

Abstract
Regulatory change has long been hypothesized to drive the delineation of the human phenotype from other closely related primates. Here we provide evidence that CpG dinucleotides play a special role in this process. CpGs enable epigenome variability via DNA methylation, and this epigenetic mark functions as a regulatory mechanism. Therefore, species-specific CpGs may influence species-specific regulation. We report non-polymorphic species-specific CpG dinucleotides (termed “CpG beacons”) as a distinct genomic feature associated with CpG island (CGI) evolution, human traits and disease. Using an inter-primate comparison, we identified 21 extreme CpG beacon clusters (≥ 20/kb peaks, empirical p < 1.0 × 10−3) in humans, which include associations with four monogenic developmental and neurological disease related genes (Benjamini-Hochberg corrected p = 6.03 × 10−3). We also demonstrate that beacon-mediated CpG density gain in CGIs correlates with reduced methylation in these species in orthologous CGIs over time, via human, chimpanzee and macaque MeDIP-seq. Therefore mapping into both the genomic and epigenomic space the identified CpG beacon clusters define points of intersection where a substantial two-way interaction between genetic sequence and epigenetic state has occurred. Taken together, our data support a model for CpG beacons to contribute to CGI evolution from genesis to tissue-specific to constitutively active CGIs.
Keywords: epigenetics, epigenomics, CpG islands, gene regulation, evolution, human disease
Introduction
The CpG dinucleotide is unique for its ability to carry both genetic and epigenetic information in the genome of a differentiated mammalian cell.1,2 Variation in DNA methylation, facilitated by this two base pair motif, influences gene expression, and thereby enables tissue-specific function.3-6 However, this dinucleotide is substantially depleted, to one fifth of the expected level, due to the hypermutability (~11-fold) of cytosines when methylated.7-9 Yet, a minority of CpGs is retained against this strong tide of loss by a variable combination of: evasion of methylation in the germline, functional importance or chance. These are predominantly in CpG dense regions.10 Additionally, new CpGs are created by mutation through base substitution and as a by-product of the increase of GC in regions of biased gene conversion (BGC).11
A high density region of unmethylated CpGs can recruit CpG binding proteins, such as Cfp1 and KDM2A, which modify histone tails.12,13 Thomson et al. have shown that the experimental inclusion of a cluster of unmethylated CpGs, is sufficient to establish domains of H3K4me3.12 This histone modification leads to genomic three-dimensional structure change and the acquisition of permissive chromatin regions within the expanse of repressed genome.14 CpG clusters, termed CpG Islands (CGIs), co-locate with 60–70% of human gene promoters, often those of housekeeping genes that are hypomethylated in the germ line,15,16 but also 40% that are tissue-specific.10,17 Methylation of CGI promoters acts as a durable silencing mechanism. However, the majority of CGIs are unmethylated in differentiated cells independently of their transcriptional activity.10,18 The methylation state of CGI is strongly correlated with its CpG content, with high density CGIs being predominately constitutively unmethylated and “weak” low density islands the preferred target for tissue-specific methylation.10,19 CpG gain that shifts an island from weak to strong status therefore affects its dynamic ability for methylation change.
CpGs located in the lower density regions surrounding islands, termed CpG shores (~2kb up- or down-stream), identify significant tissue-, cancer- and reprogramming-specific methylation variability.6,20-22 Therefore, shore accretion and island erosion by subtle modulation in CpG density within these regions may have a disproportionate influence on the methylation levels and locations of these flanking regions. Additionally, an increase in methylation variance has been proposed to have an evolutionary important role, as well as being a potential influence on disease susceptibility.23
The genetic loss and gain of CpG dinucleotides over evolutionary time will impact upon the epigenome. Genome-wide variation in GC content at the megabase scale led to the formation of isochores before mammalian radiation24,25 with an increase in CpGs occurring ~90 million years ago (MYA).26 A subsequent clock-like loss of CpGs, due to the time- not generation-dependent substitution rate of cytosine deamination,27 has led to roughly similarly numbered, but differing sets of CpGs in primates. The mutability of individual CpGs can be determined by accounting for the influence of surrounding CpG density, as well as by sequence context and nucleosome position.28,29 Regions of CpGs that remain hyperconserved have been found to co-locate with polycomb repressive complex 2 binding domains and developmental genes.28
On the other hand, GC increase is influenced by primate recombination rates.30,31 So much so, that regions of extreme substitutional divergence in the human genome32-34 co-locate with recombination-associated BGC.35-39 This process therefore negates or obscures any potential evidence of weak selection.39,40 BGC can lead to the formation of CGIs11 and, furthermore, Cohen et al. have recently shown that CGIs can evolve without the requirement of selective pressure,41 although a possible subtle influence on CpGs via gene body methylation may exist.42
Cytosine deamination is consequently the predominant single nucleotide mutational force, occurring at one order of magnitude higher in the genome than other single base substitutions. Conversely, a highly localized BGC-mediated increase of CpGs occurs, associated with recombination.36,40 To discover the locations of potential species-specific regulatory modulation, due to CpG dinucleotide change, we identified a subset of human CpGs that were only present, either uniquely maintained or gained, in that lineage. While there are approximately ~40 million genetic differences between human and chimpanzee, the vast majority are due to random genetic drift.43 Divergence at CpG sites between these two species is estimated to be at 15.2%, compared with 0.92% for other nucleotide substitutions.43 Therefore, we used the additive collective power of multiple closely related species in a six-species inter-primate comparison.44,45
The CpG sequence can itself act as a genomic signaling molecule via combinatorial transcription factor binding specificity,1,46 facilitate epigenomic variation by influencing CGI promoter amenable chromatin structure and gene body methylation.47,48 Consequently we hypothesized that by identifying human-specific CpGs we may find potential regions of species-specific differential regulation.5,42,49,50 The sequence comparative approach would be blind to any potential causative mechanisms. The novel CpG clusters identified may highlight genes where a species-specific shift in epigenetic control has been enabled by this genetic change. These regions would potentially be enriched for human traits, as well as the possibility of associations with disease susceptibilities that have arisen as a by-product of human evolution.
Results
Human-Specific CpGs
To uncover the subset of CpGs present only in the human lineage, an inter-primate comparison was performed by examining the Enredo-Pecan-Ortheus Whole-Genome Multiple Alignments Sequences for human, chimpanzee, gorilla, orang-utan, rhesus macaque and common marmoset (Ensembl Compara.6_primates_EPO).51,52 This set of sequences contains 19,198 blocks and has been able to align 84.54% of the human genome. We parsed the blocks of this alignment requiring non-duplicated sequence in both human and chimpanzee and sequence of at least one other primate, which reduced our quasi-genomic set (referred to as h1c1o1: human, chimpanzee and other primate) to 79.99% of the human genome. This contained 25,100,205 or ~88.95% of the total haploid human CpGs. Each of these remaining CpGs was then interrogated with the requirement that at its precise position none of the other primates had a CpG dinucleotide present. Furthermore the chimpanzee sequence and the closest nearest other primate present in the alignment block (96.6% Gorilla) were required to have aligned sequence at this position i.e. was not N or -. This led to an initial human-specific subset of 1,820,319 CpGs. These CpGs were then conservatively filtered for polymorphism utilizing 1000 genomes data removing any CpG with any evidence of variation, as a SNP, or within a copy number or structural variant,53 leading to a final estimate of 1,192,484 human-specific CpGs.
CpG beacons
We define “beacons” as species-specific non-polymorphic DNA motifs able to carry both genetic and epigenetic information. According to the above analysis, we estimated the number of CpG beacons to be ~1.19 million in the human genome. In the future a definitive set will be able to be established following mass whole genome sequencing in a large number of these primates. However this current calculation will already be enriched for “true” human CpG beacons that can facilitate unique species-specific epigenomic variation. A user interface to view the human CpG beacons in the UCSC genome browser in the context of existing annotation is available at www2.cancer.ucl.ac.uk/medicalgenomics/humanCpGBeacons/trackList.php.
The density distribution of the human beacons in 1 kb windows was estimated, which showed more than half were singletons, ~2% were ≥ 5 beacons/kb, and 0.03% were ≥ 20 beacons/kb. To assess the significance of this long tail with higher density, we performed 1,000 permutations by choosing a set of random beacons from the CpG locations in the h1c1o1 genomic set. This simulation never exceeded the number of peaks that are ≥ 20 CpG beacons/kb in the observed genome set (peaks ≥ 20 CpG beacons/kb: simulation peaks range = 0–7, simulation average = 1.527 peak per genome, observed peaks = 21, empirical p < 1 × 10−3).
Extreme CpG beacon clusters
Taking this ≥ 20 CpG beacons/kb as an initial threshold (which reflects an increase of ≥ 4% in CpG density per kb in human compared with the other primates) we identified 21 extreme genomic outliers of human CpG beacon density (see Table S1). Beacon density distribution is displayed across the genome in Figure 1. This initial observation revealed that the third highest peak on Chromosome 20 co-located with the promoter CGI of the HAR1A gene, a non-coding gene significant in cortical development discovered by Pollard et al.(Fig. S1).32 HAR1A was identified to be co-expressed by Cajal-Retzius neurons, with Reelin, a secreted glycoprotein that is fundamental in specifying the six-layer structure of human cortex.32 This gene had been first identified by mammalian and vertebrate comparative genomics for regions of high conservation but outlying substitution of any type in the human genome, with an extreme region of 118 bp containing 18 human changes since the Homo-Pan split. In fact eight of these substitutions are CpG beacon creating, from the total of 35 in this cluster that spans ~1.8 kb. This locus would still be a genomic CpG beacon cluster outlier with a peak of 24/kb even with this 118bp region removed. Therefore this critical non-coding gene was able to be identified without any recourse to longstanding vertebrate or mammalian conservation but purely by focusing on inter-primate CpG density change. Larger regions of bias identified across this locus have implicated recombination hotspot drift over time.38,54

Figure 1. Human CpG beacons by 1 kb density score and genomic location. Loci greater than or equal to a threshold of 20/kb are indicated. A telomeric bias in peaks is evident, as well as in the historic chromosome 2q13 fusion point.36
We were therefore interested in the significance of the other 20 CpG beacon clusters identified in the human genome. We preformed gene set enrichment with Ingenuity Pathway Analysis (IPA, ©2011 Ingenuity Systems, Inc.) on these genes overlapping an extreme beacon cluster (70%), or within 100 kb 5′ or 50 kb 3′ of their transcript, and found highest significant enrichment for the categories of developmental and genetic disorder, inflammatory response, reproductive system development and function, and neurological disease genes [p value with Benjamini-Hochberg correction (PB-H) = 6.03 × 10−3, Fig. S2). These 20 clusters included the causative genes when mutated for four different monogenic mental retardation disorders (ANKRD11,55 CHL1,56 EHMT157 and VLDLR58,59) and genes implicated in complex traits through GWAS and CNV analyses to phenotypes such as behavioral and psychiatric disorders including autism and bipolar disorder (ANKRD11,60 DLGAP261 and DPP1060,62); synaptic transmission (SYT163); as well as total cerebral brain volume in a radiological examination (CDH464) (see Table S1). To take into consideration any possible gene size bias, we also performed an analysis using the regional based binomial test included in the GREAT gene enrichment analysis tool65 (using default cut-offs but reducing the maximum distal extension from 1 Mb to 100 kb/ see methods) for these 20 CpG beacon clusters, excluding the known HAR1A result. This was significant for only three categories of biological process with an FDR Q ≤ 0.15: which included the categories cognition (binomial raw p = 1.43 × 10−5, FDR Q = 1.26 × 10−1), and behavior (binomial raw p = 2.25 × 10−5, FDR Q = 9.87 × 10−2). Furthermore, as a negative control, we also calculated the locations of the Chimpanzee CpG beacon clusters that exceed the 20/kb threshold and these identified no genes implicated with developmental delay or mental retardation (see Table S2) and as well was non-significant with GREAT analysis (via liftOver to human).
The human CpG beacon clusters represent regions of potential regulatory modulation or change to the nearby genes that is human-specific. This correlation, and not causation within these regions, is of interest particularly as these monogenic disease genes have shown that genetic mutation within them is not lethal but carries significant developmental and neurological pathology. These important genes could therefore be the plausible targets of significant regulatory change between human and other closely related primates due to the similarity of their proteomes.66
Biased-gene conversion overlap
The human extreme beacon clusters showed very strong overlap with the top 200 regions of BGC identified by Drezser et al. (57.1% of ≥ 20 CpG beacons/kb clusters, χ2 p < 2.20 × 10−16),36 thus implicating localized GC rise, which is thought to be a neutral process, with a consequent increase in CpGs. Therefore this implies the CpG beacon clusters associate with a recombination driven CpG increase in human, as opposed to regions of high CpG mutability in other primates. Moreover, we also identified the majority of these clusters in telomeric regions (52.3% in terminal chromosome bands), which are known to have elevated rates of recombination in males,67 with hotspots associated with BGC.40 A 15% greater divergence in terminal ends of chromosomes was identified in the chimpanzee sequencing project.8
Cohen et al. recently reclassified CGIs using evolutionary modeling into those that were classical hypo deaminated islands, with ~80% of these 10 kb from a transcription start site (TSS) and with strong overlap with H3K4me3, and those that had arisen as a by-product of BGC that were typically constitutively hypermethylated.41 However, on examination of the available sperm methylome data via MeDIP-seq,68 which includes data for 18 of these extreme beacon cluster regions that co-located with CGI, these were found to be predominately hypomethylated. The average methylation level was 26.38%, therefore aiding the retention of CpGs by reduced mutability, enabling potential regulation by methylation to occur.10
Positive CpG beacon clusters
To differentiate between specific CpG increase, as opposed to generalized regions of GC rise, we controlled by the concurrent formation of the exact inverse dinucleotide GpC; which lacks methylation ability. We defined Positive CpG Beacon Clusters (PBCs) as regions where CpG beacons outweighed their local human-specific GpC content. BGC increases regional GC content and therefore passively CpGs, but if CpGs are methylated in the germ line their continual loss will eventually lead to the acquired GpCs outweighing CpGs over time. We calculated this via a sliding window analysis with a window size of 1 kb and slide of 100 bp across the genome (see Fig. 2). The vast majority of the extreme beacon clusters were genomic outliers of PBC score, i.e. EHMT1 and CDH4 and all except two possessed positive scores (see Table S1). These two extreme negative scores were identified in loci known for extensive and continual gene conversion, the olfactory receptors, with PBC score peaks of -23/kb and -16/kb for OR2T3 and OR2T12, respectively.

Figure 2. Human positive CpG beacon scores calculated across the genome in 1 kb windows with 100 bp slide. Extreme positive or negative loci are indicated.
Extreme CpG beacon clusters appear to be strongly driven by BGC; therefore, PBCs indicate regions where the gained CpGs beacons are not as hypermutable as would be expected, likely due to a loss of methylation in germ line. By retaining from the 20 clusters only those with at least a +5 PBC score, more significant p values in both biological category enrichments of cognition and behavior were obtained (binomial p = 7.19 × 10−6 and 9.41 × 10−6, Q FDR value = 6.3 × 10−2 and 4.1 × 10−2, respectively). To explore the potential of this CpG beacon-specific increase genome-wide, we identified all the PBC ≥ +5 loci comprising 2,601 regions, that account for ~0.1% of the human genome. IPA analysis of associated PBC genes showed significant results for a large number of common disease categories (PB-H < 1 × 10−20) (data not shown), although this result will be biased disproportionally with larger gene regions. Examining these PBC loci with GREAT (genomic regions enrichment of annotation tool) analysis, which corrects for this issue of potential genomic space available to input signal, we identified a number of significant results for potential human phenotypes and traits (see Table S3, FDR Q < 0.05), such as cortical gyral simplification (binomial FDR Q-value = 1.94 × 10−4), atrophy/degeneration affecting the central nervous system (Q = 1.69 × 10−2), abnormality of the cerebral cortex (Q = 2.23 × 10−2) and poor speech (Q = 4.66 × 10−2). A large number of biological processes were implicated as significant, including the nucleus accumbens development (Q = 6.31 × 10−7), nose development (Q = 4.52 × 10−6) and neurotransmitter transport (Q = 1.27 × 10−2) (see Table S4).
These PBCs regions were also enriched in intragenic islands (CGI within transcripts but not at the classical 5′ promoter region), being found twice as commonly within these regions as would be expected for their genomic size (see Fig. S3, χ2 p < 2.20 × 10−16). In fact, with increasing CpG beacon density this intragenic enrichment became stronger (see Fig. S4, Beacons ≥ 10, χ2 p < 2.20 × 10−16). These islands have been implicated in a number of other studies for their significant role in developmentally important isoforms.5,69,70 Examination of repeat classes identified enrichment in the hominid-specific SVA subclass71,72 (see Fig. S5, χ2 p < 2.20 × 10−16), which arose ~20 MYA and has been extremely prolific during evolution of the primate genome.73,74
Correlation between CpG density and CGI hypomethylation
While specific genetic methylation-determining regions (MDRs75) have been identified within CGIs, a correlation with CpG density and hypomethylation has also previously been recognized.10 Therefore, CpG beacon clusters will lead to species-specific CpG density increases which may be associated with increased CGI hypomethylation and formation of permissive chromatin.76 A CpG density of ~20% CpGs (or 10% methylatable cytosines) was proposed by Eckhardt et al.19 as a threshold beyond which CGI are highly likely to be constitutively unmethylated across all differentiated tissues. Examining the available data from two bisulphite sequencing experiments from Li et al.77 in peripheral blood cells and Lister et al.2 from fibroblasts, we find methylation within CGIs is strongly correlated in these sets (r2 = 0.84), despite being confounded by different experiments, tissues and cell line effects. Furthermore, the same significant trend of reduced methylation when CGIs were categorized into subgroups of all, ≥ 15%, ≥ 20%, ≥ 25% CpG density is seen using both the Ensembl CGI definition and an alternate CGI set by Wu et al. identified via hidden Markov models.78 (Kruskal-Wallis p < 2.2 × 10−16) (Fig. 3; Fig. S6A‒C).

Figure 3. CpG density influence on methylation in Wu et al. CpG Islands from Li et al. bisulphite methylome data from peripheral blood mononuclear cell DNA (Kruskal-Wallis rank sum test p value < 2.2 × 10–16).77,78
Next, we generated peripheral blood cell methylome data by MeDIP-seq of pooled samples from chimpanzee and rhesus macaque as well as pooled human samples. Examination of these data also supported the inverse correlation of CpG density with methylation in CGI across all three species in the Ensembl CGI set (see Fig. S7A, p < 2.2 × 10−16) and as well the Wu et al. CGIs that are proposed to have improved trans-species CGI prediction (Fig. S7B, p < 2.2 × 10−16).78
We then investigated whether the influence on methylation change was still apparent in CpG density that changed over time in orthologous CGI between these species. We identified the orthologous CGI set between human and chimpanzee (Ensembl n = 13,999, Wu et al. n = 34,053), and human and macaque (Ensembl n = 4,654, Wu et al. n = 19,200) and chimpanzee and macaque (Ensembl n = 4,004, Wu et al. n = 18,747). For example, the orthologous DPP10 CpG beacons extreme cluster CGI, showed average methylation (RPM) of 0.51 and 4.80 in human and chimpanzee respectively, but not enough CpG density in macaque for a CGI to be defined even by the Wu et al. methodology. The subset of these orthologous islands that were ≥ 20% CpG density in one species and ≤ 19% in the other was then obtained. A significant difference was identified in the Ensembl set for human vs. chimpanzee CGI (Wilcoxon p = 1.954 × 10−12; data not shown) and in the larger Wu et al. set these groupings showed a small but significant reduction in methylation [expressed as average reads per million (RPM)] consistently in the higher density CGI group across all species comparisons (see Figure 4, all p Wilcoxon < 2.2 × 10−16).

Figure 4. Comparison of methylation in subset of orthologous CGI set from Wu et al.78 The RPM in these islands was compared by subtraction. First Human[RPM] – Chimpanzee[RPM], in islands Human (Hs) > 20% CpG and Chimpanzee (Pt) < 19% CpG then Chimpanzee > 20% CpG and Human < 19% CpG. Then Human[RPM] – Macaque[RPM], Human (Hs) > 20% CpG and Macaque (Mm) < 19% CpG and Macaque > 20% CpG and Human < 19% CpG. Finally Chimpanzee [RPM] – Macaque [RPM] in islands Chimpanzee > 20% CpG and Macaque < 19% CpG then Macaque > 20% CpG and Chimpanzee < 19% CpG. All show a consistent pattern where the less CpG dense island (< 19%) is more methylated than the more CpG dense island (> 20%).
Therefore, we have shown that varying CpG density changes the methylation potential within CGIs. This change in likelihood of methylation between low and high density CGIs was found to occur across and between the three species. The most extreme human-specific genomic eruptions of CpGs occur in the identified “CpG beacon” clusters, which in turn have highlighted genes associated with human traits and disease.
Discussion
The proteome is similar across placental mammals; therefore, the creation of new protein coding genes is rare,79 although not unknown.80 Regulatory modification has long been proposed as critical in human acquired traits66 and genomic data acquired in the last decade supports the hypothesis that species evolution is predominately via novel regulatory adaptation and subsequent altered gene expression.79,81 Recently, the identification of human-specific loss of regulatory DNA revealed insights into human evolutionary divergence.82
Epigenetic mechanisms, including DNA methylation, are critical in genome regulation and viable mammalian development requires the ability to methylate cytosines.83 This chemical modification can lead to disparate effects depending on genomic location; repressive in CGI,18 activating in gene bodies47 and splicing influence in CTCF binding sites.84 Changes in developmental timing are significant in species-specific differences85 and the epigenetic modulation of intragenic islands may direct developmentally critical isoforms.5,86 Thus, this epigenomic extra layer of control enables additional axes to the adaptive landscape and aids in the evolution of complex phenotypes.23,87 Cytosine methylation has also been suggested to be significant in karyotype evolution.88 Even simply focusing on human higher cognitive functioning, notwithstanding all the other phenotypic differences, levels of brain tissue-specific imprinting,89,90 distinctive neuronal DNA methylation profiles91,92 and potential role in synaptic plasticity,93 as well as pathogenic Methyl Binding Domain gene mutations in post-natal brain development disorders,94,95 all postulate that the gain and loss of CpG may be fundamental in the human-specific phenotype.
We therefore identified a subset of species-specific CpGs by inter-primate comparison, impartial to mechanistic cause, which we have termed CpG beacons. Focusing initially on extreme human CpG beacon clusters, we showed they are enriched for neurological disease genes and, additionally, co-locate with the evolutionary accelerated HAR1A nc-RNA gene. A strong correlation between accelerated genomic loci and bias toward increased GC content was observed previously,35 due to the effects of recombination.30 Fine scale recombination hotspots show high diversity between human and chimpanzee, as they are short-lived relative to divergence times,96,97 potentially strongly influenced by the variation in zinc finger binding of PRDM9.98,99 GC-coupled CpG increase due to recombination has been suggested to have played a considerable role in CGI formation11 and thus may be a strong driver in the formation of CpG beacon clusters and thus species-specific regulation.
However, on top of the localized strong effect of BGC on increased GC, multiple subtle substitutions have been shown to have a morphological evolution effect altering the timing and level of expression.100 We looked for potential CpG-specific signatures by identifying where human-specific CpG exceeded human-specific GpC, defining Positive CpG Beacon Clusters, which identified potential human traits that may have arisen during human evolution.101
Recent comparative methylome analyses have revealed species-specific differences.102,103 Molaro et al. examined chimpanzee and human sperm and supported the link between genome and epigenome, by identifying strong CpG decay correlated with methylation over brief evolutionary periods. They also found extensive species-specific methylation differences in SVA repeats, with significantly increased numbers of orthologous hypomethylated SVAs within humans. Interestingly, this is the same subtype in which we identified high enrichment of positive beacon clusters, which could be driving this hypomethylation. Additionally SVAs have recently been identified to be involved in active somatic retrotranspositon in human brain.104
Lienart et al. have recently identified the existence of methylation-determining regions (MDRs) due to sequence characteristics within CGI, but also acknowledged the necessity of a critical CpG density within these regions.75 Increasing CpG density forms low density CpG islands which are more likely to have variable methylation and hence to be tissue-specific or developmental time-dependent. Further density rise will create high density islands that eventually will become increasingly likely to become constitutively hypomethylated above a threshold identified earlier by Eckhardt et al.19 This correlation can be seen by re-examination of the Li et al.77 and Lister et al.2 bisulphite sequencing data and is supported across three species in this study via MeDIP-seq. Either MDR-induced CpG retention, BGC created CpGs, or potentially both can lead to clusters of CpGs resulting in hypomethylated islands that then recruit factors such as Cfp1. While this process does not lead to explicit expression per se, it establishes open permissive chromatin via H3K4me3 marked domains.12,76 Therefore, CpG beacons have the capacity to move, or indicate the movement of, these loci along a continuum from no island to tissue-specific island to constitutively active island, as illustrated in the model shown in Figure 5.

Figure 5. Model for beacon-mediated CGI evolution and example for possible association with disease. The left panel illustrates a model for CpG beacon-mediated increase of CpG density. A moderate CpG beacon increase lead to the formation of low density CGIs which are predominantly methylated and prone to tissue-specific methylation. Further CpG beacon increase can eventually lead to high density CGIs with increased likelihood of becoming constitutively hypomethylated. Variation in such CGIs could then result in species-specific phenotypic changes as discussed in this study for CGIs containing extreme clusters of > 20 beacons per kb. CpG beacon gain may indicate reduced mutability due to acquisition of new methylation determining regions75, direct gain due to accumulated substitution or biased gene conversion, or a combination of all of these processes. The right panel shows an example of one of the identified extreme CpG beacon cluster containing CGIs that is associated with the human ANKRD11 gene, implicated in Autism. In this case, the CpG density and methylation state of the orthologous CGIs in human, chimpanzee and macaque are concordant with the proposed model. Methylation values were determined by MeDIP-seq and are given as reads per million (RPM).
In conclusion, the CpG dinucleotide is vital for regulation and not only transmits genomic data but also enables epigenomic variation. Thus, genomic change in this dynamic dinucleotide required for DNA methylation, influencing CGI methylation, gene body methylation, imprinting and splicing, is fundamental to understanding our evolutionary acquired traits and vulnerabilities to disease.
Methods
CpG beacon identification
CpG loss is time- not replication-dependent; therefore, there are almost equal counts of CpG in human and chimpanzee.43 Recent estimates from whole genome sequencing for mutation rate is ~1 × 10−8 per generation105 with the CpG dinucleotide approximately ten times this. Due to the rapid turnover in regulatory elements,106 most are too weakly conserved to mouse to distinguish45 which will particularly be the case with the highly mutable CpGs. By utilizing the additive collective divergence of multiple primates44,45 a CpG’s human-specific state was attributed. The comparatively inferior sequence quality of these individual non-human primate sequences was balanced by the combined multispecies comparison vs. human. Assignment of ancestral state by use of chimpanzee alone was found to have an error rate of 0.65% utilizing macaque as a second out-group.107 Furthermore, most SNPs have been calculated to be < 1 million years of age, compared with the minimum divergence time of chimpanzee and human which is estimated at 5 million years.108
Therefore, to identify the human-specific changes we utilized the Ensembl Compara.6_primates_EPO six primates alignment51,52 build 58. This includes the species Homo sapiens, Pan troglodytes, Gorilla gorilla, Pongo abelii, Macaca mulatta and Callithrix jacchus. The builds included are human GRCh37, chimpanzee Mar. 2006 (CGSC 2.1/pan Trog2), gorilla (gorGor3); orang-utan Jul. 2007 (WUSTL version Pongo_albelii-2.0.2) ; macaque Jan 2006 (MGSC Merged 1.0/rheMac2) ; and marmoset Mar. 2009 (WUGSC 3.2 (GCA_000004665.1)) . The Ensembl Compara.6_primates_EPO alignment blocks was then reduced in a stepwise fashion due to the requirement of: unique human sequence, i.e. does not align to greater than one location in human genome (82.71% of human genome remaining); unique chimpanzee sequence (80.54%); and contains at least one other primate (79.99%) in order to utilize the strength of the inter-primate comparison.45 Within this ~80% of the human genome that was able to be aligned, an algorithm was devised to identify the location of each human CpG site within these blocks and then compared with the corresponding bases in other species. To be identified as a potential human-specific CpG, the requirement in chimpanzee sequence was that it did not match CG and did not contain N or –. All other species sequences at this position also did not match CpG and the closest other primate (gorilla in 96.64% of sites) did not contain N or –. If this was the case then it was recorded as a human-specific CpG site, which led to a set of 1,820,319 CpGs. All human cluster locations are given in build Human GRCh37 coordinates. The chimpanzee beacons were calculated using the exact same methodological algorithm as the human, but instead changing the focused species to Pan troglodytes.
Polymorphic filter
Any CpG with evidence of polymorphism from 1,000 Genomes data for SNP, CNV and Indel was then removed from the set. The December 2010 Data update Full Project Genotype Release from calls on 629 individuals from the 20100804 sequence was used. Indel data was from the February 2011 update, which were calls from Dindel on the same 629 individuals from the 20100804 sequence and alignment, and also available CNV data from 179 unrelated individuals.109 This resulted in a non-polymorphic human set of 1,192,484 CpGs.
CpG beacon density calculation and permutations
Initial density permutations were calculated for each CpG beacon by counting the number of CpG beacons within a region of 499bp downstream and 500 bp upstream. Random beacon sets of the same number 1,192,484 were generated from the total set of locations of CpGs in the h1c1o1 genome set of 20,207,732 and density calculated.
Positive CpG beacon clusters
Positive beacon clusters were calculated, via a sliding window of 1 kb and slide of 100 bases across the genome. The total non-polymorphic human-specific GpCs within the 1kb region was subtracted from the total CpG beacons within this region.
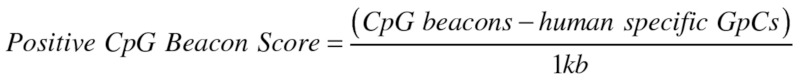 |
Gene set enrichment analyses
Ingenuity Pathway Analysis ©IPA was performed for gene set enrichment. The location of clusters was assigned to genes if it mapped to within 100k 5′ and 50k 3′ of the transcript. The following IPA analysis settings were used: Reference set: Ingenuity Knowledge Base (Genes Only), Relationship to include: Direct and Indirect, Includes Endogenous Chemicals. Optional Analyses: My Pathways My List. Filter Summary: Consider only molecules and/or relationships where (species = Human) AND (confidence = Experimentally Observed). Benjamini-Hochberg multiple test corrected p values are shown only. The region-based binomial analysis of GREAT analysis 2.0.165 was utilized to identify genome regional enrichments from the location of the extreme beacon clusters, as well as the moderate positive beacons clusters ≥ 5. This takes into consideration the bias of potential genomic space available compared with the traditional hypergeometric test. The parameters used were association by Basal plus extension with default values of proximal 5 kb upstream, 1 kb downstream, but a reduced distal limit to 100 kb from 1 Mb and significance assessed by the regional based Binomial test FDR Q value.
Publicly available bisulphite sequencing data
The Lister et al. fibroblast methylome data was downloaded from http://neomorph.salk.edu/human_methylome/data.html. The Li et al. PBMC methylome data was downloaded from the NCBI Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE17972). For the Lister et al. and Li et al. data sets individual CpG methylation was calculated by combining the reads from the two strands and subsequently requiring a 5 read minimum coverage for inclusion.
Comparative MeDIP-seq
DNA was extracted from peripheral blood of 5 chimpanzees (Pan troglodytes, 3 males, 2 females) and 5 rhesus macaques (Macaca mulatta, 3 males, 2 females). Samples were taken from captive individuals at Tierpark Nordhorn, Basel Zoo, Leipzig Zoo and at the German Primate Center during routine health checks and not specifically for this study. Microsatellite analysis conducted at the German Primate Center verified that respective individuals are not related. Sample collection adhered to the American Society of Primatologists (ASP) Principles for the Ethical Treatment of Non-Human Primates (www.asp.org/society/resolutions/EthicalTreatmentOfNonHumanPrimates.cfm).
To obtain averaged methylomes and reduce individual genotype effects, DNAs were pooled for each species at equal concentration for each individual. MeDIP was then executed according to Auto-MeDIP-seq protocol as described previously110 and sequenced on Illumina GAIIx. This was performed with paired end reads of 36 bp with average fragment sizes of: 197 bp in human, 222 bp in chimpanzee, and 217 bp in macaque. The corresponding methylome data are available from the authors on request. A comprehensive analysis of these methylomes will be described elsewhere (Wilson G.A. et al., manuscript in preparation).
MeDIP-seq data was processed using MeDUSA (Methylated DNA Utility for Sequence Analysis).111 This computational pipeline performs a full analysis of MeDIP-seq data by utilizing a number of freely available software packages. Raw sequence data in fastq format were aligned to the reference genomes (Human GRCh37, panTro2 and rheMac2) using alignment software BWA (v0.5.8),112 with default parameters, to generate a SAM format alignment file. Aligned reads were filtered using SAMtools (v0.1.9)113 to remove reads that failed to form a correctly aligned pair (forward and reverse templates). Further filtering based on mapping score was also performed (read pair must contain read with mapping quality ≥ 10). Potential PCR artifacts were removed by discarding all but one read within groups of non-unique reads (i.e., reads aligned to the exact same start and stop position on the same chromosome). FastQC (www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) was used to determine sequence data was of acceptable quality and the Bioconductor (v2.7)114 package MEDIPS (v1.0.0)115 performed enrichment and coverage analyses. Reads per million (RPM) was calculated within regions as (reads/total reads) times 106 for each species (total human reads = 40,797,356, chimpanzee = 32,910,189 and macaque = 24,933,164).
Genetic influence on the DNA methylome
The MEDIPS package was used to approximate absolute methylation scores from relative MeDIP results.115 This enables regional methylation to be compared over features, i.e., CpG Islands utilizing the appropriate genome sequence for each species. LiftOver116 was used to calculate orthologous CGI sets with overlap of at least 95% required. Greater than 20% and less than 19% orthologous sets were chosen from the orthologous island sets for each grouping with the following numbers: Hs ≥ 20 and Pt ≤ 19, Ensembl = 375, Wu = 557; Pt ≥ 20 and Hs ≤ 19, Ensembl = 150, Wu = 614; Hs ≥ 20 and Mm ≤ 19, Ensembl = 248, Wu = 605; Mm ≥ 20 and Hs ≤ 19 Ensembl = 62, Wu = 1530; Pt ≥ 20 and Mm ≤ 19, Ensembl = 256, Wu = 700; Mm ≥ 20 and Pt ≤ 19 Ensembl = 118, Wu = 1451 (Hs = Homo sapiens, Pt = Pan troglodytes and Mm = Macaca mulata).
Statistical analysis
Statistical calculations were performed in the R statistical environment.117 Empirical p values were calculated as the excess of simulation vs. observed values. Kruskal-Wallis rank sum test was used to compare methylation in CGI density sets and Wilcoxon test for comparison between average RPM values for orthologous island sets. Chi-square calculations for enrichments were performed for PBC by bases covered of PBC vs. total bases of category and CpG beacon vs. total CpGs.
Supplementary Material
Acknowledgments
We thank the staff of the Tierpark Nordhorn, Basel Zoo, Leipzig Zoo and the German Primate Center for providing blood samples of chimpanzees and rhesus macaques. Work in the Beck lab was supported by the Wellcome Trust (084071), Royal Society Wolfson Research Merit Award (WM100023) and EU-FP7 projects HEROIC (018883), EPIGENESYS (257082) and BLUEPRINT (282510).
Glossary
Abbreviations:
- BGC
biased gene conversion
- CGI
CpG islands
- MeDIP
methylated DNA immunoprecipitation
- PBC
positive beacon clusters
- PBMC
peripheral blood mononuclear cells
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
Conceived and designed the experiments: C.G.B., G.A.W. and .SB. Performed the experiments: C.G.B., L.M.B. and G.A.W. Analyzed the data: C.G.B. and G.A.W. Contributed reagents/materials/analysis tools: C.R. and L.W. Wrote the paper: C.G.B. and S.B.
Supplemental Material
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/22127
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/22127
References
- 1.Bird A. The dinucleotide CG as a genomic signalling module. J Mol Biol. 2011;409:47–53. doi: 10.1016/j.jmb.2011.01.056. [DOI] [PubMed] [Google Scholar]
- 2.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–70. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 4.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 5.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–1. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 8.Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 9.Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–9. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–66. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 11.Polak P, Arndt PF. Long-range bidirectional strand asymmetries originate at CpG islands in the human genome. Genome Biol Evol. 2009;1:189–97. doi: 10.1093/gbe/evp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–6. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ. CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell. 2010;38:179–90. doi: 10.1016/j.molcel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, et al. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16:564–71. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 15.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–7. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Illingworth RS, Bird AP. CpG islands--‘a rough guide’. FEBS Lett. 2009;583:1713–20. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 17.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 19.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1757–64. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nekrutenko A, Li WH. Assessment of compositional heterogeneity within and between eukaryotic genomes. Genome Res. 2000;10:1986–95. doi: 10.1101/gr.10.12.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eyre-Walker A, Hurst LD. The evolution of isochores. Nat Rev Genet. 2001;2:549–55. doi: 10.1038/35080577. [DOI] [PubMed] [Google Scholar]
- 26.Arndt PF, Petrov DA, Hwa T. Distinct changes of genomic biases in nucleotide substitution at the time of Mammalian radiation. Mol Biol Evol. 2003;20:1887–96. doi: 10.1093/molbev/msg204. [DOI] [PubMed] [Google Scholar]
- 27.Hwang DG, Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci U S A. 2004;101:13994–4001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanay A, O’Donnell AH, Damelin M, Bestor TH. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc Natl Acad Sci U S A. 2007;104:5521–6. doi: 10.1073/pnas.0609746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tolstorukov MY, Volfovsky N, Stephens RM, Park PJ. Impact of chromatin structure on sequence variability in the human genome. Nat Struct Mol Biol. 2011;18:510–5. doi: 10.1038/nsmb.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meunier J, Duret L. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol. 2004;21:984–90. doi: 10.1093/molbev/msh070. [DOI] [PubMed] [Google Scholar]
- 31.Romiguier J, Ranwez V, Douzery EJ, Galtier N. Contrasting GC-content dynamics across 33 mammalian genomes: relationship with life-history traits and chromosome sizes. Genome Res. 2010;20:1001–9. doi: 10.1101/gr.104372.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–72. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 33.Prabhakar S, Noonan JP, Pääbo S, Rubin EM. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- 34.Bird CP, Stranger BE, Liu M, Thomas DJ, Ingle CE, Beazley C, et al. Fast-evolving noncoding sequences in the human genome. Genome Biol. 2007;8:R118. doi: 10.1186/gb-2007-8-6-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, et al. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2006;2:e168. doi: 10.1371/journal.pgen.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dreszer TR, Wall GD, Haussler D, Pollard KS. Biased clustered substitutions in the human genome: the footprints of male-driven biased gene conversion. Genome Res. 2007;17:1420–30. doi: 10.1101/gr.6395807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berglund J, Pollard KS, Webster MT. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 2009;7:e26. doi: 10.1371/journal.pbio.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katzman S, Kern AD, Pollard KS, Salama SR, Haussler D. GC-biased evolution near human accelerated regions. PLoS Genet. 2010;6:e1000960. doi: 10.1371/journal.pgen.1000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratnakumar A, Mousset S, Glémin S, Berglund J, Galtier N, Duret L, et al. Detecting positive selection within genomes: the problem of biased gene conversion. Philos Trans R Soc Lond B Biol Sci. 2010;365:2571–80. doi: 10.1098/rstb.2010.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galtier N, Duret L. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 2007;23:273–7. doi: 10.1016/j.tig.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 41.Cohen NM, Kenigsberg E, Tanay A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145:773–86. doi: 10.1016/j.cell.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 42.Branciamore S, Chen ZX, Riggs AD, Rodin SN. CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors. Proc Natl Acad Sci U S A. 2010;107:15485–90. doi: 10.1073/pnas.1010506107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 44.Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, et al. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–4. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- 45.Wang QF, Prabhakar S, Chanan S, Cheng JF, Rubin EM, Boffelli D. Detection of weakly conserved ancestral mammalian regulatory sequences by primate comparisons. Genome Biol. 2007;8:R1. doi: 10.1186/gb-2007-8-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kadonaga JT. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell. 2004;116:247–57. doi: 10.1016/S0092-8674(03)01078-X. [DOI] [PubMed] [Google Scholar]
- 47.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–3. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 49.Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–92. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulz R, Proudhon C, Bestor TH, Woodfine K, Lin CS, Lin SP, et al. The parental non-equivalence of imprinting control regions during mammalian development and evolution. PLoS Genet. 2010;6:e1001214. doi: 10.1371/journal.pgen.1001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008;18:1814–28. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paten B, Herrero J, Fitzgerald S, Beal K, Flicek P, Holmes I, et al. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 2008;18:1829–43. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, et al. 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coop G, Wen X, Ober C, Pritchard JK, Przeworski M. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science. 2008;319:1395–8. doi: 10.1126/science.1151851. [DOI] [PubMed] [Google Scholar]
- 55.Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89:289–94. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frints SG, Marynen P, Hartmann D, Fryns JP, Steyaert J, Schachner M, et al. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet. 2003;12:1463–74. doi: 10.1093/hmg/ddg165. [DOI] [PubMed] [Google Scholar]
- 57.Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006;79:370–7. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77:477–83. doi: 10.1086/444400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/S0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 60.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Djurovic S, Gustafsson O, Mattingsdal M, Athanasiu L, Bjella T, Tesli M, et al. A genome-wide association study of bipolar disorder in Norwegian individuals, followed by replication in Icelandic sample. J Affect Disord. 2010;126:312–6. doi: 10.1016/j.jad.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 63.Poskanzer KE, Marek KW, Sweeney ST, Davis GW. Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature. 2003;426:559–63. doi: 10.1038/nature02184. [DOI] [PubMed] [Google Scholar]
- 64.Seshadri S, DeStefano AL, Au R, Massaro JM, Beiser AS, Kelly-Hayes M, et al. Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham Study. BMC Med Genet. 2007;8(Suppl 1):S15. doi: 10.1186/1471-2350-8-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–16. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 67.Yu A, Zhao C, Fan Y, Jang W, Mungall AJ, Deloukas P, et al. Comparison of human genetic and sequence-based physical maps. Nature. 2001;409:951–3. doi: 10.1038/35057185. [DOI] [PubMed] [Google Scholar]
- 68.Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26:779–85. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Illingworth R, Kerr A, Desousa D, Jørgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deaton AM, Webb S, Kerr AR, Illingworth RS, Guy J, Andrews R, et al. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011;21:1074–86. doi: 10.1101/gr.118703.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang H, Xing J, Grover D, Hedges DJ, Han K, Walker JA, et al. SVA elements: a hominid-specific retroposon family. J Mol Biol. 2005;354:994–1007. doi: 10.1016/j.jmb.2005.09.085. [DOI] [PubMed] [Google Scholar]
- 72.Warnefors M, Pereira V, Eyre-Walker A. Transposable elements: insertion pattern and impact on gene expression evolution in hominids. Mol Biol Evol. 2010;27:1955–62. doi: 10.1093/molbev/msq084. [DOI] [PubMed] [Google Scholar]
- 73.Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005;309:1850–4. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- 75.Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schübeler D. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. 2011;43:1091–7. doi: 10.1038/ng.946. [DOI] [PubMed] [Google Scholar]
- 76.Bock C, Walter J, Paulsen M, Lengauer T. CpG island mapping by epigenome prediction. PLoS Comput Biol. 2007;3:e110. doi: 10.1371/journal.pcbi.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using hidden Markov models. Biostatistics. 2010;11:499–514. doi: 10.1093/biostatistics/kxq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–97. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 80.Wu DD, Irwin DM, Zhang YP. De novo origin of human protein-coding genes. PLoS Genet. 2011;7:e1002379. doi: 10.1371/journal.pgen.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008;134:25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 82.McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011;471:216–9. doi: 10.1038/nature09774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 84.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–9. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Somel M, Franz H, Yan Z, Lorenc A, Guo S, Giger T, et al. Transcriptional neoteny in the human brain. Proc Natl Acad Sci U S A. 2009;106:5743–8. doi: 10.1073/pnas.0900544106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, et al. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010;6:6. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson LJ, Tricker PJ. Epigenomic plasticity within populations: its evolutionary significance and potential. Heredity (Edinb) 2010;105:113–21. doi: 10.1038/hdy.2010.25. [DOI] [PubMed] [Google Scholar]
- 88.Carbone L, Harris RA, Vessere GM, Mootnick AR, Humphray S, Rogers J, et al. Evolutionary breakpoints in the gibbon suggest association between cytosine methylation and karyotype evolution. PLoS Genet. 2009;5:e1000538. doi: 10.1371/journal.pgen.1000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wilkinson LS, Davies W, Isles AR. Genomic imprinting effects on brain development and function. Nat Rev Neurosci. 2007;8:832–43. doi: 10.1038/nrn2235. [DOI] [PubMed] [Google Scholar]
- 90.Gregg C, Zhang J, Butler JE, Haig D, Dulac C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science. 2010;329:682–5. doi: 10.1126/science.1190831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Iwamoto K, Bundo M, Ueda J, Oldham MC, Ukai W, Hashimoto E, et al. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011;21:688–96. doi: 10.1101/gr.112755.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schroeder DI, Lott P, Korf I, LaSalle JM. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res. 2011;21:1583–91. doi: 10.1101/gr.119131.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–73. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- 94.McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89:551–63. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arnheim N, Calabrese P. Understanding what determines the frequency and pattern of human germline mutations. Nat Rev Genet. 2009;10:478–88. doi: 10.1038/nrg2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Winckler W, Myers SR, Richter DJ, Onofrio RC, McDonald GJ, Bontrop RE, et al. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–11. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]
- 98.Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, et al. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–40. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N, et al. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 2009;5:e1000753. doi: 10.1371/journal.pgen.1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frankel N, Erezyilmaz DF, McGregor AP, Wang S, Payre F, Stern DL. Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature. 2011;474:598–603. doi: 10.1038/nature10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crespi BJ. The origins and evolution of genetic disease risk in modern humans. Ann N Y Acad Sci. 2010;1206:80–109. doi: 10.1111/j.1749-6632.2010.05707.x. [DOI] [PubMed] [Google Scholar]
- 102.Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell. 2011;146:1029–41. doi: 10.1016/j.cell.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martin DI, Singer M, Dhahbi J, Mao G, Zhang L, Schroth GP, et al. Phyloepigenomic comparison of great apes reveals a correlation between somatic and germline methylation states. Genome Res. 2011;21:2049–57. doi: 10.1101/gr.122721.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011;479:534–7. doi: 10.1038/nature10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, et al. 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–4. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–40. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gojobori J, Tang H, Akey JM, Wu CI. Adaptive evolution in humans revealed by the negative correlation between the polymorphism and fixation phases of evolution. Proc Natl Acad Sci U S A. 2007;104:3907–12. doi: 10.1073/pnas.0605565104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jiang C, Zhao Z. Directionality of point mutation and 5-methylcytosine deamination rates in the chimpanzee genome. BMC Genomics. 2006;7:316. doi: 10.1186/1471-2164-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. 1000 Genomes Project Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Butcher LM, Beck S. AutoMeDIP-seq: a high-throughput, whole genome, DNA methylation assay. Methods. 2010;52:223–31. doi: 10.1016/j.ymeth.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wilson G, Dhami P, Feber A, Cortazar D, Suzuki Y, Schulz R, et al. Resources for methylome analysis suitable for gene knockout studies of potential epigenome modifiers. GigaScience. 2012;1:3. doi: 10.1186/2047-217X-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chavez L, Jozefczuk J, Grimm C, Dietrich J, Timmermann B, Lehrach H, et al. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010;20:1441–50. doi: 10.1101/gr.110114.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39(Database issue):D876–82. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ihaka R, Gentleman RR. A Language for Data Analysis and Graphics. J Comput Graph Statist. 1996;5:299–314. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.