

Abstract
Although recent studies in patients with paternal uniparental disomy 14 [upd(14)pat] and other conditions affecting the chromosome 14q32.2 imprinted region have successfully identified underlying epigenetic factors involved in the development of upd(14)pat phenotype, several matters, including regulatory mechanism(s) for RTL1 expression, imprinting status of DIO3 and placental histological characteristics, remain to be elucidated. We therefore performed molecular studies using fresh placental samples from two patients with upd(14)pat. We observed that RTL1 expression level was about five times higher in the placental samples of the two patients than in control placental samples, whereas DIO3 expression level was similar between the placental samples of the two patients and the control placental samples. We next performed histological studies using the above fresh placental samples and formalin-fixed and paraffin-embedded placental samples obtained from a patient with a maternally derived microdeletion involving DLK1, the-IG-DMR, the MEG3-DMR and MEG3. Terminal villi were associated with swollen vascular endothelial cells and hypertrophic pericytes, together with narrowed capillary lumens. DLK1, RTL1 and DIO3 proteins were specifically identified in vascular endothelial cells and pericytes, and the degree of protein staining was well correlated with the expression dosage of corresponding genes. These results suggest that RTL1as-encoded microRNA functions as a repressor of RTL1 expression, and argue against DIO3 being a paternally expressed gene. Furthermore, it is inferred that DLK1, DIO3 and, specially, RTL1 proteins, play a pivotal role in the development of vascular endothelial cells and pericytes.
Keywords: Upd(14)pat, expression dosage, histopathology, imprinting, microdeletion, placenta
Introduction
Human chromosome 14q32.2 region carries a cluster of imprinted genes including protein coding paternally expressed genes (PEGs) such as DLK1 and RTL1 (alias PEG11) and non-coding maternally expressed genes (MEGs) such as MEG3 (alias GTL2) and RTL1as (RTL1 antisense encoding microRNAs).1,2 The 14q32.2 imprinted region also harbors two differentially methylated regions (DMRs), i.e., the germline-derived primary DLK1-MEG3 intergenic DMR (IG-DMR) and the postfertilization-derived secondary MEG3-DMR.1,2 Both DMRs are hypermethylated after paternal transmission and hypomethylated after maternal transmission in the body, whereas in the placenta the IG-DMR alone remains as a DMR and the MEG3-DMR is rather hypomethylated.2 We have previously revealed that the hypomethylated IG-DMR and MEG3-DMR of maternal origin function as imprinting control centers in the placenta and the body, respectively, and that the IG-DMR functions hierarchically as an upstream regulator for the methylation pattern of the MEG3-DMR on the maternally inherited chromosome in the body, but not in the placenta.3
Consistent with these findings, paternal uniparental disomy 14 [upd(14)pat] results in a unique phenotype characterized by facial abnormality, small bell-shaped thorax with coat hanger appearance of the ribs, abdominal wall defects, placentomegaly and polyhydramnios.2,4 We have studied multiple patients with upd(14)pat and related conditions, such as epimutations of the maternally derived DMRs and various types of microdeletions involving the maternally inherited imprinted region, suggesting that markedly increased RTL1 expression is the major underlying factor for the development of upd(14)pat-like phenotype.2 The notion of excessive RTL1 expression is primarily based on the following mouse data indicating a trans-acting repressor function of Rtl1as-encoded microRNAs for Rtl1 expression: (1) targeted deletion of the maternally derived IG-DMR causes maternal to paternal epigenotypic switch of the imprinted region, with ~4.5 times rather than ~2 times of Rtl1 expression as well as ~2 times of Dlk1 expression and nearly absent Megs expression, in the presence of two functional copies of Pegs and no functional copy of Megs5 and; (2) targeted deletion of the maternally derived Rtl1as results in 2.5–3.0 times of Rtl1 expression, in the presence of a single functional copy of Rtl1.6 Similarly, in the human, typical upd(14)pat phenotype is observed in patients with epimutations that are likely associated with markedly increased RTL1 expression because of the combination of two functional copies of RTL1 and no functional copy of RTL1as, whereas relatively mild upd(14)pat-like phenotype is found in patients with maternally inherited microdeletions involving RTL1as that are likely accompanied by moderately elevated RTL1 expression because of the combination of a single functional copy of RTL1 and no functional copy of RTL1as.2
Human imprinting disorders are usually associated with placental abnormalities. For example, Beckwith-Wiedemann syndrome (BWS) and upd(14)pat are associated with placentomegaly,4,7 and Silver-Russell syndrome is accompanied by hypoplastic placenta.8Similarly, mouse imprinting aberrations also usually affect placental growth and development.9 In agreement with this, virtually all the imprinted genes studied to date are expressed in the placenta and play a pivotal role in the placental growth and development,10 although placental structure is more or less different between placental animals.11
However, several matters remain to be clarified in upd(14)pat and related conditions. For example, it is unknown whether human RTL1 expression is actually elevated in the absence of functional RTL1as-encoded microRNAs. It is also unknown whether DIO3 is a PEG, although mouse Dio3 has been shown to undergo partial imprinting.12 In this regard, while we examined fresh blood cells, cultured skin fibroblasts and formalin-fixed and paraffin-embedded placental and body samples obtained from patients with upd(14)pat-like phenotype, precise assessment of RTL1 and DIO3 expression levels was impossible because of extremely low RTL1 and DIO3 expression levels in fresh blood cells and cultured skin fibroblasts and poor quality of RNAs extracted from paraffin-embedded tissues.2,3 In addition, while cSNP genotyping has demonstrated paternal DLK1 and RTL1 expression and maternal MEG3 expression in the body and the placenta,2,3 no informative cSNP data showing paternal DIO3 expression have been obtained.2,3 Furthermore, although standard light microscopic (LM) examinations have been performed using formalin-fixed and paraffin-embedded placental samples, fine placental histopathological studies, such as electron microscopic (EM) examinations and immunohistochemical (IHC) examinations, remain to be performed.
To examine these unresolved matters, fresh placental tissues are highly useful, because precise quantitative real-time PCR (q-PCR) analyses and EM studies can be performed with fresh placentas. Thus, we performed q-PCR analyses and EM studies, as well as IHC studies with RTL1 antibodies produced by ourselves and commercially available DLK1 and DIO3 antibodies, using fresh placental samples obtained from two previously reported patients with prenatally diagnosed upd(14)pat.13,14 We also performed IHC studies using formalin-fixed and paraffin-embedded placental samples obtained from a previously reported patient with a microdeletion involving DLK1, but not RTL1 and DIO3,2 to compare the placental protein expression levels between upd(14)pat and the microdeletion. Furthermore, we also studied a hitherto unreported patient with an unbalanced translocation involving the 14q32.2 imprinted region, to obtain additional data regarding the RTL1-RTL1as interaction and the primary factor for the development of upd(14)pat phenotype.
Results
Patients and samples
This study consisted of three previously reported patients with typical body and placental upd(14)pat phenotype and a normal karyotype (cases 1–3),2,13-15 and a new patient with various non-specific features and a 46,XX,der(17)t(14;17)(q31;p13) karyotype accompanied by three copies of the distal 14q region and a single copy of the terminal 17p region (case 4) . Clinical phenotypes of cases 1–4 are summarized in Table S1. In brief, cases 1 and 2 were suspected to have upd(14)pat phenotype including bell-shaped thorax by prenatal ultrasound studies performed for polyhydroamnios, and were confirmed to have upd(14)pat by microsatellite analysis after birth. Case 3 was found to have typical upd(14)pat phenotype during infancy and was shown to have a maternally derived microdeletion affecting the chromosome 14q32.2 imprinted region. Case 4 had growth failure, developmental delay, multiple non-specific anomalies, and omphalocele. There was no history of polyhydramnios or placentomegaly. Thus, except for omphalocele, case 4 had no upd(14)pat-like phenotype. The parental karyotype was normal, indicating a de novo occurrence of the unbalanced translocation.
We obtained fresh placental samples immediately after birth from prenatally diagnosed cases 1 and 2 for molecular studies using genomic DNA and RNA, and fresh leukocyte samples from cases 1, 2 and 4 and their parents for molecular studies using genomic DNA. The fresh placental samples of cases 1 and 2 were also utilized for histopathological examinations, together with formalin-fixed and paraffin-embedded placental samples of case 3. For controls, we obtained three fresh placentas at 37 weeks of gestation, and fresh leukocytes from three adult subjects; for molecular studies using placentas, we prepared pooled samples consisting of an equal amount of DNA or RNA extracted from each placenta.
Molecular studies in cases 1 and 2
We performed microsatellite analysis for 19 loci on chromosome 14 and bisulfite sequencing for the IG-DMR (CG4 and CG6) and the MEG3-DMR (CG7), using placental and leukocyte genomic DNA samples; while microsatellite analysis had been performed for 15 loci in case 1 and 16 loci in case 2, only leukocyte genomic DNA samples were examined in the previous study.15 Consequently, we identified two peaks for D14S609 and single peaks for the remaining loci in case 1 (the combination of paternal heterodisomy and isodisomy), and single peaks for all the examined loci in case 2 (apparently full paternal isodisomy) (Table S2). Furthermore, no trace of maternally inherited peak was identified in both placental and leukocyte genomic DNA samples (Fig. 1). Bisulfite sequencing showed that both the IG-DMR and the MEG3-DMR were markedly hypermethylated in the leukocytes of cases 1 and 2, whereas in the placental samples the IG-DMR was obviously hypermethylated and the MEG3-DMR was grossly hypomethylated to an extent similar to that identified in control placentas (Fig. 2). Furthermore, q-PCR analysis for placental RNA samples revealed that DLK1, RTL1, and DIO3 expression levels were 3.3 times, 6.1 times and 1.9 times higher in the placental samples of case 1 than in the control placental samples, respectively, and were 3.1 times, 9.4 times and 1.7 times higher in the placental samples of case 2 than in the control placental samples, respectively (Fig. 3A). By contrast, the expressions of all MEGs examined were virtually absent in the placental samples of cases 1 and 2. PCR products were sufficiently obtained after 30 cycles for the fresh placental as well as leukocyte samples, consistent with high quality of DNA and RNA obtained from fresh materials.
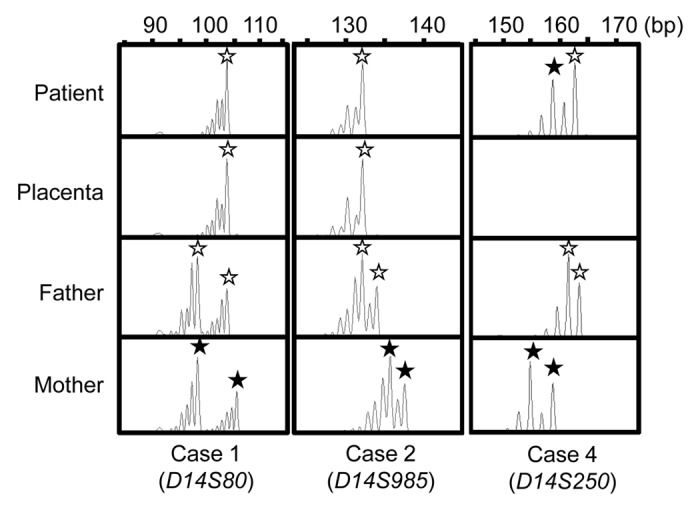
Figure 1. Representative results of microsatellite analysis, using leukocyte genomic DNA samples of the patient and the parents and placental genomic DNA samples. In cases 1 and 2, one of the two paternal peaks is inherited by the patients and the placentas, and no trace of maternal peaks is identified. In case 4, both paternally and maternally derived peaks are found in the patient, with the paternally derived long peak being larger than the maternally inherited short peak.

Figure 2. Bisulfite sequencing analysis of the IG-DMR (CG4 and CG6) and the MEG3-DMR (CG7), using leukocyte and placental genomic DNA samples. Filled and open circles indicate methylated and unmethylated cytosines at the CpG dinucleotides, respectively. Upper part: structure of CG4, CG6, and CG7. Pat, paternally derived chromosome; Mat, maternally derived chromosome. The PCR products for CG4 (311 bp) harbor 6 CpG dinucleotides and a G/A SNP (rs12437020), those for CG6 (428 bp) carry 19 CpG dinucleotides and a C/T SNP (rs10133627) and those for CG7 (168 bp) harbor 7 CpG dinucleotides. Lower part: the results of cases 1, 2, 4 and a control subject. Each horizontal line indicates a single subcloned allele. The control data represent the methylation patterns obtained with a leukocyte genomic DNA sample extracted from a single subject heterozygous for the G/A SNP (rs12437020) (body) and those obtained with a pooled DNA sample consisting of an equal amount of genomic DNA extracted from three control placentas homozygous for that SNP.

Figure 3. Quantitative real-time PCR analysis using placental samples. For a control, a pooled RNA sample consisting of an equal amount of total RNA extracted from three fresh control placentas was utilized. (A) Relative mRNA expression levels for DLK1, RTL1, and DIO3 against GAPDH (mean ± SE) and lack of MEGs expression (indicated by arrows) (miR433 and miR127 are encoded by RTL1as) in the placental samples of cases 1 and 2. (B) Relative mRNA expression levels for DLK1, RTL1, and DIO3 against GAPDH (mean ± SE), in the equal amount of expression positive placental cells (vascular endothelial cells and pericytes) of cases 1 and 2 (corrected for the difference in the relative proportion of expression positive cells between the placental samples of cases 1 and 2 and the control placental samples, on the assumption that the DLK1 expression level is ″simply doubled″ in the expression positive placental cells of case 1 and 2).
Molecular studies in case 3
Detailed molecular findings have already been reported previously.2 In brief, microsatellite analysis revealed biparentally derived homologs of chromosome 14, and a deletion analysis demonstrated a maternally inherited 108,768 bp microdeletion involving DLK1, the IG-DMR, the MEG3-DMR, and MEG3, but not affecting RTL1/RTL1as. Since loss of the DMRs causes maternal to paternal epigenotypic alteration,2 it is predicted that case 3 has a single functional copy of DLK1 and two functional copies of RTL1 and DIO3, as well as no functional copy of RTL1as and other MEGs. Bisulfite sequencing showed that both the IG-DMR and the MEG3-DMR were markedly hypermethylated in leukocytes, whereas in the formalin-fixed and paraffin-embedded placental samples the IG-DMR was obviously hypermethylated and the MEG3-DMR was comprised of roughly two-thirds of hypermethylated clones and roughly one-third of hypomethylated clones. In addition, RT-PCR analysis for such placental samples indicated positive PEGs (especially RTL1) expression and absent MEGs expression. For the formalin-fixed and paraffin-embedded placental samples, PCR products could be obtained only after 35 cycles, because of poor quality (severe degradation) of DNA and RNA.
Molecular findings in case 4
We examined the presence or absence of the 14q32.2 imprinted region on the der(17) chromosome (Fig. 4). Oligoarray comparative genomic hybridization (CGH) indicated three copies of a ~19.6 Mb 14q31–qter region, and FISH analysis for four segments around the chromosome 14q32.2 imprinted region delineated positive signals on the der(17) chromosome as well as on the normal chromosome 14 homologs. This demonstrated the presence of the 14q32.2 imprinted region on the der(17) chromosome. In addition, similar oligoarray CGH and FISH analysis revealed loss of a ~455 kb region from the distal chromosome 17p (Fig. S1).

Figure 4. Array CGH and FISH analysis for the distal chromosome 14 region in case 4. In CGH analysis, the black, the red, and the green dots denote signals indicative of the normal, the increased (> +0.5), and the decreased (< –1.0) copy numbers, respectively. In FISH analysis, red signals (arrows) are derived from the probes detecting the various parts of the 14q32.2 imprinted region (the physical positions are indicated with yellow bars), and the green signals (arrowheads) are derived from an RP11–566I2 probe for 14q11.2 used as an internal control.
Thus, we investigated the parental origin of the translocated 14q distal region. Microsatellite analysis for D14S250 and D14S1007 on the translocated 14q distal region delineated biparentally derived two peaks, with paternally derived long PCR products showing larger peaks than maternally derived short PCR products (Fig. 1; Table S2). Since short products are usually more easily amplified than long products, this indicated paternal origin of the der(17) chromosome harboring the chromosome14q32.2 imprinted region. Consistent with this, bisulfite sequencing showed moderate hypermethylation of the IG-DMR and the MEG3-DMR (Fig. 2).
Placental histopathological studies
We performed LM and EM studies, and IHC examinations (Fig. 5). LM examinations showed proliferated chorionic villi in cases 1–3. Capillary lumens were irregularly dilated with thickened endothelium in the stem to intermediate villi, but not in the terminal villi. Immature villi were present in case 3, probably because of 30 weeks of gestational age. Chorangioma was also identified in case 3. There was no villous chorangiosis, edematous change of villous stroma, or mesenchymal dysplasia characterized by grapelike vesicles in cases 1–3. Although the terminal villi exhibited no definitive abnormalities in the LM studies, EM examinations revealed swelling of vascular endothelial cells and hypertrophic change of pericytes in the terminal villi, together with narrowed capillary lumens, in cases 1 and 2.
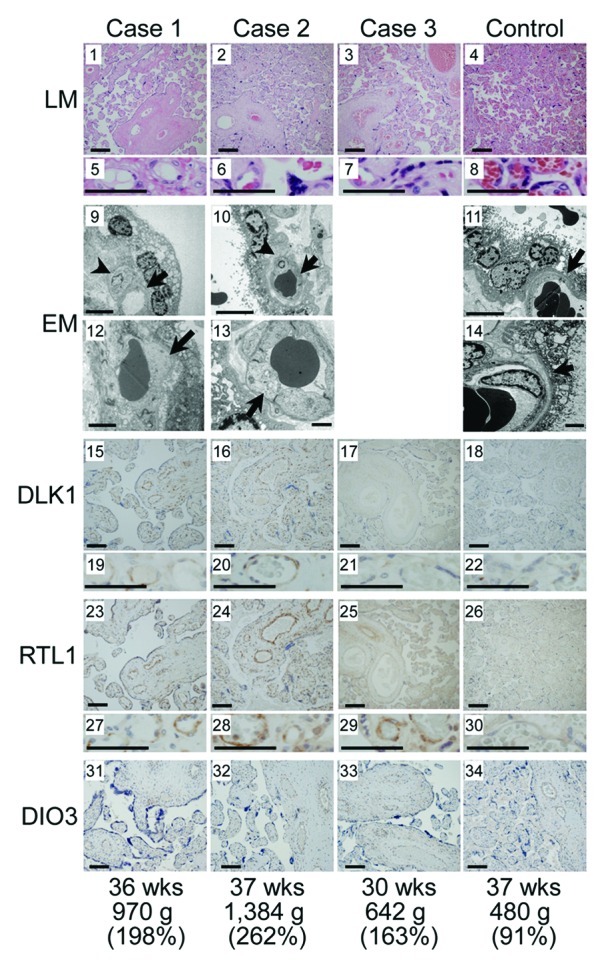
Figure 5. Histological examinations. LM, light microscopic examinations; EM, electron microscopic examinations; DLK1, RTL1 and DIO3, immunohistochemical examinations for the corresponding proteins. The arrows and arrowheads in the EM findings indicate endothelial cells and pericytes, respectively. Scale bars represent 100 μm for 1–4, 15–18, 23–26 and 31–34, 50 μm for 5–8, 19–22 and 27–30, 5 μm for 9–11 and 2 μm for 12–14. Gestational age, placental weight, and % placental weight assessed by the gestational age-matched Japanese references for placental weight4,22are described.
IHC examinations identified RTL1, DLK1 and DIO3 protein expressions in the vascular endothelial cells and pericytes of chorionic villi, but not in the cytotrophoblasts, syncytiotrophoblasts, and stromal cells, in the placentas of cases 1–3 and in the control placenta. The PEGs protein expression level was variable in the control placenta, with moderate DLK1 expression, high RTL1 expression, and low DIO3 expression. Furthermore, DLK1 protein expression was apparently stronger in the placentas of cases 1 and 2 than in the placenta of case 3 and the control placenta, RTL1 protein expression was obviously stronger in the placentas of cases 1–3 than in the control placenta, and DIO3 protein expression was apparently similar between the placentas of cases 1–3 and the control placenta.
Discussion
We studied placental samples obtained from cases 1–3 with typical body and placental upd(14)pat phenotype. In this regard, the microsatellite data suggest that upd(14)pat with heterodisomic and isodisomic loci in case 1 was caused by trisomy rescue or gamete complementation, and that upd(14)pat with isodisomic loci alone in case 2 resulted from monosomy rescue or post-zygotic mitotic error, although it is possible that heterodisomic locus/loci remained undetected in case 2.15Notably, there was no trace of a maternally inherited locus indicative of the presence of trisomic cells or normal cells with biparentally inherited chromosome 14 homologs in the placentas as well as in the leukocytes of cases 1 and 2. In addition, the microdeletion of case 3 has been shown to be inherited from the mother with the same microdeletion.2 These findings imply that the placental tissues as well as the leukocytes of cases 1–3 almost exclusively, if not totally, consisted of cells with upd(14)pat or those with the microdeletion.
The q-PCR analysis was performed for the fresh placental samples of cases 1 and 2. In this context, two matters should be pointed out. First, the proportion of vascular endothelial cells and pericytes expressing DLK1, RTL1, and DIO3 would be somewhat variable among samples, because only a small portion of the placenta was analyzed. This would be relevant to the some degree of difference in the expression levels between the placental samples of cases 1 and 2. Second, the relative proportion of vascular endothelial cells and pericytes expressing DLK1, RTL1, and DIO3 would be higher in the placental samples of cases 1 and 2 than in the control placental samples, because the placentas of cases 1 and 2 were accompanied by proliferation of the chorionic villi with such expression positive cells. Thus, it would be inappropriate to perform a simple comparison of relative expression levels against GAPDH between the placental samples of cases 1 and 2 and the control placental samples. Indeed, although a complex regulatory mechanism(s), as implicated for the RTL1 expression,1,2 is unlikely to be operating for the DLK1 expression, the relative DLK1 expression level was 3.3 times and 3.1 times, not 2 times, higher in the placental samples of cases 1 and 2 than in the control placental samples, respectively (Fig. 3A). Assuming that DLK1 expression level is simply doubled in expression positive cells of cases 1 and 2, it is predicted that the relative proportion of such expression positive cells is 1.65 times (3.3 ÷ 2.0) and 1.55 times (3.1 ÷ 2.0) larger in the placental samples of cases 1 and 2 than in the control placental samples, respectively. Thus, the expression level against GAPDH in the equal amount of expression positive cells is estimated as 3.69 times (6.1 ÷ 1.65) increased for RTL1 and 1.15 times (1.9 ÷ 1.65) increased for DIO3 in case 1, and as 6.06 times (9.4 ÷ 1.55) increased for RTL1 and 1.09 times (1.7 ÷ 1.55) increased for DIO3 in case 2 (Fig. 3B).
Thus, the expression data are summarized as follows (Fig. 6). First, it is inferred that the relative RTL1 expression level is markedly (~5 times) increased in the expression positive cells of the placentas with upd(14)pat, as compared with the control placentas. This degree of elevation is grossly similar to that identified in the body of mice with the targeted deletion of the maternally derived IG-DMR (~4.5 times).5 Such a markedly increased RTL1 expression would be explained by assuming that RTL1as-encoded microRNAs (e.g., miR433 and miR127) function as a repressor for RTL1 expression through the RNAi mechanism, as has been indicated for the mouse Rtl1-Rtl1as interaction.16,17 Second, it is unlikely that DIO3 is solely expressed from the paternally inherited allele, although it remains to be determined whether DIO3 undergoes partial imprinting like mouse Dio312 or completely escapes imprinting. In either case, the results would explain why patients with upd(14)pat and upd(14)mat lack clinically recognizable thyroid disorders,2 although DIO3 plays a critical role in the inactivation of thyroid hormones.18

Figure 6. Schematic representation of the chromosome 14q32.2 imprinted region in a control subject, cases 1 and 2 with upd(14)pat, case 3 with a microdeletion (indicated by stippled rectangles), and case 4 with two copies of the imprinted region of paternal origin and a single copy of the imprinted region of maternal origin. This figure has been constructed using the present results and the previous data.2,3 P, paternally derived chromosome; M, maternally derived chromosome. Filled and open circles represent hypermethylated and hypomethylated DMRs, respectively; since the MEG3-DMR is grossly hypomethylated and regarded as non-DMR in the placenta, it is painted in gray. PEGs (DLK1 and RTL1) are shown in blue, MEGs (MEG3, RTL1as, MEG8, snoRNAs and miRNAs) in red, a probably non-imprinted gene (DIO3) in black, and non-expressed genes in white. Thick arrows for RTL1 in cases 1–3 represent increased RTL1 expression that is ascribed to loss of functional microRNA-containing RTL1as as a repressor for RTL1.
This study provides further support for a critical role of excessive RTL1 expression in the development of upd(14)pat phenotype (Fig. 6). Indeed, markedly (~5 times) increased RTL1 expression is shared in common by cases 1–3 with typical upd(14)pat body and placental phenotype. In this context, it is notable that case 4 had no clinically recognizable upd(14)pat body and placental phenotype, except for omphalocele. This would imply that a single copy of RTL1as can almost reduce the RTL1 expression dosage below the threshold level for the development of upd(14)pat phenotype by exerting a trans-acting repressor effect on the two functional copies of RTL1. By contrast, the relevance of DLK1 to upd(14)pat phenotype is unlikely, because case 3 exhibited typical upd(14)pat phenotype in the presence of a single functional copy of DLK1, and case 4 showed no upd(14)pat phenotype except for omphalocele in the presence of two functional copies of DLK1. Similarly, if DIO3 were more or less preferentially expressed from paternally inherited allele, the relevance of DIO3 to upd(14)pat phenotype would also remain minor, if any. Case 4 had no upd(14)pat phenotype except for omphalocele in the presence of with two copies of DIO3 of paternal origin. It should be pointed out, however, that the absence of MEGs expression may have a certain effect on the development of upd(14)pat phenotype.
The placental histological examinations revealed several informative findings. First, DLK1, RTL1, and DIO3 proteins were specifically identified in vascular endothelial cells and pericytes of chorionic villi in the control placenta, with RTL1 protein being most strongly expressed. These results, together with abnormal LM and EM findings of such cells in cases 1–3, suggest that these proteins, especially RTL1 protein, plays a pivotal role in the development of endothelial cells and pericytes. In this regard, it may be possible that the endothelial thickening and resultant narrowing the capillary lumens in the terminal villi have resulted in the dilatation of the stem to intermediate portions of the chorionic villi.
Second, the degree of protein staining was well correlated with the expression dosage of corresponding genes. In this regard, since characteristic macroscopic and microscopic placental features were identified in cases 1–3 who shared markedly elevated RTL1 protein expression, this is consistent with the notion that upd(14)pat phenotype is primarily caused by the markedly elevated RTL1 expression.2 Indeed, DLK1 protein expression was not exaggerated in case 3 with typical upd(14)pat phenotype, and DIO3 protein expression was not enhanced in cases 1–3. It may be possible, however, that the abnormality of placental structures may have resulted in a difference in immunostaining without an actual change in gene expression. This point awaits further investigations.
Third, villous chorangiosis, stromal expansion, and mesenchymal dysplasia were not identified in the placental samples of cases 1–3, although such a lesion(s) may have existed in non-examined portions. Notably, such lesions are frequently observed in placentas of patients with BWS.19-21 Thus, while both upd(14)pat and BWS are associated with placentomegaly and polyhydroamnios, characteristic histological findings appear to be different between upd(14)pat and BWS.
This study would also provide useful information on the methylation patterns of the MEG3-DMR in the placenta. Our previous studies using formalin-fixed and paraffin-embedded placental samples revealed that roughly two-thirds of clones were hypermethylated and the remaining roughly one-third of clones were hypomethylated in case 3 as well as in the previously reported patients with upd(14)pat (not cases 1 and 2) and epimutation (hypermethylation of the IG-DMR and the MEG3-DMR of maternal origin), and that roughly one-third of clones were hypermethylated and the remaining roughly two-thirds of clones were hypomethylated in control placental samples (see Fig. S2C in ref. 2). However, this study showed that the MEG3-DMR was grossly hypomethylated in the fresh placental samples of cases 1 and 2, with an extent similar to that identified in the fresh control placental samples. In this regard, it is notable that PCR products could be obtained only after 35 cycles for the formalin-fixed and paraffin-embedded placental samples and were sufficiently obtained after 30 cycles for the fresh placental samples. Thus, several specific clones may have been selectively amplified in the previous study. Furthermore, it may be possible that efficacy of bisulfite treatment (conversion of unmethylated cytosine into uracils and subsequently thymines) may be insufficient for the formalin-fixed and paraffin-embedded placental samples. Thus, it appears that the present data denote precise methylation patterns of the MEG3-DMR in the placenta.
In summary, the present study provides useful clues for the clarification of regulatory mechanism for the RTL1 expression, imprinting status of DIO3 and characteristic placental histological findings in patients with upd(14)pat and related conditions. Further studies will help improve our knowledge about upd(14)pat and related conditions.
Methods
Ethical approval
This study was approved by the Institutional Review Board Committees of each investigator, and performed after obtaining written informed consent.
Primers
Primers utilized in this study are summarized in Table S3.
Sample preparation for molecular studies
Genomic DNA samples were obtained from leukocytes using FlexiGene DNA Kit (Qiagen) and from placental samples using ISOGEN (Nippon Gene). Transcripts of DLK1, MEG3, RTL1, MEG8 and DIO3 were isolated with ISOGEN (Nippon Gene), and microRNAs were extracted with mirVanaTM miRNA Isolation Kit (Ambion). After DNase treatment, cDNA samples for DLK1, MEG3, MEG8 and DIO3 were prepared with oligo(dT) primers from 1 μg of RNA using Superscript III Reverse Transcriptase (Invitrogen), and those of microRNAs were synthesized from 300 ng of RNA using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). For RTL1, 3'-RACE was utilized to prevent amplification of RTL1as; cDNA was synthesized from 1 μg of RNA using Superscript III Reverse Transcriptase with a long primer hybridizing to poly A site and introducing the adaptor sequence. Lymphocyte metaphase spreads for FISH analysis were prepared from leukocytes using colcemide (Invitrogen).
Molecular studies
Microsatellite analysis for 19 loci on chromosome 14, methylation analysis for the IG-DMR and the MEG3-DMR, and FISH analyses for the 14q32.2 region were performed as described previously.2,3 For FISH analysis of 17p13.3, a 17p sub-telomere probe and an RP11–411G7 probe for the 17p13.3 region were utilized, together with a CEP17 probe for the 17p11.1 region utilized as an internal control. The 17p sub-telomere probe was detected according to the manufacture’s protocol, the RP11–411G7 probe was labeled with digoxigenin and detected by rhodamine anti-digoxigenin, and the CEP17 control probe was labeled with biotin and detected by avidin conjugated to fluorescein isothiocyanate. Quantitative real-time PCR analysis was performed on an ABI PRISM 7000 (Applied Biosystems) using TaqMan real-time PCR probe primer mixture for the following genes (assay No: Hs00171584 for DLK1, Hs00292028 for MEG3, Hs00419701 for MEG8 and Hs00704811 for DIO3; assay ID: 001028 for miR433 and 000452 for miR127). For RTL1, q-PCR analysis was performed with a forward primer hybridized to the sequence of RTL1 and a reverse primer hybridized to the adaptor sequence. Fifty nanongrams of cDNA in a 50 μl reaction mixture contacting 2 × KOD FX buffer (Toyobo), 2.0 mM dNTP mixture (Toyobo), KOD FX (Toyobo), SYBR Green I (Invitrogen), and primer set for RTL1 were subjected to the ABI PRISM 7000. Data were normalized against GAPDH (catalog No: 4326317E) for DLK1, MEG3, MEG8, RTL1, and DIO3, and against RNU48 (assay ID: 0010006) for microRNAs. The expression studies were performed three times for each sample. Oligoarray CGH was performed using 1 × 1M format Human Genome Array (Catalog No G4447A) (Agilent Technologies).
Histopathogical analysis
Placental samples were fixed with 20% buffered formaldehyde at room temperature and embedded in paraffin wax according to standard protocols for LM examinations. Then, sections of 3 μm thick were stained with hematoxylin-eosin. For EM examinations, fresh placental tissues were fixed with phosphate-buffered 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide, and embedded in Epon 812 (catalog No. R3245, TAAB). Semithin sections were stained with 1% methylene blue, and ultrathin sections were double-stained with uranyl acetate and lead citrate. Subsequently, they were examined with a Ninhon Denshi JEM-1230 electron microscope.
For IHC analysis, sections of 3 μm thick were prepared by the same methods utilized for the LM examinations, and were examined with rabbit anti human DLK1 polyclonal antibody at 1:100 dilutions (catalog No 10636-1-AP, ProteinTech Group), rabbit anti human RTL1 polyclonal antibody at 1:200 dilutions, and rabbit anti human DIO3 polyclonal antibody at 1:50 dilutions (catalog No ab102926, abcam); anti human RTL1 polyclonal antibody was produced by immunizing rabbits with the synthesized RTL1 peptide (NH2-RGFPRDPSTESG-COOH) in this study. Sections were dewaxed in xylene and rehydrated through graded ethanol series and, subsequently, incubated in 10% citrate buffer (pH 6.0) for 40 min in a 98°C water bath, for antigen retrieval. Endogenous peroxidase activity was quenched with 1% H2O2 and 100% methanol for 20 min. To prevent non-specific background staining, sections are incubated with Protein Block Serum-Free (Dako corporation) for 10 min at room temperature. Then, sections were incubated overnight with primary antibody at 4°C and, subsequently, treated with the labeled polymer prepared by combining amino acid polymers with peroxidase and anti- rabbit polyclonal antibody (Histofine Simple Stain MAX PO MULTI, Nichirei). Peroxidase activities were visualized by diaminobenzidine staining, and the nuclei were stained with hematoxylin.
Supplementary Material
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (A) (22249010) and Research (B) (21028026) from the Japan Society for the Promotion of Science (JSPS), by Grants-in-Aid for Scientific Research on Innovative Areas (22132004-A04) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), by Grants for Research on Intractable Diseases (H22–161) from the Ministry of Health, Labor and Welfare (MHLW), by Grant for National Center for Child Health and Development (23A-1), and by Grant from Takeda Science Foundation and from Novartis Foundation.
Glossary
Abbreviations:
- PEGs
paternally expressed genes
- MEGs
maternally expressed genes
- DMRs
differentially methylated regions
- IG-DMR
DLK1-MEG3 intergenic DMR
- RTL1as
RTL1 antisense
- upd(14)pat
paternal uniparental disomy 14
- BWS
Beckwith-Wiedemann syndrome
- q-PCR
quantitative real-time PCR
- CGH
oligoarray comparative genomic hybridization
- LM
light microscopic
- EM
electron microscopic
- IHC
immunohistochemical
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/21937
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/21937
References
- 1.da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24:306–16. doi: 10.1016/j.tig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 2.Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40:237–42. doi: 10.1038/ng.2007.56. [DOI] [PubMed] [Google Scholar]
- 3.Kagami M, O’Sullivan MJ, Green AJ, Watabe Y, Arisaka O, Masawa N, et al. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 2010;6:e1000992. doi: 10.1371/journal.pgen.1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kagami M, Yamazawa K, Matsubara K, Matsuo N, Ogata T. Placentomegaly in paternal uniparental disomy for human chromosome 14. Placenta. 2008;29:760–1. doi: 10.1016/j.placenta.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, et al. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet. 2003;35:97–102. doi: 10.1038/ng1233. [DOI] [PubMed] [Google Scholar]
- 6.Sekita Y, Wagatsuma H, Nakamura K, Ono R, Kagami M, Wakisaka N, et al. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet. 2008;40:243–8. doi: 10.1038/ng.2007.51. [DOI] [PubMed] [Google Scholar]
- 7.Lage JM. Placentomegaly with massive hydrops of placental stem villi, diploid DNA content, and fetal omphaloceles: possible association with Beckwith-Wiedemann syndrome. Hum Pathol. 1991;22:591–7. doi: 10.1016/0046-8177(91)90237-J. [DOI] [PubMed] [Google Scholar]
- 8.Yamazawa K, Kagami M, Nagai T, Kondoh T, Onigata K, Maeyama K, et al. Molecular and clinical findings and their correlations in Silver-Russell syndrome: implications for a positive role of IGF2 in growth determination and differential imprinting regulation of the IGF2-H19 domain in bodies and placentas. J Mol Med (Berl) 2008;86:1171–81. doi: 10.1007/s00109-008-0377-4. [DOI] [PubMed] [Google Scholar]
- 9.Georgiades P, Watkins M, Burton GJ, Ferguson-Smith AC. Roles for genomic imprinting and the zygotic genome in placental development. Proc Natl Acad Sci U S A. 2001;98:4522–7. doi: 10.1073/pnas.081540898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fowden AL, Sibley C, Reik W, Constancia M. Imprinted genes, placental development and fetal growth. Horm Res. 2006;65(Suppl 3):50–8. doi: 10.1159/000091506. [DOI] [PubMed] [Google Scholar]
- 11.Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta. 2002;23:3–19. doi: 10.1053/plac.2001.0738. [DOI] [PubMed] [Google Scholar]
- 12.Tsai CE, Lin SP, Ito M, Takagi N, Takada S, Ferguson-Smith AC. Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr Biol. 2002;12:1221–6. doi: 10.1016/S0960-9822(02)00951-X. [DOI] [PubMed] [Google Scholar]
- 13.Yamanaka M, Ishikawa H, Saito K, Maruyama Y, Ozawa K, Shibasaki J, et al. Prenatal findings of paternal uniparental disomy 14: report of four patients. Am J Med Genet A. 2010;152A:789–91. doi: 10.1002/ajmg.a.33247. [DOI] [PubMed] [Google Scholar]
- 14.Suzumori N, Ogata T, Mizutani E, Hattori Y, Matsubara K, Kagami M, et al. Prenatal findings of paternal uniparental disomy 14: Delineation of further patient. Am J Med Genet A. 2010;152A:3189–92. doi: 10.1002/ajmg.a.33719. [DOI] [PubMed] [Google Scholar]
- 15.Kagami M, Kato F, Matsubara K, Sato T, Nishimura G, Ogata T. Relative frequency of underlying genetic causes for the development of UPD(14)pat-like phenotype. Eur J Hum Genet. 2012;20:928–32. doi: 10.1038/ejhg.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, et al. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet. 2003;34:261–2. doi: 10.1038/ng1171. [DOI] [PubMed] [Google Scholar]
- 17.Davis E, Caiment F, Tordoir X, Cavaillé J, Ferguson-Smith A, Cockett N, et al. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr Biol. 2005;15:743–9. doi: 10.1016/j.cub.2005.02.060. [DOI] [PubMed] [Google Scholar]
- 18.Köhrle J. Thyroid hormone transporters in health and disease: advances in thyroid hormone deiodination. Best Pract Res Clin Endocrinol Metab. 2007;21:173–91. doi: 10.1016/j.beem.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Kraus FT, Redline RW, Gersell DJ, Nelson DM, Dicker JM. Disorders of placental Development. Placental Pathology (Atlas of Noutumor Pathology). Washington, DC: American Registry of Pathology, 2004:59-68. [Google Scholar]
- 20.Fox HE, Sebire NJ. The placenta in abnormalities and disorders of the fetus. Pathology of the Placenta. Third edition, Philadelphia, PA: SAUNDERS, 2007:262-3. [Google Scholar]
- 21.Parveen Z, Tongson-Ignacio JE, Fraser CR, Killeen JL, Thompson KS. Placental mesenchymal dysplasia. Arch Pathol Lab Med. 2007;131:131–7. doi: 10.5858/2007-131-131-PMD. [DOI] [PubMed] [Google Scholar]
- 22.Nakayama M. Placental pathology. Tokyo, Igaku Shoin, 2002:106-7 (in Japanese). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.