

Abstract
The two DNA strands of the nuclear genome are replicated asymmetrically using three DNA polymerases, α, δ, and ε. Current evidence suggests that DNA polymerase ε (Pol ε) is the primary leading strand replicase, whereas Pols α and δ primarily perform lagging strand replication. The fact that these polymerases differ in fidelity and error specificity is interesting in light of the fact that the stability of the nuclear genome depends in part on the ability of mismatch repair (MMR) to correct different mismatches generated in different contexts during replication. Here we provide the first comparison, to our knowledge, of the efficiency of MMR of leading and lagging strand replication errors. We first use the strand-biased ribonucleotide incorporation propensity of a Pol ε mutator variant to confirm that Pol ε is the primary leading strand replicase in Saccharomyces cerevisiae. We then use polymerase-specific error signatures to show that MMR efficiency in vivo strongly depends on the polymerase, the mismatch composition, and the location of the mismatch. An extreme case of variation by location is a T-T mismatch that is refractory to MMR. This mismatch is flanked by an AT-rich triplet repeat sequence that, when interrupted, restores MMR to >95% efficiency. Thus this natural DNA sequence suppresses MMR, placing a nearby base pair at high risk of mutation due to leading strand replication infidelity. We find that, overall, MMR most efficiently corrects the most potentially deleterious errors (indels) and then the most common substitution mismatches. In combination with earlier studies, the results suggest that significant differences exist in the generation and repair of Pol α, δ, and ε replication errors, but in a generally complementary manner that results in high-fidelity replication of both DNA strands of the yeast nuclear genome.
Author Summary
The stability of complex and highly organized nuclear genomes partly depends on the ability of mismatch repair (MMR) to correct a variety of different mismatches generated as the leading and lagging strand templates are copied by three polymerases, each with different fidelity. Here we provide the first comparison, to our knowledge, of the efficiency of MMR of leading and lagging strand replication errors. We first confirm that Pol ε is the primary leading strand replicase, complementing earlier assignment of Pols α and δ as the primary lagging strand replicases. We then show that MMR efficiency in vivo strongly depends on the polymerase that generates the mismatch and on the composition and location of mismatches. In one extreme case, a flanking triplet repeat sequence eliminates MMR altogether. Overall, MMR is most efficient for mismatches generated at the highest rates and having the most deleterious potential, thereby ultimately achieving high-fidelity replication of both DNA strands.
Introduction
Three processes operate to ensure faithful replication of the eukaryotic nuclear genome [1], [2]. The first is the ability of DNA polymerases α, δ and ε to selectively insert correct rather than incorrect nucleotides onto correctly aligned rather than misaligned primer-templates. The second is proofreading, the 3′ exonucleolytic excision of errors from the primer terminus during replication. The third is mismatch repair (MMR) of errors that escape proofreading (reviewed in [3]–[7]). MMR begins when a mismatch is recognized by homologues of the bacterial MutS homodimer, either Msh2-Msh6 (MutSα) or Msh2-Msh3 (MutSβ). This recognition initiates a series of steps that ultimately remove the replication error from the nascent strand and allow new DNA to be synthesized accurately.
The origin and nature of the strand discrimination signal used for MMR in vivo remains uncertain. MMR requires the presence of a discontinuity in the newly synthesized strand. At least in vitro, this discontinuity can be a nick or gap located either 3′ or 5′ to the mismatch, with the protein requirements for MMR differing somewhat depending on the location of the DNA ends relative to the mismatch. This provides an attractive possibility (reviewed in [3]), namely that MMR may be directed to the nascent strand by the 3′ ends of growing chains at the replication fork and/or by the 5′ ends of Okazaki fragments that are transiently present during lagging strand replication. That the latter could provide a higher signal density for MMR of lagging strand replication errors was suggested in an earlier study of MMR of a damaged (8-oxo-G-A) mismatch [8]. This leads to a previously unexplored question addressed by the present study, i.e., is the efficiency of MMR similar or different for mismatches generated during leading and lagging strand replication?
Investigation of this question is complicated by the fact that DNA polymerases α, δ and ε (Pols α, δ and ε, respectively) are all required to efficiently replicate the nuclear genome [9], and these polymerases have different error rates and error specificities [2], [10]. Over the years, multiple models have been considered for the division of labor among these three polymerases during replication (reviewed in [9]–[12]). Among these models, recent evidence [2], [13], [14] suggests that under normal circumstances, the leading strand template is primarily replicated by Pol ε, while the lagging strand template is replicated by Pol α-primase and Pol δ.
Although MMR corrects errors made by all three polymerases [2], [13], [15]–[21], it has only recently become possible to determine the extent to which MMR efficiency, and possibly MMR enzymology, varies depending on the replicase that made the error, the nascent strand containing the error and/or the location of the error within a DNA strand. We are investigating these variables using Saccharomyces cerevisiae strains containing mutant alleles of the POL1 (Pol α), POL2 (Pol ε) and POL3 (Pol δ) genes. These mutant alleles, pol1-L868M [18], [19], pol2-M644G [13] and pol3-L612M ([2] and references therein), encode enzymes with single animo acid replacements at the polymerase active site that reduce the fidelity of DNA synthesis. As a consequence, strains harboring these alleles have elevated spontaneous mutation rates, thereby allowing assignment of responsibility for most in vivo errors to a chosen mutator polymerase, rather than its wild type counterparts [2], [13]. In strains containing these mutator polymerases, URA3 mutation rates and mutational spectra can be determined and used to calculate the rates for specific mutations, e.g., single base substitutions and insertions/deletions (indels) in various sequence contexts. Comparison of these rates in MMR-proficient yeast strains to strains that lack MSH2-dependent MMR yields a calculation of the apparent MSH2-dependent MMR efficiency for a variety of replication errors generated during replication in vivo.
Using this approach, we recently described the efficiency of repairing lagging strand replication errors generated by L868M Pol α and L612M Pol δ [21]. Here we extend the effort using yeast strains encoding M644G Pol ε, allowing the comparison of MMR correction efficiencies for replication errors made by each of the three eukaryotic replicative polymerases. The results indicate that on average, MMR balances the fidelity of leading and lagging strand DNA replication, but with exceptions that place some base pairs at high risk of mutation from replication infidelity even in cells with normal MMR.
Results
The present study presents what to our knowledge is the first direct comparison of MMR efficiency for errors made by all three replicases in vivo, thereby providing insights into the contribution of MMR to leading and lagging strand replication fidelity. This comparison is a continuation of efforts to examine the possibility that MMR may be directed to the nascent strand by the 3′ ends of growing chains at the replication fork [22], and/or by the 5′ ends of Okazaki fragments that are transiently present during lagging strand replication [8].
Pol ε preferentially incorporates rNMPs into the nascent leading strand
Our previous inference that Pol ε is a leading strand replicase was based on patterns of rare mutations in one gene (URA3) at one locus (AGP1) [13]. Two recent studies have made it feasible to test Pol ε strand assignment using a different biomarker, ribonucleotide incorporation into nuclear DNA. The first study demonstrated that, in addition to reduced fidelity for single base mismatches, M644G Pol ε also has reduced sugar discrimination, i.e., it incorporates rNTPs into DNA much more readily than does wild-type Pol ε [23]. In that study, rNMPs incorporated into nascent DNA during replication by M644G Pol ε were detected as alkali-sensitive sites in the nuclear genome of a pol2-M644G rnh201Δ strain, which lacks the ability to repair rNMPs in DNA due to deletion of the RNH201 gene encoding the catalytic subunit of RNase H2. A more recent study exploited this fact to probe the genomic DNA of a homologous S. pombe polε-M630F rnh201Δ mutant strain by strand-specific Southern blotting [14]. When strand-specific probes flanking ARS3003/3004 were used, the results revealed that more rNMPs were incorporated into the nascent leading strand than into the nascent lagging strand. This led to the interpretation that, as in budding yeast, fission yeast Pol ε is also the primary leading strand replicase [14]. Using this same strategy, we examined the strand specificity of rNMP incorporation in S. cerevisiae pol2-M644G rnh201Δ strains with the URA3 reporter in one of two possible orientations, using alkali treatment and subsequent probing for either the nascent leading or lagging strand with strand-specific URA3 probes (Figure 1A). One of the two strands from each pol2-M644G rnh201Δ strain was preferentially sensitive to alkaline hydrolysis (Figure 1B). In each case, this corresponded to the nascent leading strand products of replication (probe A in orientation 2 and probe B in orientation 1). These results strongly support the idea that Pol ε preferentially replicates the leading strand template. Note that the distribution of ribonucleotides within the two strands across the whole genome remains to be determined and could differ.
Figure 1. Strand-specific incorporation of rNMPs into genomic DNA.
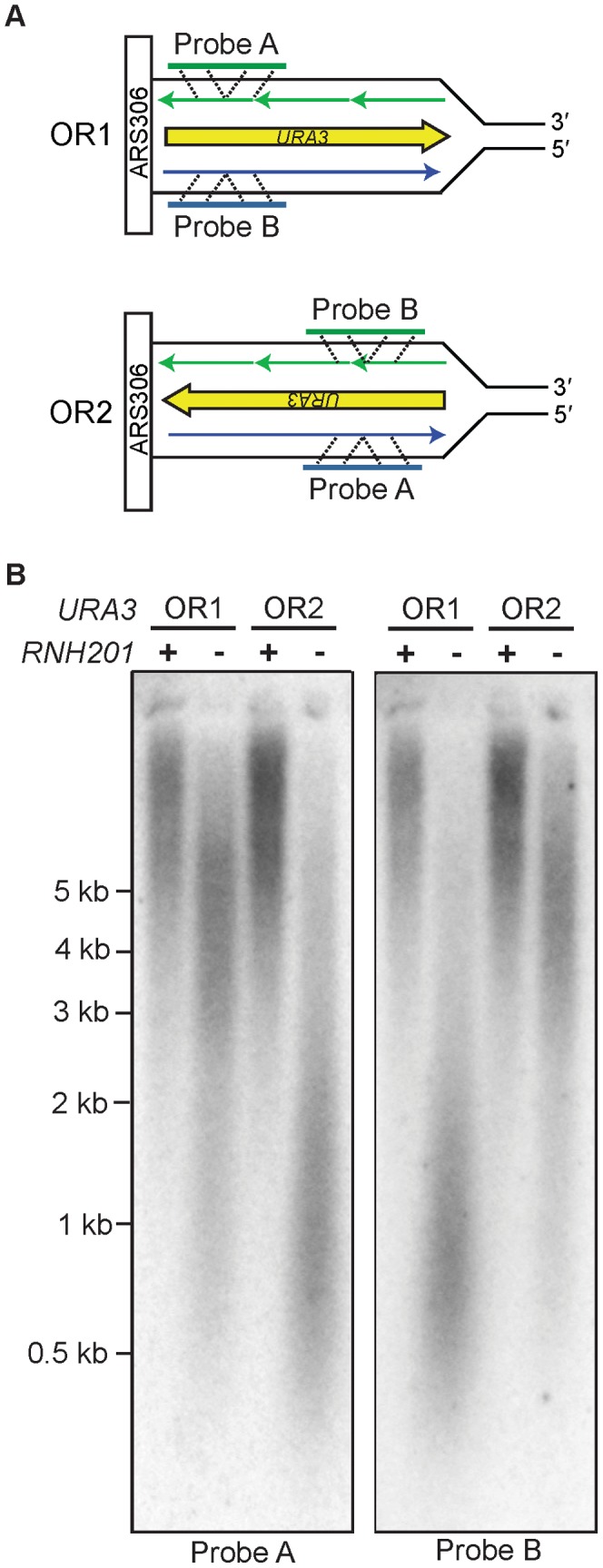
A. The orientation of the URA3 reporter with respect to coding sequence is indicated as orientation 1 (OR1) or orientation 2 (OR2). DNA template strands are in black, the nascent leading strand is in blue and the nascent lagging strand is in green. B. Detection of alkali-sensitive sites in yeast genomic DNA reveals a strand bias for incorporation of ribonucleotides. Following alkaline hydrolysis and alkaline agarose-electrophoresis, the DNA was transferred to a nylon membrane and processed for Southern analysis. The indicated region of the URA3 reporter gene was examined using strand-specific radiolabeled probes that anneal to either the nascent leading or nascent lagging strand. The sizes of DNA markers are indicated on the left. All strains harbor the pol2-M644G mutator allele. Increased DNA mobility is indicative of alkali-sensitivity due to the presence of ribonucleotides in the nascent DNA strand.
Mutagenesis in MMR–proficient pol2-M644G strains
The strategy used here to study strand-specific MMR involves measuring spontaneous mutation rates in yeast strains with the URA3 reporter gene present in either of two orientations, both proximal to ARS306, a well-characterized, early-firing replication origin [24]. In our initial study of the role of Pol ε in replication [13], we compared mutation rates in MMR proficient (MSH2 +) strains with wild type Pol ε (encoded by the POL2 gene) to rates in strains with the pol2-M644G mutation. The pol2-M644G strains had elevated mutation rates [13], an observation that is reproduced here (Table 1). The majority of 5-FOA resistant mutants had single-base mutations in the URA3 gene. In orientation 1, these were predominantly A-T to T-A mutations at base pairs 279 and 686. These mutations were rare in orientation 2 (partial spectra in [13], complete spectra in Figure S1A). This strong orientation bias, and the fact that the in vitro error rate for template T-dTMP mismatches by M644G Pol ε is much higher than the error rate for template A-dAMP mismatches, implies that Pol ε participates in leading strand DNA replication [13]. Two later studies [2], [25] indicated that Pol δ primarily acts as a lagging strand polymerase and has a less substantial role in leading strand replication. This further implied that Pol ε not only participates in leading strand DNA replication, but that it is the major leading strand replicase.
Table 1. Mutation rates and sequencing data for pol2-M644G ± MSH2 strains.
Some 5-FOA resistant mutants had no sequence change in the 804 base pair URA3 open reading frame. These mutants were not investigated further, but they may result from epigenetic silencing, they may contain sequence changes in the promoter or 3′ untranslated region of URA3, or they may contain mutations in other genes that result in 5-FOA resistance.
Expanded from [21].
Expanded from [13].
Mutation rates and specificity in pol2M644G msh2Δ strains
The pol2-M644G msh2Δ mutants have strongly elevated mutation rates relative to the MSH2 + strains (Table 1), indicating that the vast majority of the mutations are made by M644G Pol ε. In the absence of mismatch repair, most 5-FOA resistant mutants contained single base changes that were widely scattered throughout the URA3 coding sequence (Figure S1B). As compared to MMR proficient pol2-M644G strains, base pairs 279 and 686 in pol2-M644G msh2Δ strains did not stand out as hotspots for A-T to T-A transversions in orientation 1, even though base substitution and single base deletion hotspots were observed at several other locations (Figure S1B).
MMR correction factors
The data in Table 1 and Figure S1 were used to calculate rates for single base mutations in the MMR-proficient and msh2Δ strains (Table S2). The ratio of these rates reflects the apparent MMR correction efficiency for each type of error, and the results can be compared (see discussion) to those reported earlier [21] for replication errors made by L868M Pol α and L612M Pol δ. As noted previously [21], [26]–[29], certain correction factors could be higher if some mismatches in the MMR proficient strains are not subject to MMR, either because they are damaged or because they are generated during DNA transactions that occur outside of replication.
Conclusions about the overall balance of repair between strands and polymerases derive from collective consideration of all single base mismatches. In the pol2-M644G strain background, the MMR correction factor for all single base mismatches is 250-fold (Table 2; Figure 2A, blue bar; Table S2), i.e., on average, 249 of 250 single base replication errors generated by M644G Pol ε are corrected by MMR. This correction factor is higher than for L612M Pol δ (Table 2; Figure 2A, green diamond), but lower than for L868M Pol α (Table 2; Figure 2A, red diamond). As a consequence, the mutation rates for all three variant polymerase strains are similar when MMR is operative (top line in Table 2). Average correction factors are high for each of the four classes of single base changes generated by M644G Pol ε (Figure 2A), in the following order: deletions (1,500-fold), insertions (1,100-fold), transitions (440-fold) and transversions (72-fold). Correction factors vary widely between specific positions in the URA3 open reading frame. Figure 2B–2D show eight locations where it is possible to compare MMR of the same mismatch generated by M644G Pol ε during leading strand replication (blue bars) or by Pol α (red diamonds) and δ (green diamonds) during lagging strand replication (expanded from [21]). In order to maintain equivalent template context, leading strand errors found in one URA3 orientation in the pol2-M644G strains are always compared to lagging strand errors found in the other URA3 orientation in the pol1-L868M and pol3-L612M strains. For example, the correction factors in Figure 2B for deleting an A-T pair from the three longest runs of A-T pairs in the URA3 coding sequence (base pairs 174–178, 201–205 and 255–260, Figure S1) are each inferred to involve a single unpaired T. The comparative MMR correction factors and their implications are considered in the Discussion.
Table 2. Mutation rates and correction factors for all single-base mismatches in three mutator polymerase backgrounds.
Figure 2. Correction factors for various mismatches made by each mutator polymerases.

Mismatch repair correction factors for errors created by M644G Pol ε (blue columns), L868M Pol α (red diamonds), and L612M Pol δ (green diamonds) [21]. All correction factors are significant (p≤0.05) unless otherwise noted. (A) Correction factors for six classes of mutations across all URA3 sequence positions. URA3-orientations 1 and 2 correction factors are averaged (geometric mean). Mutation class abbreviations are shown in parentheses. In panels B, C and D, for specific mutations, the inferred mismatch and the surrounding sequence context are shown below the chart. Mutation positions are shown to the left. The nascent (above) and template (below) strands are shown to the right. Triangles indicate synthesis direction. The coding strand is green, the non-coding strand is blue, and mismatched bases are red. (B) Correction factors for unpaired T bases at specific URA3 positions, as compared to averages and general frameshift mutations. (C) Correction factors for C-dT mismatches at specific URA3 positions, as compared to averages and general transversion mutations. Only positions 345 and 679 had sufficient observations to allow significant calculations for all three polymerases. (D) Correction factors for G-dT mismatches at specific URA3 positions, as compared to averages and general transition mutations. URA3 positions 310, 608, and 764 had sufficient observations. x– Average of relevant sequence positions and both URA3 orientations. ≤ Upper bound, estimated by increasing msh2Δ observation count from 0 to 1 for purposes of correction factor calculations. a Calculated from only one URA3 orientation due to insufficient observations in the other. b p>0.05.
A-T to T-A transversions at base pair 686
In contrast to the efficient repair of most single mismatches, the rate of A-T to T-A transversions at base pair 686 in orientation 1 (Figure S1 and Table S3) is no higher in the pol2-M644G msh2Δ strain than in the MMR-proficient pol2-M644G strain (Table S2). This indicates that T-T mismatches generated at base pair 686 during leading strand replication by M644G Pol ε are not efficiently corrected by MMR (Figure 3, “T-dT, 686,” dark blue bar). This contrasts with an average of 41-fold correction (dark blue bar on left) of the same mismatch inferred at all other A-T base pairs in URA3, i.e., A to T substitutions in orientation 1 and T to A transversions in orientation 2 (Figure S1). Adjacent to base pair 686 is a triplet repeat sequence, 5′-ATT ATT ATT gTT (designated here as ATT3). For several reasons (see Discussion), we speculated that this sequence might suppress MMR at base pair 686. To test this, we constructed strains in which ATT3 was modified to 5′-ATA ATC ATA gTT (designated ATT0, see Figure 3), with the three (underlined) changes interrupting the repeat units without changing the amino acid sequence. We then measured spontaneous mutation rates and generated mutational spectra (Figure S2) to determine if the flanking sequence changes allowed MMR of T-T mismatches at base pair 686. The results (Table S3) indicate that this is indeed the case. The MMR correction factor at base pair 686 increased to 35-fold (Figure 3, p≤0.001), indicating that 97% of T-T mismatches are repaired when base pair 686 is flanked by ATT0.
Figure 3. Restoration of T-dT repair at position 686 by removing a flanking triplet repeat.

Mismatch repair correction factors for errors created by M644G Pol ε in ATT3 (dark blue columns) and ATT0 (light blue columns) URA3 sequences. The T-dT mismatch at position 686 and the surrounding sequence context are shown below the chart: nascent strand above; template strand below. Triangles indicate the direction of synthesis. The three silent mutations made to convert ATT3 into ATT0 URA3 are underlined. Note that the polymerase active site encounters the triplet repeat in the ATT3 template strand after making the T-dT mismatch at position 686. ≤ Upper bound, estimated by increasing msh2Δ observation count from 0 to 1 for purposes of correction factor calculations.
Single base-base mismatches are repaired by MutSα (Msh2-Msh6) but not by MutSβ (Msh2-Msh3) [3]–[7], implying that the ATT3 sequence is suppressing repair of the T-T mismatch that would normally occur via MutSα. However, given evidence that MutSβ can bind to a non-B-DNA structure that can form in a triplet repeat sequence and promote triplet repeat expansion (reviewed [30]), we examined whether suppression of MMR at base pair 686 might depend on MutSβ. This was done by calculating the A-T to T-A mutation rate at base pair 686 in URA3 orientation 1 in a pol2-M644G msh3Δ rnh201Δ strain [31]. The calculated A-T to T-A rate is 17×10−8, which is no lower than observed here in the Msh3+ strain (6.8×10−8, Table S3). Thus suppression of MMR by ATT3 is independent of MutSβ.
Discussion
This study provides new insights into relationships between the intrinsic asymmetry of DNA replication and MMR in yeast.
Ribonucleotides are “biomarkers” of Pol ε action in vivo
We previously inferred that Pol ε participates in leading strand replication using base substitutions as biomarkers for leading strand replication. These events are rare, occurring approximately once per 10 million incorporations. The present study uses ribonucleotides as an independent and much more abundant biomarker. The preferential presence of ribonucleotides in the nascent leading strand observed here in pol2-M644G rnh201Δ strains (URA3 orientation 1 and orientation 2; Figure 1) strongly supports the inference that Pol ε primarily participates in leading strand replication. This does not preclude occasional Pol ε participation in lagging strand replication. The interpretation that Pol ε primarily participates in leading strand replication lends credibility to the interpretations presented below regarding the efficiency of repairing mismatches made by Pol ε during leading strand replication as compared to mismatches of similar composition made by Pols α and δ during lagging strand replication. An additional notable point here is that the sizes of the nascent leading strand fragments resulting from alkaline hydrolysis of DNA from the pol2-M644G rnh201Δ strains (Figure 1) indicate that approximately one ribonucleotide may be incorporated for every 1,000 deoxyribonucleotides. This density of ribonucleotide incorporation into DNA is about four orders of magnitude higher than for A-T- to T-A transversions. Thus ribonucleotides mapped by deep sequencing techniques could serve as high density, genome-wide biomarkers of Pol ε action in vivo during replication and possibly during repair and recombination.
Variations in repairing M644G Pol ε replication errors
The average MMR correction factors for errors made by M644G Pol ε are highest for indels, intermediate for transitions and lowest for transversions (Figure 2A). This rank order is common to E. coli [28], [29], [32] and to errors made by yeast Pols α and δ [21], suggesting that MMR has conserved the ability to most efficiently correct the most potentially deleterious errors (indels), and also the base-base mismatches made at the highest rates by both bacterial and eukaryotic replicases. This general principal is qualified by the observation that MMR efficiency varies, even for the same inferred mismatch (e.g., either an extra T, a C-dT or a G-dT mismatch, Figure 2B, 2C and 2D, respectively) made by the same polymerase (M644G Pol ε) during replication of the same (leading) strand. Most sequence-dependent variations in MMR efficiency seen here are in the 2- to 10-fold range (Figure 2) depending on the comparison. That such variations are typically small is perhaps expected, since MMR is needed to preserve the stability of nuclear genomes despite their enormous sequence complexity.
Variations due to mismatch composition and location are consistent with biochemical studies showing differences in MMR in vitro [33] and with mutational studies in vivo in which the identity of the replicase that made the mismatch was unknown. Several explanations for variations in eukaryotic MMR efficiency can be explored in the future. For example, the efficiency with which E. coli repairs transversion mismatches in phage λ increases with increasing G-C content in neighboring nucleotides [32], and recognition of certain mismatches by MutSα is influenced by a 6-nucleotide region surrounding the mismatch [34]. Thus it may be that flanking sequences, such as those shown in Figure 2, influence eukaryotic MMR efficiency in vivo by modulating (i) mismatch binding by MutSα, which contacts several base pairs on either side of the mismatch [35], (ii) base pair stacking, since a MutSα-bound mismatched base stacks with a conserved phenylalanine in Msh6, and/or (iii) DNA flexibility, since MutSα-bound mismatched DNA is kinked, and a transition between bent and unbent DNA may be critical for limiting MMR to processing of mismatched as compared to matched base pairs [36]. Variations in MMR efficiency might also depend on proteins that operate downstream of mismatch binding, such as MutLα or exonucleases, or they may reflect other variables, such as the timing of nucleosome reloading behind the replication fork, nucleosome dynamics and/or chromatin remodeling.
A natural DNA sequence that suppresses MMR
A striking observation here is the apparent absence of MMR of the A-T to T-A transversion at base pair 686 (Figure 3), which is inferred to result from a T-T mismatch made by M644G Pol ε during leading strand replication. This lack of repair contrasts sharply with efficient repair at many other locations. For example, the deletion mismatch at base pairs 255–260, which is predicted to involve a mismatch containing a single unpaired T in the template (Figure 2B), has an approximately 6000-fold higher correction factor than for the T-T mismatch at base pair 686. Lack of repair at base pair 686 is not due to a general inability to correct A-T to T-A transversion mismatches, because the average correction factor for these events elsewhere in URA3 is 41-fold (Figure 3). The absence of correction at position 686 led us to test whether MMR was inhibited by the adjacent 5′-ATTATTATTgTT sequence. There were several reasons to suspect that this could be the case. The sequence is A-T rich and may have unusual helical parameters that could diminish MMR. For example, sequences containing larger numbers of ATT repeats can form a non-hydrogen bonded structure [37], and can be induced into hairpins by the DNA minor groove binding ligand DAPI (4′,6-diamidino-2-phenylindole) [38], [39]. Triplet repeat sequences can form non-B-DNA structures that bind MMR proteins (reviewed in [30]), and they are often associated with genome instability (reviewed in [40]), albeit characterized by indels rather than base substitutions. In addition, recent studies have demonstrated that nucleosomes influence the behavior of MMR proteins and visa versa (e.g., see [41]–[44]), and nucleosome binding to DNA is influenced by DNA sequence, with A-T-rich dinucleotides such as those present in ATT3 having an important role in nucleosome positioning (e.g., see [45], [46] and references therein).
For these reasons, we examined MMR at base pair 686 after changing the flanking sequence to eliminate the triplet repeats and decrease A-T content by one base pair. The results indicate that these changes allowed correction of 97% of the mismatches generated by M644G Pol ε at base pair 686 (88% correction at the lower 95% confidence limit, Figure 3). This suggests that the ATT3 flanking sequence is a natural cis-acting suppressor of the normal MSH2-dependent MMR machinery. Suppression does not decrease upon deletion of MSH3, and thus is MutSβ independent, unlike triplet repeat expansion [30]. Collectively, position 686 and ATT3 are an example of what has been called an “At Risk sequence Motif” [47], i.e., a naturally occurring DNA sequence that results in inefficient operation of a DNA transaction required for genome stability. The fact that one such sequence exists in the 804 base pair open reading frame of URA3 leads one to wonder how many natural suppressors of MMR might be present in nuclear genomes. This issue is currently being investigated using the deep sequencing approach previously used to infer that Pol δ is a lagging strand replicase across the yeast genome [25]. Experiments are also planned to examine which (if any) of the possibilities mentioned in the preceding section may be relevant to inefficient MMR at base pair 686.
Correcting leading and lagging strand replication errors
We previously suggested that MMR may be directed to the nascent strand by the 3′ ends of growing chains at the replication fork [22], and/or by the 5′ ends of Okazaki fragments that are transiently present during lagging strand replication [8]. The 5′ ends of Okazaki fragments, and perhaps the PCNA required to process these ends, could potentially provide a higher signal density for MMR of lagging strand replication errors as compared to errors generated during leading strand replication, which is thought to be more continuous. If so, then MMR might be more efficient in correcting lagging strand errors. In an initial test of this hypothesis, we found that mutagenesis due to a mismatch formed at one particular G-C base pair during replication of unrepaired 8-oxo-G in ogg1-deficient yeast was lower for lagging as compared to leading strand replication, and importantly, that this bias was largely eliminated in MMR defective strains [8]. Among several possible explanations that we considered for loss of the strand bias, one was that 8-oxo-G-dA mismatches made during lagging strand replication may be more efficiently corrected than are 8-oxo-G-dA mismatches made during leading strand replication. A major goal of the present study was to test this hypothesis for multiple, natural (i.e., undamaged) mismatches generated at different locations during replication of a larger target sequence. The present study accomplishes this, and allows the first direct comparison of MMR efficiency for errors made by all three replicases, to our knowledge, thereby providing insights into the contribution of MMR to leading and lagging strand replication fidelity.
From the results in Figure 2, we conclude that in general, mismatches made by all three replicases are repaired very efficiently. This is logical given the need to preserve genetic information in both DNA strands. This conclusion is independent of various models regarding which DNA polymerase replicates which strand (reviewed in [11], [12]). Other implications derive from the model wherein Pols α and δ are the primary lagging strand replicases and Pol ε is the primary leading strand replicase. In our earlier report [21], we pointed out that correction factors were higher for mismatches made by Pol α than for the same mismatches made by Pol δ, suggesting that the 5′ ends of Okazaki fragments may be strand discrimination signals and that MMR efficiency may be related to the proximity of a mismatch to that signal. This is interesting given that DNA polymerase ε is highly processive, at least as processive as DNA polymerase δ, and that leading strand replication is thought to be largely continuous [48], [49], [50]. It is of course conceivable that leading strand replication may not be as continuous as current models imply. If leading strand replication is indeed largely continuous, then the fact that MMR corrects most Pol ε errors about as efficiently as it corrects errors made by Pols α and δ (Figure 2) implies the existence of MMR signals other than the 5′ ends of Okazaki fragments, and these can very efficiently direct MMR to the nascent leading strand. Possible signals for leading strand replication include the above-mentioned 3′ ends of growing chains at the replication fork [22], [51], [52], nicks introduced into the nascent leading strand by nucleases, and/or asymmetrically bound PCNA [8], [53]. PCNA is a particularly attractive possibility for differentially modulating the efficiency of MMR of errors made by the three replicases, because it is involved in early steps in MMR (see [3]–[7] for review]), it does not influence DNA synthesis by Pol α, and it does stimulate DNA synthesis by both Pol δ and Pol ε, albeit through different PCNA-polymerase interactions (see [9] and references therein).
The results in Figure 2 further suggest that, even for the same mismatch (extra T, G-dT or C-dT) in a common sequence context, MMR efficiency varies depending on which polymerase made the error. In two of three instances involving deletion of a single template T (Figure 2B), the repair of mismatches made by Pol δ is higher than for mismatches made by Pol ε. This correlates with the observation that Pol δ generates this mismatch in vitro at a higher rate than does Pol ε [54]. Similarly, transitions and transversions (Figure 2A) and several site-specific base substitutions (Figure 2C and 2D) generated by Pol α are corrected more efficiently than are mismatches generated by Pol δ and Pol ε. Pol α lacks an intrinsic proofreading exonuclease activity and is less accurate than proofreading-proficient Pols δ and ε (reviewed in [10], [55]). Thus the present study of mismatches generated by Pol ε extends the idea that MMR has evolved to most efficiently correct the most deleterious mismatches (i.e., indel mismatches). Within classes of similar deleterious potential (base-base mismatches), evolution has produced the highest efficiency versus the most frequently generated mismatches. In a model wherein Pol ε is the major leading strand replicase and Pols α and δ conduct about 10% and 90% of lagging strand replication [2], respectively, the results (Table 2; Figure 2A, average repair for single base errors) further suggest that MMR balances the fidelity of replication of the two strands despite the use of replicases with substantially different fidelity and error specificity.
Materials and Methods
Strains, mutation rates, and analysis of ura3 mutants
The strains used in this study, the measurements of spontaneous mutation rates and the sequencing of URA3 mutants were as previously described [2], [13], [21], save that MSH2 was deleted from haploid pol2-M644G strains rather than diploid. The ATT3 to ATT0 conversion was made via site-directed mutagenesis and integration pop-out [56] in a strain with wild type polymerases. PCR product containing the ATT0 URA3 allele was then transformed into msh2Δ backgrounds and proper insertion verified via sequencing.
Probing for alkali-sensitive sites in genomic DNA
Genomic DNA was isolated from exponentially growing cultures (grown in YPDA at 30°C) using the Epicentre Yeast DNA purification kit. Five µg of DNA was treated with 0.3 M KOH for 2 h at 55°C and subjected to alkaline-agarose electrophoresis as described [23]. Following neutralization, DNA was transferred to a charged nylon membrane (Hybond N+) by capillary action and probed by Southern analysis. Strand-specific radiolabeled probes were prepared from a PCR-amplified fragment of URA3 template, using a previously described procedure and probe design [14].
Statistical analysis
See Text S1.
Supporting Information
Mutational spectra in pol2-M644G and pol2-M644G msh2Δ strains. The URA3 reporter was present in either orientation 1 (OR1) or orientation 2 (OR2) at position AGP1. The coding strand of the URA3 open reading frame is shown, with every 10th base indicated by a dot and mutations depicted above (OR1) and below (OR2) the wild type URA3 sequence. Single letters represent base substitutions, open triangles represent single base deletions, and closed triangles represent single base additions. Indels in homonucleotide runs are shown at the 5′-most position of the run. (A) Spectra in MSH2 strains [13]. (B) Spectra in msh2Δ strains.
(TIF)
Mutational spectra in pol2-M644G and pol2-M644G msh2Δ strains with ATT0 URA3. As for Figure S1, with spectra for the pol2-M644G msh2Δ and pol2M644G MSH2 strains shown above and below the ATT0 URA3 sequence, respectively. The three bases that differ between the ATT3 and ATT0 URA3 sequences are shown in bold (positions 690, 693, and 696).
(TIF)
Multi-base mutations omitted from spectrum figures.
(DOCX)
Mutation counts, mutation rates, and MMR correction factors.
(XLSX)
Mutation Rates for pol2-M644G ± msh2Δ and ATT3/ATT0 URA3 Orientation 1 strains. a Expanded from [21]. b Expanded from [13].
(DOCX)
The supporting information includes methods for calculating and statistical analyses of mutation rates and correction factors.
(DOC)
Acknowledgments
We thank Jeffrey Stumpf and Steven Roberts for thoughtful comments on the manuscript, and the NIEHS DNA Sequencing Core and the Molecular Genetics Core Facilities for sequence analysis of 5-FOA-resistant mutants.
Funding Statement
This work was supported by Project Z01 ES065089 to TAK, from the Division of Intramural Research of the National Institutes of Health, National Institute of Environmental Health Sciences. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kunkel TA (2004) DNA replication fidelity. J Biol Chem 279: 16895–16898. [DOI] [PubMed] [Google Scholar]
- 2. Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA (2008) Division of labor at the eukaryotic replication fork. Mol Cell 30: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kunkel TA, Erie DA (2005) DNA mismatch repair. Annu Rev Biochem 74: 681–710. [DOI] [PubMed] [Google Scholar]
- 4. Iyer RR, Pluciennik A, Burdett V, Modrich PL (2006) DNA mismatch repair: functions and mechanisms. Chem Rev 106: 302–323. [DOI] [PubMed] [Google Scholar]
- 5. Hsieh P, Yamane K (2008) DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev 129: 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiricny J (2006) The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 7: 335–346. [DOI] [PubMed] [Google Scholar]
- 7. Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18: 85–98. [DOI] [PubMed] [Google Scholar]
- 8. Pavlov YI, Mian IM, Kunkel TA (2003) Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr Biol 13: 744–748. [DOI] [PubMed] [Google Scholar]
- 9. Burgers PM (2009) Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem 284: 4041–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCulloch SD, Kunkel TA (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 18: 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kunkel TA, Burgers PM (2008) Dividing the workload at a eukaryotic replication fork. Trends Cell Biol 18: 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pavlov YI, Shcherbakova PV (2010) DNA polymerases at the eukaryotic fork-20 years later. Mutat Res 685: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA (2007) Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 317: 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyabe I, Kunkel TA, Carr AM (2011) The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet 7: e1002407 doi:10.1371/journal.pgen.1002407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morrison A, Bell JB, Kunkel TA, Sugino A (1991) Eukaryotic DNA polymerase amino acid sequence required for 3′–5′ exonuclease activity. Proc Natl Acad Sci U S A 88: 9473–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morrison A, Johnson AL, Johnston LH, Sugino A (1993) Pathway correcting DNA replication errors in Saccharomyces cerevisiae. Embo J 12: 1467–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morrison A, Sugino A (1994) The 3′–>5′ exonucleases of both DNA polymerases delta and epsilon participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol Gen Genet 242: 289–296. [DOI] [PubMed] [Google Scholar]
- 18. Niimi A, Limsirichaikul S, Yoshida S, Iwai S, Masutani C, et al. (2004) Palm mutants in DNA polymerases alpha and eta alter DNA replication fidelity and translesion activity. Mol Cell Biol 24: 2734–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, et al. (2006) Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr Biol 16: 202–207. [DOI] [PubMed] [Google Scholar]
- 20. Albertson TM, Ogawa M, Bugni JM, Hays LE, Chen Y, et al. (2009) DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A 106: 17101–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nick McElhinny SA, Kissling GE, Kunkel TA (2010) Differential correction of lagging-strand replication errors made by DNA polymerases {alpha} and {delta}. Proc Natl Acad Sci U S A 107: 21070–21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Umar A, Buermeyer AB, Simon JA, Thomas DC, Clark AB, et al. (1996) Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 87: 65–73. [DOI] [PubMed] [Google Scholar]
- 23. Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, et al. (2010) Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol 6: 774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poloumienko A, Dershowitz A, De J, Newlon CS (2001) Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol Biol Cell 12: 3317–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Larrea AA, Lujan SA, Nick McElhinny SA, Mieczkowski PA, Resnick MA, et al. (2010) Genome-wide model for the normal eukaryotic DNA replication fork. Proc Natl Acad Sci U S A 107: 17674–17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kramer B, Kramer W, Fritz HJ (1984) Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system of E. coli. Cell 38: 879–887. [DOI] [PubMed] [Google Scholar]
- 27. Dohet C, Wagner R, Radman M (1985) Repair of defined single base-pair mismatches in Escherichia coli. Proc Natl Acad Sci U S A 82: 503–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schaaper RM, Dunn RL (1991) Spontaneous mutation in the Escherichia coli lacI gene. Genetics 129: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schaaper RM (1993) Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J Biol Chem 268: 23762–23765. [PubMed] [Google Scholar]
- 30. McMurray CT (2010) Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11: 786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clark AB, Lujan SA, Kissling GE, Kunkel TA (2011) Mismatch repair-independent tandem repeat sequence instability resulting from ribonucleotide incorporation by DNA polymerase epsilon. DNA Repair (Amst) 10: 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones M, Wagner R, Radman M (1987) Repair of a mismatch is influenced by the base composition of the surrounding nucleotide sequence. Genetics 115: 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Su SS, Lahue RS, Au KG, Modrich P (1988) Mispair specificity of methyl-directed DNA mismatch correction in vitro. J Biol Chem 263: 6829–6835. [PubMed] [Google Scholar]
- 34. Marsischky GT, Kolodner RD (1999) Biochemical characterization of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 complex and mispaired bases in DNA. J Biol Chem 274: 26668–26682. [DOI] [PubMed] [Google Scholar]
- 35. Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, et al. (2007) Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell 26: 579–592. [DOI] [PubMed] [Google Scholar]
- 36. Wang H, Yang Y, Schofield MJ, Du C, Fridman Y, et al. (2003) DNA bending and unbending by MutS govern mismatch recognition and specificity. Proc Natl Acad Sci U S A 100: 14822–14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ohshima K, Kang S, Larson JE, Wells RD (1996) TTA.TAA triplet repeats in plasmids form a non-H bonded structure. J Biol Chem 271: 16784–16791. [DOI] [PubMed] [Google Scholar]
- 38. Trotta E, Del Grosso N, Erba M, Paci M (2000) The ATT strand of AAT.ATT trinucleotide repeats adopts stable hairpin structures induced by minor groove binding ligands. Biochemistry 39: 6799–6808. [DOI] [PubMed] [Google Scholar]
- 39. Trotta E, Del Grosso N, Erba M, Melino S, Cicero D, et al. (2003) Interaction of DAPI with individual strands of trinucleotide repeats. Effects of replication in vitro of the AAT x ATT triplet. Eur J Biochem 270: 4755–4761. [DOI] [PubMed] [Google Scholar]
- 40. Lopez Castel A, Cleary JD, Pearson CE (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11: 165–170. [DOI] [PubMed] [Google Scholar]
- 41. Li F, Tian L, Gu L, Li GM (2009) Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. J Biol Chem 284: 33056–33061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Javaid S, Manohar M, Punja N, Mooney A, Ottesen JJ, et al. (2009) Nucleosome remodeling by hMSH2-hMSH6. Mol Cell 36: 1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gorman J, Plys AJ, Visnapuu ML, Alani E, Greene EC (2010) Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat Struct Mol Biol 17: 932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kadyrova LY, Blanko ER, Kadyrov FA (2011) CAF-I-dependent control of degradation of the discontinuous strands during mismatch repair. Proc Natl Acad Sci U S A 108: 2753–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Widom J (2001) Role of DNA sequence in nucleosome stability and dynamics. Q Rev Biophys 34: 269–324. [DOI] [PubMed] [Google Scholar]
- 46. Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, et al. (2006) A genomic code for nucleosome positioning. Nature 442: 772–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gordenin DA, Resnick MA (1998) Yeast ARMs (DNA at-risk motifs) can reveal sources of genome instability. Mutat Res 400: 45–58. [DOI] [PubMed] [Google Scholar]
- 48. Syvaoja J, Linn S (1989) Characterization of a large form of DNA polymerase delta from HeLa cells that is insensitive to proliferating cell nuclear antigen. J Biol Chem 264: 2489–2497. [PubMed] [Google Scholar]
- 49. Chui G, Linn S (1995) Further characterization of HeLa DNA polymerase epsilon. J Biol Chem 270: 7799–7808. [DOI] [PubMed] [Google Scholar]
- 50. Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, et al. (2007) The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic Acids Res 35: 6588–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tran HT, Gordenin DA, Resnick MA (1999) The 3′–>5′ exonucleases of DNA polymerases delta and epsilon and the 5′–>3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol Cell Biol 19: 2000–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kleczkowska HE, Marra G, Lettieri T, Jiricny J (2001) hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev 15: 724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, et al. (2010) PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci U S A 107: 16066–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fortune JM, Pavlov YI, Welch CM, Johansson E, Burgers PM, et al. (2005) Saccharomyces cerevisiae DNA polymerase delta: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J Biol Chem 280: 29980–29987. [DOI] [PubMed] [Google Scholar]
- 55. Nick McElhinny SA, Pavlov YI, Kunkel TA (2006) Evidence for extrinsic exonucleolytic proofreading. Cell Cycle 5: 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scherer S, Davis RW (1979) Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci U S A 76: 4951–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mutational spectra in pol2-M644G and pol2-M644G msh2Δ strains. The URA3 reporter was present in either orientation 1 (OR1) or orientation 2 (OR2) at position AGP1. The coding strand of the URA3 open reading frame is shown, with every 10th base indicated by a dot and mutations depicted above (OR1) and below (OR2) the wild type URA3 sequence. Single letters represent base substitutions, open triangles represent single base deletions, and closed triangles represent single base additions. Indels in homonucleotide runs are shown at the 5′-most position of the run. (A) Spectra in MSH2 strains [13]. (B) Spectra in msh2Δ strains.
(TIF)
Mutational spectra in pol2-M644G and pol2-M644G msh2Δ strains with ATT0 URA3. As for Figure S1, with spectra for the pol2-M644G msh2Δ and pol2M644G MSH2 strains shown above and below the ATT0 URA3 sequence, respectively. The three bases that differ between the ATT3 and ATT0 URA3 sequences are shown in bold (positions 690, 693, and 696).
(TIF)
Multi-base mutations omitted from spectrum figures.
(DOCX)
Mutation counts, mutation rates, and MMR correction factors.
(XLSX)
Mutation Rates for pol2-M644G ± msh2Δ and ATT3/ATT0 URA3 Orientation 1 strains. a Expanded from [21]. b Expanded from [13].
(DOCX)
The supporting information includes methods for calculating and statistical analyses of mutation rates and correction factors.
(DOC)