryonic eyelid closure in woe2 embryos. At E15.5 WT (A, A', A") and woe2 (B, B', B") embryos manifested open eyes of similar shape and with no distinct morphological difference. The accumulation of rounded peridermal cells at the leading edge of both WT (A', A") and woe2 (B', B") eyelids was evident (arrowhead). Differences between the WT and woe2 eyelids were evident at E16.5. The eyelids of WT embryos were completely fused (C, C', C") at the midline with flattened cells at the junction margins (C” arrow). In contrast, failure of eyelid closure was apparent in the eyes of woe2 mice at this time (D, D', D"). At this time point, very few peridermal cells were evident at the eyelid margin of woe2 eyelids (D” arrow).
PPP1R13L expression in the developing eyelids
Expression of PPP1R13L in mouse skin has been previously reported [16,19,20], although expression of PPP1R13L in the eyelids and anterior segment has never been investigated. In order to better understand the role of PPP1R13L during embryonic eyelid closure, the expression pattern of PPP1R13L during mouse eyelid closure was pursued. Immunohistological analysis showed PPP1R13L is expressed in the cells of the epithelial sheet of the developing eyelid including palpebral epidermis, palpebral and bulbar conjunctiva (Figure 6A and 6C). Following eyelid closure, PPP1R13L remained expressed in the cells of the eyelid junction (Figure 6E). In P0.5 eyelids, PPP1R13L was also identified as expressed in the developing hair follicle and in the cells of the developing meibomian glad (Figure 6G). In addition to the eyelid, immunohistological analysis identified expression of PPP1R13L in the corneal epithelium (Figure 6A and 6C), lens epithelium and the retina (not shown).
Figure 6.

PPP1R13L expression in the developing eyelid. Immunostaining for PPP1R13L (green) showed expression of PPP1R13L at E14.5-P0.5 (A, C, E, G) in the palpebral epidermis, palpebral conjunctiva, bulbar conjunctiva, corneal epithelium, lens epithelium and retina. DAPI (blue) was used as a nuclear stain. As eyelid closure begins, at E15.5 (C) PPP1R13L is also expressed in the leading edge of the migrating eyelid. As eyelid closure reaches completion at E16.5 (E), expression is maintained in the eyelid junction. At P0.5 (G), PPP1R13L is expressed in the developing meibomian gland (G arrow) and hair follicle (G arrowhead). Scale bars = 50 μm.
Discussion
In this study we characterized woe2, a novel mouse mutation which arose spontaneously. Genetic analysis of the woe2 locus identified a 1308 bp deletion in the Ppp1r13l gene encompassing exons 9 through 11 resulting in an aberrant Ppp1r13lΔexon9-11 transcript. The putative woe2 PPP1R13L protein encoded by the Ppp1r13lΔexon9-11 transcript lacks 223 amino acids from the C-terminal end. PPP1R13L, at the C-terminal end, is characterized with ankyrin (AKN) repeats, and a SRC homology (SH3) domain [23]. The biochemical analyses of PPP1R13L have established that the ANK repeats and the SH3 domain are essential for the PPP1R13L binding with its partners: p53 [17], p65RelA [18], and p63 [24]. The 223 amino acids missing from the mutant PPP1R13L from woe2 encompass both the ANK repeats and the SH3 domain. Although we did not functionally evaluate the truncated woe2 PPP1R13L protein, we expect that a loss of 223 amino acids may compromise the stability of the mutant woe2 PPP1R13L protein. Even if the mutant woe2 PPP1R13L protein remains s, the loss of the ANK repeats and the SH3 domain most likely would result in a complete loss of PPP1R13L function. This is consistent with the findings previously reported for wa3 mice that carry a 14-bp deletion in Ppp1r13l resulting in a loss of the SH3-domain from the C-terminal end [14]. The mutant PPP1R13L protein from wa3 mice exhibited a loss of binding to the p65RelA subunit of NF-кB [14], a previously established PPP1R13L binding partner [16]. As such, wa3 was established as a Ppp1r13l loss-of-function mutation [14]. These findings collectively suggest that woe2 is a novel Ppp1r13l loss-of-function mutation.
The wavy fur and cardiac phenotypes identified in woe2 resemble the cardiac and wavy fur phenotypes previously characterized in the wa3 mice [14]. Abnormalities in the fur and heart were also reported for cattle affected with cardiomyopathy and wooly hair coats (CWH) syndrome that carry a frame-shift mutation in the bovine ortholog of the Ppp1r13l gene [25]. Although the EOB phenotype was also reported for wa3 mice, it was not further investigated [14]. As a part of this study we established that in woe2 the EOB phenotype is due to a defect in embryonic eyelid closure. Even though the morphological analysis of woe2 embryonic eyelids did not identify any obvious abnormalities in the formation of the primitive eyelid, the formation of the leading edge or accumulation of peridermal cells at the leading edge, the woe2 embryonic eyelids failed to close. Immunohistochemical analysis showed that PPP1R13L was expressed in the eyelid epithelial sheet including epidermis, palpebral and bulbar conjunctiva, as well as in the cells of the leading edge. It has been shown before that the embryonic eyelid closure process depends on the movements of the epithelial sheet of the developing eyelids in a morphogenic process similar to dorsal closure in Drosophila [25]. In the developing epidermis of the skin, via its interaction with p63, PPP1R13L regulates the expression of genes that play an essential role in cell-matrix adhesion, as well as in epithelial cell junctions [17]. In addition, PPP1R13L was identified to regulate cell senescence and is required for epithelial stratification [20]. Given that the role of PPP1R13L during embryonic eyelid closure has never been investigated, our current hypothesis is that PPP1R13L interacts with p63 in a similar way as it does in the developing epidermis of the skin and regulates the expression of the adhesion genes and/or genes involved in the differentiation of the cells of the leading edge. Our hypothesis is further supported by the observation that p63−/− mice also exhibit the EOB phenotype even though the development of the eyelids proceeds normally prior to the embryonic eyelid closure [26]. Although our current hypothesis implicates a role of PPP1R13L via interactions with p63, we cannot exclude the possibility that PPP1R13L may also be involved in molecular processes via interactions with the two other partners p65RelA and/or p53. Therefore, the role of PPP1R13L, as well as which molecular pathways are affected during embryonic eyelid closure requires further investigation.
Results from this study also identified the absence of meibomian glands in the woe2 eyelids. Meibomian glands along with hair, teeth, nails and exocrine glands are epidermal appendages formed from an ectodermal placode [27]. Ectodermal dysplasia syndromes comprise a group of greater than 175 highly diverse disorders characterized by abnormalities of at least two ectodermal tissues where at least one involves hair, teeth, nails or sweat glands. In ectodermal dysplasia patients, other structures exhibiting abnormalities may include the mammary glands, thyroid gland, thymus, anterior pituitary, adrenal medulla, central nervous system, external ear, melanocytes, cornea, conjunctiva, lacrimal gland and lacrimal duct and meibomian glands [28]. Interestingly, alterations of or the absence of meibomian glands was identified as one of the most reliable clinical ocular indications in ectodermal dysplasia patients [29]. In addition to the absence of meibomian glands, evaluation of woe2 eyes revealed abnormalities of the cornea and the anterior structures. The corneal epithelium is a tissue also originating from the surface ectoderm [30]; proper maturation of the corneal epithelium plays an essential role in ocular development, especially for the development of the anterior segment [30]. Taken together, these findings, along with the structural hair abnormalities previously identified as responsible for the wavy fur in wa3 mice [14], prompted us to morphologically evaluate other epidermal appendages in woe2 mice. However, our analysis did not identify any obvious defects in teeth, nails, lacrimal, salivary, mammary, sebaceous and sweat glands in woe2 mice. Our results suggest that PPP1R13L may have unique roles in the development of meibomian glands, cornea and hair follicles although PPP1R13L may have overlapping roles in the development of other ectodermal appendages.
It should be noted that the abnormalities in the cornea and anterior segment structures identified in woe2 mice may be solely related to the premature exposure to the environment consequent to the failure of the embryonic eyelid closure. Furthermore, the absence of meibomian glands may be further contributing to the abnormalities of the cornea--especially the corneal epithelium. Meibomian glands produce a lipid-rich secretion called meibum that is released from the orifices of the glands; the meibum is spread across the ocular surface and mix with tears produced by lacrimal glands to produce tear film that covers the entire ocular surface and has a protective, lubricatory, nutritional, and antimicrobial roles [31]. A dysfunction of meibomian glands results in abnormalities of the tear film and consequently ocular irritation, inflammation and ocular surface disease [32]. At this point, the molecular etiology of the anterior segment defects in woe2 remains unclear. It should be also noted that woe2 EOB and ocular phenotypes reported in this study are very similar to phenotypes reported for mice with defects in EGFR signaling pathway [5-14]. While no functional relationship between PPP1R13L and EGFR signaling has been reported, the similarities in the eyelid and ocular phenotypes as well as hair phenotypes identified in woe2 and mice with EGFR signaling defects suggest a common molecular pathway. Taken together, woe2 mice provide an excellent resource for determining the role of PPP1R13L especially during development as well as for elucidating the molecular mechanisms associated with PPP1R13L function.
Conclusions
The woe2 mouse is a novel spontaneous autosomal recessive mutation exhibiting EOB and wavy fur phenotypes. Genetic analysis identified a deletion in the Ppp1r13l gene. A detailed morphological evaluation showed that EOB in woe2 mice is due to a defect in embryonic eyelid closure. In addition to EOB, woe2 mice exhibited defects in the eye anterior segment development and the absence of meibomian glands. The precise role of PPP1R13L during the embryonic eyelid closure, development of meibomian glands and anterior segment structures requires further investigation. We anticipate that woe2 mice will provide a useful model in elucidating the etiology and underlying mechanisms of PPP1R13L function.
Competing interests
The authors of this manuscript declare that they have no competing interests.
Authors’ contributions
Design of the study, data analysis and critical revision of the manuscript was conducted by DJS and JAT. The original draft of the manuscript and experimentation was conducted by JAT. LL contributed to experimentation, analysis and interpretation of the data. This manuscript has been read and approved by all authors.
Supplementary Material
Sequences of PCR primers used in this study.
Contributor Information
Joseph Toonen, Email: jtoonen@mcw.edu.
Lina Liang, Email: lianglina2001@gmail.com.
Duska J Sidjanin, Email: djsidjani@mcw.edu.
Acknowledgments
We thank Dr. Joseph Besharse at the Medical College of Wisconsin for generously providing us with woe2 mice used in this study. We also thank Mr. Joseph Fogerty at the Medical College of Wisconsin for his help in preliminary studies. We would like to thank Mr. Doug Holmyard at the Mount Sinai Hospital, Toronto, Canada for his assistance with scanning electron microscopy. This work was supported in part by the National Institutes of Health grants EY18872 (D.J.S) and EY02414 (J.C.B.).
References
- Findlater GS, McDougall RD, Kaufman MH. Eyelid development, fusion and subsequent reopening in the mouse. J Anat. 1993;183(Pt 1):121–129. [PMC free article] [PubMed] [Google Scholar]
- Teraishi T, Yoshioka M. Electron-microscopic and immunohistochemical studies of eyelid reopening in the mouse. Anat Embryol (Berl) 2001;204(2):101–107. doi: 10.1007/s004290100189. [DOI] [PubMed] [Google Scholar]
- Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, Wisdom RM, Johnson RS. c-Jun is essential for organization of the epidermal leading edge. Dev Cell. 2003;4(6):865–877. doi: 10.1016/S1534-5807(03)00159-X. [DOI] [PubMed] [Google Scholar]
- Weng J, Luo J, Cheng X, Jin C, Zhou X, Qu J, Tu L, Ai D, Li D, Wang J. et al. Deletion of G protein-coupled receptor 48 leads to ocular anterior segment dysgenesis (ASD) through down-regulation of Pitx2. Proc Natl Acad Sci U S A. 2008;105(16):6081–6086. doi: 10.1073/pnas.0708257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann GB, Fowler KJ, Gabriel A, Nice EC, Williams RL, Dunn AR. Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell. 1993;73(2):249–261. doi: 10.1016/0092-8674(93)90227-H. [DOI] [PubMed] [Google Scholar]
- Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73(2):263–278. doi: 10.1016/0092-8674(93)90228-I. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179(5):2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- Hassemer EL, Le Gall SM, Liegel R, McNally M, Chang B, Zeiss CJ, Dubielzig RD, Horiuchi K, Kimura T, Okada Y. et al. The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics. 2010;185(1):245–255. doi: 10.1534/genetics.109.113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN. et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282(5392):1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, Jenkins NA, Lee DC. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 1994;8(4):399–413. doi: 10.1101/gad.8.4.399. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC. et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269(5221):230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376(6538):337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- Du X, Tabeta K, Hoebe K, Liu H, Mann N, Mudd S, Crozat K, Sovath S, Gong X, Beutler B. Velvet, a dominant Egfr mutation that causes wavy hair and defective eyelid development in mice. Genetics. 2004;166(1):331–340. doi: 10.1534/genetics.166.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Cross SH, Strunk KE, Morgan JE, Bailey CL, Jackson IJ, Threadgill DW. Wa5 is a novel ENU-induced antimorphic allele of the epidermal growth factor receptor. Mamm Genome. 2004;15(7):525–536. doi: 10.1007/s00335-004-2384-2. [DOI] [PubMed] [Google Scholar]
- Mine N, Iwamoto R, Mekada E. HB-EGF promotes epithelial cell migration in eyelid development. Development. 2005;132(19):4317–4326. doi: 10.1242/dev.02030. [DOI] [PubMed] [Google Scholar]
- Herron BJ, Rao C, Liu S, Laprade L, Richardson JA, Olivieri E, Semsarian C, Millar SE, Stubbs L, Beier DR. A mutation in NFkB interacting protein 1 results in cardiomyopathy and abnormal skin development in wa3 mice. Hum Mol Genet. 2005;14(5):667–677. doi: 10.1093/hmg/ddi063. [DOI] [PubMed] [Google Scholar]
- Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK, O'Connor DJ, Zhong S, Campargue I, Tomlinson ML. et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33(2):162–167. doi: 10.1038/ng1070. [DOI] [PubMed] [Google Scholar]
- Yang JP, Hori M, Sanda T, Okamoto T. Identification of a novel inhibitor of nuclear factor-kappaB, RelA-associated inhibitor. J Biol Chem. 1999;274(22):15662–15670. doi: 10.1074/jbc.274.22.15662. [DOI] [PubMed] [Google Scholar]
- Chikh A, Matin RN, Senatore V, Hufbauer M, Lavery D, Raimondi C, Ostano P, Mello-Grand M, Ghimenti C, Bahta A. et al. iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. EMBO J. 2011;30(20):4261–4273. doi: 10.1038/emboj.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notari M, Hu Y, Koch S, Lu M, Ratnayaka I, Zhong S, Baer C, Pagotto A, Goldin R, Salter V. et al. Inhibitor of apoptosis-stimulating protein of p53 (iASPP) prevents senescence and is required for epithelial stratification. Proc Natl Acad Sci U S A. 2011;108(40):16645–16650. doi: 10.1073/pnas.1102292108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liegel R, Chang B, Dubielzig R, Sidjanin DJ. Blind sterile 2 (bs2), a hypomorphic mutation in Agps, results in cataracts and male sterility in mice. Mol Genet Metab. 2011;103(1):51–59. doi: 10.1016/j.ymgme.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talamas E, Jackson L, Koeberl M, Jackson T, McElwee JL, Hawes NL, Chang B, Jablonski MM, Sidjanin DJ. Early transposable element insertion in intron 9 of the Hsf4 gene results in autosomal recessive cataracts in lop11 and ldis1 mice. Genomics. 2006;88(1):44–51. doi: 10.1016/j.ygeno.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan A, Lu X. ASPP: a new family of oncogenes and tumour suppressor genes. Br J Cancer. 2007;96(2):196–200. doi: 10.1038/sj.bjc.6603525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RA, Lu X, Jones EY, Siebold C. Biochemical and structural studies of ASPP proteins reveal differential binding to p53, p63, and p73. Structure. 2008;16(2):259–268. doi: 10.1016/j.str.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Simpson MA, Cook RW, Solanki P, Patton MA, Dennis JA, Crosby AH. A mutation in NFkappaB interacting protein 1 causes cardiomyopathy and woolly haircoat syndrome of Poll Hereford cattle. Anim Genet. 2009;40(1):42–46. doi: 10.1111/j.1365-2052.2008.01796.x. [DOI] [PubMed] [Google Scholar]
- Shalom-Feuerstein R, Lena AM, Zhou H, De La Forest Divonne S, Van Bokhoven H, Candi E, Melino G, Aberdam D. DeltaNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ. 2011;18(5):887–896. doi: 10.1038/cdd.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola ML. Genetic basis of skin appendage development. Semin Cell Dev Biol. 2007;18(2):225–236. doi: 10.1016/j.semcdb.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Visinoni AF, Lisboa-Costa T, Pagnan NA, Chautard-Freire-Maia EA. Ectodermal dysplasias: clinical and molecular review. Am J Med Genet A. 2009;149A(9):1980–2002. doi: 10.1002/ajmg.a.32864. [DOI] [PubMed] [Google Scholar]
- Kaercher T. Ocular symptoms and signs in patients with ectodermal dysplasia syndromes. Graefes Arch Clin Exp Ophthalmol. 2004;242(6):495–500. doi: 10.1007/s00417-004-0868-0. [DOI] [PubMed] [Google Scholar]
- Cvekl A, Tamm ER. Anterior eye development and ocular mesenchyme: new insights from mouse models and human diseases. Bioessays. 2004;26(4):374–386. doi: 10.1002/bies.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovich IA. The Meibomian puzzle: combining pieces together. Prog Retin Eye Res. 2009;28(6):483–498. doi: 10.1016/j.preteyeres.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Shimazaki J, Benitez-del-Castillo JM, Craig JP, McCulley JP, Den S, Foulks GN. The international workshop on meibomian gland dysfunction: report of the definition and classification subcommittee. Invest Ophthalmol Vis Sci. 2011;52(4):1930–1937. doi: 10.1167/iovs.10-6997b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of PCR primers used in this study.