

Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has been demonstrated to induce cell apoptosis in many types of tumors, while many hepatocellular carcinoma (HCC) cells display high resistance to TRAIL. Another outstanding limitation of TRAIL is the short half-life in vivo. Stem cell-based therapies provide a promising approach for the treatment of many types of tumors because of the ability of tropism. Therefore, as a new therapeutic strategy, the combination of chemotherapeutic agents and TRAIL gene modified MSCs (TRAIL-MSCs) would improve the therapeutic efficacy of HCC in vivo. This is the first time to show the potential of combination of chemotherapeutic agents and MSCs as a gene vector in the therapy of HCC.
Keywords: TRAIL, bioluminescence imaging, cisplatin, hepatocellular carcinoma, lentiviral, mesenchymal stem cells, synergistic effect
Introduction
Hepatocellular carcinoma (HCC) is among the most lethal and prevalent cancers in the world. Despite aggressive conventional therapy, including surgical resection, transcatheter arterial chemoembolization (TACE), percutaneous radiofrequency ablation or injection of ethanol, and liver transplantation, the 5-y survival rate of individuals with liver cancer is only 8.9%.1 Conventional strategies has yielded poor response rate in patients with advanced stage HCC for its invasiveness and high resistance to chemotherapy.1-3 The multikinase inhibitor (sorafenib) is the first systemic treatment to show survival benefits in patients with advanced HCC in large phase III studies and becomes a new standard treatment for HCC.4,5 However, the median survival was only 7.4 months (95% CI: 5.6, 9.2 mo) for those with advanced HCC treated with sorafenib.6 Therefore, novel and effective therapeutic strategies are required to reduce drug dosage, enhance the therapy efficacy and to diminish toxicity to normal cells.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (also known as APO-2L) is a type II transmembrane protein that belongs to the TNF superfamily.7 TRAIL might be a promising candidate for cancer therapy, and the therapeutic efficacy of TRAIL has been reported in several xenograft models of glioma,8,9 prostate10 and colorectal11 tumors in vivo. However, according to previous study, many HCC cells showed high resistance to TRAIL and TRAIL alone was unable to induce efficient apoptosis even at the highest concentration of 1,000 ng/ml, which severely limits the application of TRAIL in the treatment of HCC.12,13 The resistance of many types of cancer cells to TRAIL can be reversed by treatment with sorafenib,14,15 chemotherapeutic agents12,16-20and ionizing radiation,21,22 which suggest that this combination may be an effective strategy for anticancer therapy.
Another outstanding question of TRAIL in the therapy of tumor is the short half-life. Kelley SK studied the half-life of TRAIL in nude mice, rats, cynomolgus monkeys and chimpanzees. The half-life of TRAIL in vivo is the time taken for its concentration in plasma to decline to half its original level. The results showed that TRAIL was rapidly eliminated from the serum of these animals and the half-lives were only 3–5 min in rodents and 24–31 min in non-human primates.23,24 Therefore TRAIL cannot maintain effective concentration in blood after injection. Fortunately gene transfer approaches have been developed as an alternative method for targeted, more efficient and continuous delivery of cytokines. Thus TRAIL gene therapy might be a promising experimental and potential therapeutic tool to overcome this shortcoming.
Mesenchymal stem cells (MSCs) exhibit the ability of homing to sites of tissue damage as well as the tumor microenvironment, so MSCs are considered to be a promising platform for cell and gene therapy for tumor.25 In recent years, many studies reported that gene modified MSCs specifically targeted multiple tumor types followed by local secretion of therapeutic factors, including IL-12,26,27 pigment epithelium derived factor28 and TRAIL.29-31 MSCs expressing TRAIL exhibited short- and long-term therapeutic effects without significant complications in glioma.32-35 But there was no report about TRAIL gene modified MSCs (TRAIL-MSCs) in the therapy of HCC. As mentioned above, HCC cells showed high resistance to TRAIL, so TRAIL-MSCs alone might be difficult to achieve desired effect.
In this study, for the first time, we have combined chemotherapeutic agents (cisplatin) and TRAIL-MSCs to improve HCC treatment in vivo. The former was used to reverse the TRAIL resistance of tumor cells and the latter were engineered using the TRAIL gene to overcome the short half-life of TRAIL. In addition, noninvasive in vivo bioluminescence imaging (BLI) was utilized to determine the conditions of tumor.
Results
Cisplatin reversed TRAIL resistance through upregulation of DR5 in MHCC97-H cells, but not affected TRAIL-MSCs within a therapeutic window in vitro
We examined the effect of TRAIL combined cisplatin on the viability of MHCC97-H cells in vitro by CCK-8. TRAIL (200 ng/ml) or cisplatin (5 μg/ml) alone failed to affect the viability of MHCC97-H cells significantly. Whereas in the presence of low concentrations of cisplatin (2.5 or 5.0 μg/ml), TRAIL dose-dependently reduced the cell viability (Fig. 1A). The viability of TRAIL-MSCs at different concentrations of cisplatin (5–80 μg/ml) was also evaluated. Up to 10 μg/ml of cisplatin didn’t affect the viability of TRAIL-MSCs significantly and IC50 was 36.6 μg/ml (24h), which suggested a wide therapeutic window for TRAIL-MSCs (Fig. 1B).

Figure 1. Enhanced suppression effects of MHCC97-H cells proliferation by combination of TRAIL with cisplatin in vitro. MHCC97-H cells were treated with different concentrations of TRAIL and cisplatin for 24 h (A). The viability of TRAIL-MSCs wasn’t affected significantly in a wide therapeutic window (B). After 24 h of treatment with 5.0 μg/ml cisplatin, only the expression of DR5 in MHCC97-H cells was increased greatly (C). Cell viability was determined by CCK-8 assay. Each point represents the mean ± SD of three independent experiments. Asterisk indicates p < 0.05.
TRAIL receptors on the cell surface were investigated by FACS. MHCC97-H cells express low levels of TRAIL R1–R4. After 24 h of treatment with 5.0 μg/ml cisplatin, PE mean fluorescence intensity of TRAIL R2 (DR5) was increased greatly from 1.66–2.66 (p < 0.05), but no significant differences of the other three TRAIL receptors was observed. On the other hand, only very mild expression of TRAIL receptors was detected in TRAIL-MSCs, and there was no change of these receptors between treatment and control groups (Fig. 1C).
Establishment of TRAIL gene modified MSCs
The Lentiviral Vectors (pLenti6.3-TNFSF10-IRES-hrGFP) were titered using 293FT cells, and an MOI of ten was used for infection. DNA sequencing analysis confirmed that TRAIL gene sequence was identical to the report in Genebank and did not reveal any mutation. The transduction was measured and purified using FACS and a fluorescence microscope, which resulted in > 99% efficiency (Fig. 2). Transduction with lentivirus did not alter the morphology of MSCs, compared with untransduced cells. Like MSCs, TRAIL-MSCs were still able to differentiate into osteogenic and adipogenic cell lineages (Fig. 3). To determine whether TRAIL could be secreted efficiently in TRAIL-MSCs, TRAIL concentrations in culture supernatants were detected by ELISA. The concentrations of TRAIL were 530, 1929 and 2781 pg/ml at 24, 48 and 72 h respectively (Fig. 4). In blank control group, TRAIL wasn’t detected in supernatants of MSCs without gene modification.

Figure 2. Purified TRAIL-MSCs were analyzed by fluorescence microscopy and FACS. GFP expression was confirmed in TRAIL-MSCs (A, × 200). The transduction efficiency of MSCs was monitored by FACS and 99.11% of cells were GFP positive (B, untransduced MSCs as control; C, TRAIL-MSCs).

Figure 3. Differentiation potential of the TRAIL-MSCs. Without osteogenic induction, MSCs weren’t stained by alizarin red (A, as negative control). Whereas following osteogenic induction for 2 weeks, matrix mineralization was revealed by alizarin red staining in untransduced MSCs (B, as positive control) and TRAIL-MSCs (C, × 200). Similarly, lipid droplets weren’t detected in MSCs without adipogenic induction (D, as negative control). After 2 weeks of adipogenic induction, lipid droplets were stained by oil red O in untransduced MSCs (E, as positive control) and TRAIL-MSCs (F, ×400).
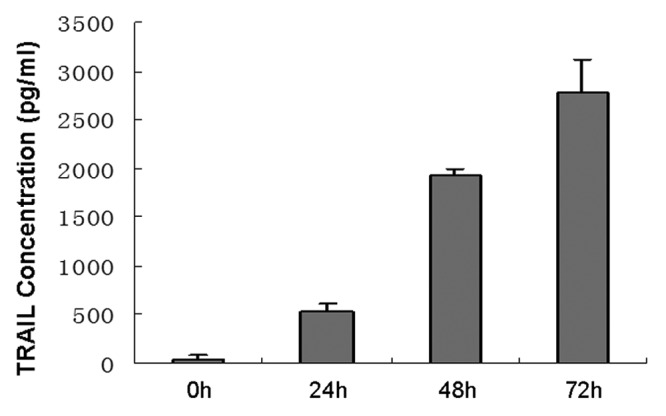
Figure 4. High-level expression of TRAIL produced by TRAIL-MSCs in vitro. The concentrations of TRAIL increased gradually with culture time. Each point represents the mean ± SD of three independent experiments.
Establishment of luciferase-RFP gene modified MHCC97-H cells
MHCC97-H cells were transduced with pLenti6.3-Firefly luciferase-IRES-RFP (mkate2) with a MOI of 30 and 8 μg/ml polybrene. After purification with FACS, the transduction efficiency was > 85% on FACS (Fig. 5).
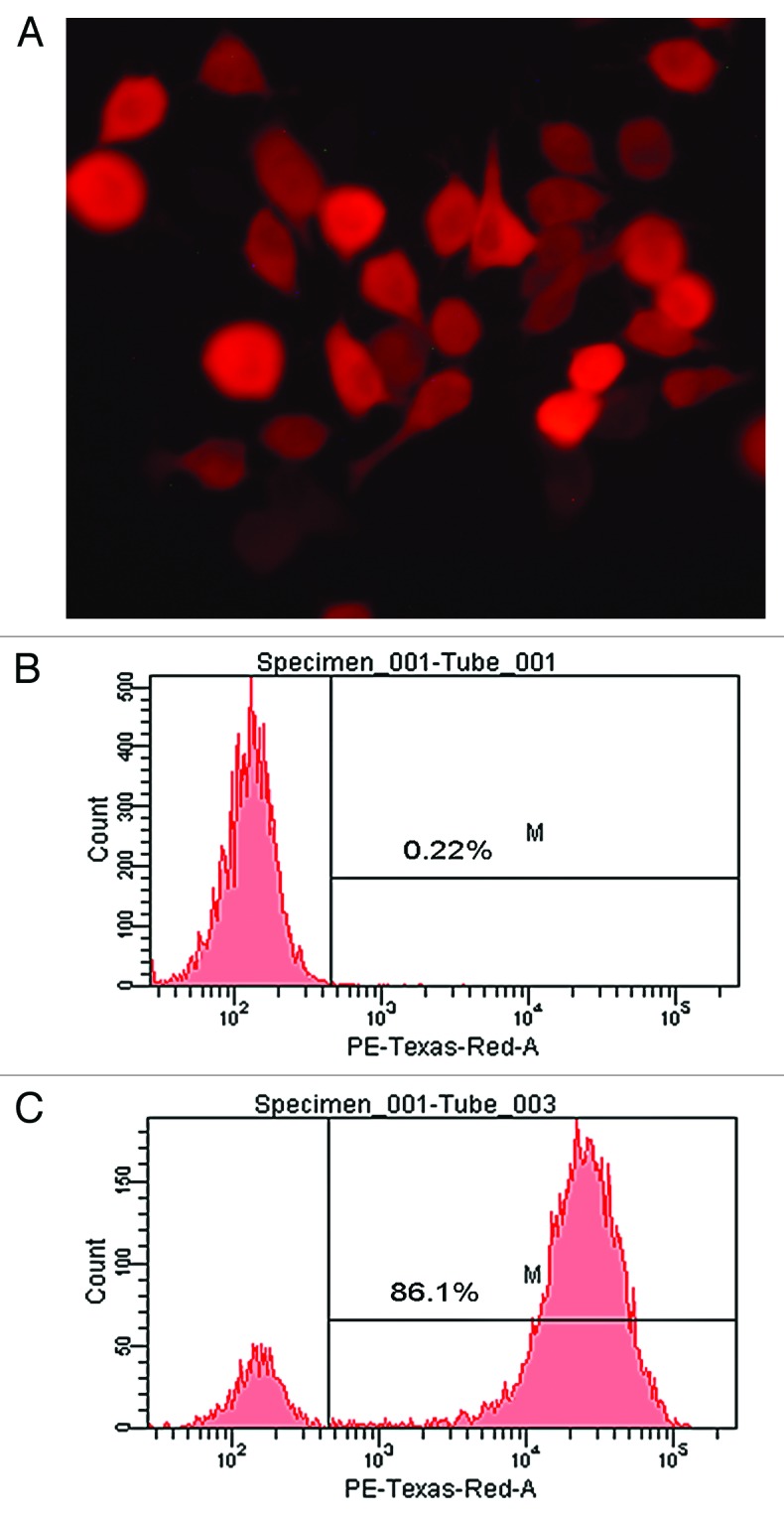
Figure 5. Live cell imaging of MHCC97-H transduced by pLenti6.3-Firefly luciferase-IRES-RFP (mkate2). RFP expression was detected in transduced MHCC97-H cells on fluorescence microscopy (A, ×400). The transduction efficiency of MHCC97-H cells was monitored by FACS and 86.1% of cells were RFP positive (B, untransduced cells as control; C, transduced MHCC97-H cells).
Effect of cisplatin combined TRAIL-MSCs on growth of MHCC97-H xenograft
To confirm whether the synergistic effect of cisplatin and TRAIL-MSCs has potentially possible clinical implications, the effect of cisplatin and TRAIL-MSCs on the growth of MHCC97-H xenograft tumors was assessed. At baseline, the tumor volume was 60.79 ± 17.73 mm3, 63.93 ± 9.32 mm3, 70.82 ± 12.06 mm3 and 71.61 ± 14.33 mm3 in control group, cisplatin group, TRAIL-MSCs group and cotreatment group respectively (p > 0.05). Tumor growth was obviously inhibited by cotreatment with cisplatin and TRAIL-MSCs for 21 d and tumor volume in cotreatment group (378.4 ± 44.5mm3) was only 30% of that in control group (1222.2 ± 245mm3) at the end of the study (p < 0.05). Treatment with cisplatin or TRAIL-MSCs alone had no significant effect on MHCC97-H tumor growth (Fig. 6A). Similarly, BLI demonstrated that the relative intensity (photons/sec) in cotreatment group was much less than that in control group at the end of the study (Fig. 6B and C).

Figure 6. In vivo therapeutic efficacy of cisplatin combined TRAIL-MSCs on MHCC97-H tumors. The tumor volume at baseline in four groups (n = 6) wasn’t statistical significant different. After 21 d observation, cotreatment with cisplatin and TRAIL-MSCs obviously suppressed the growth of tumors, while cisplatin or TRAIL-MSCs alone didn’t inhabit the growth significantly (A). The relative intensity (photons/sec) of tumors was examined by BLI. Exposure time was 0.05s. The relative intensity of tumors in cotreatment group was lower than that in other groups (B). Representative examples were shown (C). Asterisk indicates p < 0.05, co-treatment group vs. control and monoagent groups (n = 6).
In vivo migration of TRAIL-MSCs
We evaluated the migratory nature of TRAIL-MSCs toward the tumor sites in vivo by detecting the GFP-positive cells on frozen sections. Three weeks after systemic delivery, green fluorescent TRAIL-MSCs were detected within the tumor bed in TRAIL-MSCs group and combined group. While in control and cisplatin groups, no GFP-positive cells were found in tumor microenvironment (Fig. 7).

Figure 7. Migration of MSCs in HCC-bearing mice. After 3 weeks of treatment, the mice were sacrificed and the tumors were resected. Then 10-μm frozen sections were cut from representative tumors. Fluorescence microscopy revealed the red tumors cells (MHCC97-H labeled by RFP) on frozen sections. Only in TRAIL-MSCs group and combined group, the GFP positive TRAIL-MSCs (green fluorescent, arrow) were detected (×100).
Cisplatin combined TRAIL-MSCs therapy inhibited tumor angiogenesis in vivo
In addition to restricting the volume and relative intensity (photons/sec) of tumors, cotreatment with cisplatin and TRAIL-MSCs also inhibited tumor angiogenesis. This was performed with immunohistochemical staining of paraffin slides using an antibody against CD34, which is expressed on microvessel endothelial cells. Cotreatment with cisplatin combined TRAIL-MSCs reduced the CD34 count to 32.9 ± 4.5% of controls (Fig. 8).
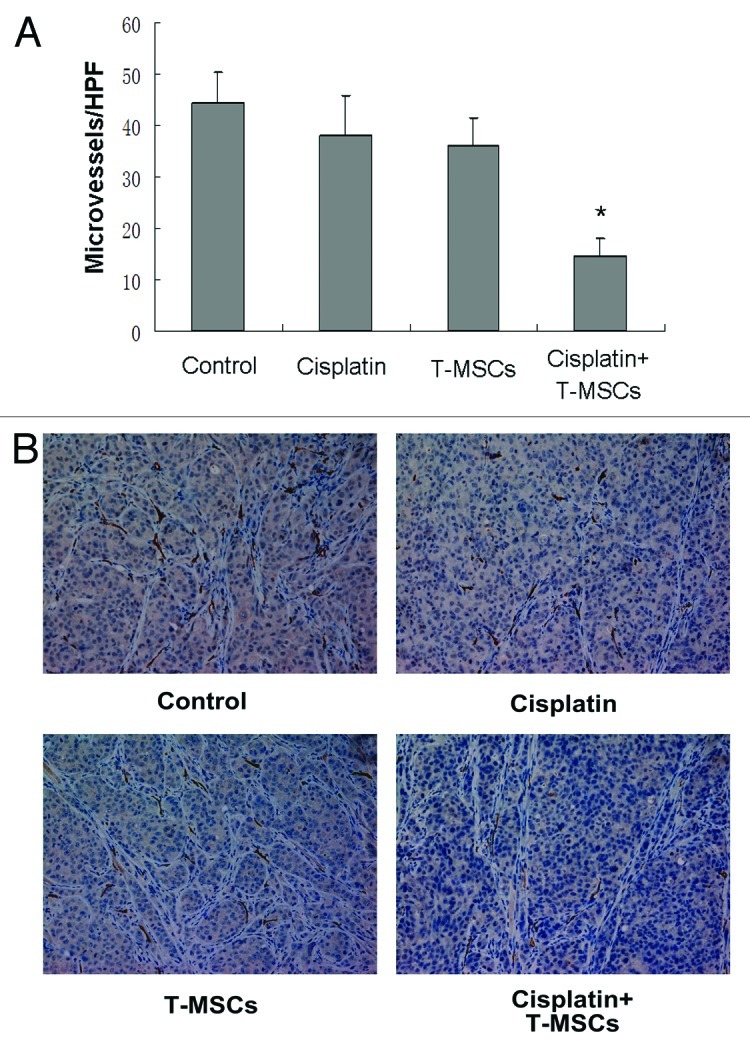
Figure 8. Cotreatment with cisplatin combined TRAIL-MSCs reduced vessel density in HCC. Tumors from control group, cisplatin group or TRAIL-MSCs group showed intense and diffuse CD34 immunoreactivity, whereas MVD from cotreatment group was significantly lower (A). Representative tumor sections stained with an anti-CD34 antibody are shown (B, ×200). Asterisk indicates p < 0.05, co-treatment group vs. control and monoagent groups (n = 6).
TRAIL expression in serum and tumor
TRAIL concentrations in serum and tumor homogenate were examined using ELISA. Results demonstrated no significant difference of TRAIL in serum within groups at different time points. However, the TRAIL concentrations of the tumor homogenate in TRAIL-MSCs group and cotreatment group were 3743.7 ± 1011.8 pg/ml and 3224.7 ± 857.3 pg/ml, more than 4-fold vs. that in control group (747.4 ± 122.1 pg/ml). There was no significant difference of TRAIL between control and cisplatin groups, as well as between TRAIL-MSCs and cotreatment groups (Fig. 9).
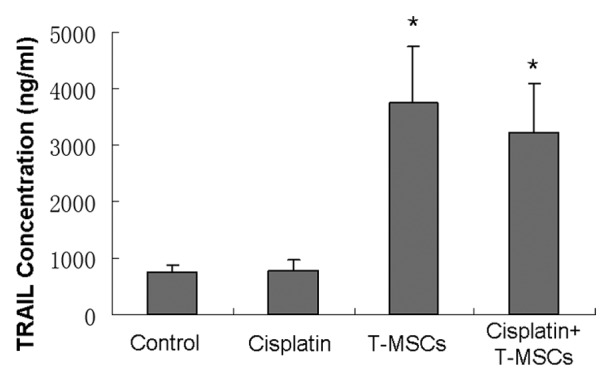
Figure 9. At the end of the study, TRAIL concentrations of the tumor homogenates (n = 6) in TRAIL-MSCs and cotreatment groups were higher than those in control and cisplatin groups (p < 0.05). Whereas no statistical difference was found between TRAIL-MSCs and cotreatment groups, as well as the other two groups (p > 0.05).
Discussion
With the ability to migrate to tumor microenvironment and to evade host immune response, MSCs represent an attractive option as a delivery vehicle for cell and gene therapy. The tumor microenvironment is considered a site of chronic inflammation.36 The level of MSCs recruitment was related to the degree of inflammation in a tumor microenvironment.37 So the migration of MSCs into the tumor stroma is thought to be mediated by high local concentrations of inflammatory chemokines and growth factors38 and seems not to be related to tumor size.39 In addition, Secchiero P reported that TRAIL played a role to promote the migration of human MSCs significantly and indicated that the TRAIL/TRAIL-R axis seemed to control the mobilization of BM-derived MSCs into the circulation.40 Our results demonstrated TRAIL-MSCs could migrate to the tumor site and GFP positive MSCs were still detected on frozen sections even 21 d after intravenous administration, and we presumed TRAIL/TRAIL-R axis might enhance this migration.
MSCs have been efficiently transduced with exogenous therapeutic genes using adenoviral,33 lentiviral,28 retroviral41 and nonviral42 vectors. In this study, we used lentiviral vectors (pLenti6.3) as the gene delivery. Though the efficiency was not very high (about 20~30%) with MOI of ten, after FACS purification, the GFP positive rate was > 99% efficiency. TRAIL gene modification did not affect the morphology and differentiation of MSCs. Moreover, TRAIL-MSCs demonstrated high resistance to cisplatin, which suggested a wide therapeutic window. Our data provided favorable evidence supporting the feasibility of the combination of MSCs modified by TRAIL gene and conventional chemotherapy in the treatment of TRAIL-resistant human cancer.
For some cytokines, such as interleukin (IL), hepatocyte growth factor (HGF) and TRAIL, the half-life is only several minutes. After intravenous injection, these cytokines cannot maintain effective concentrations in blood. Previous studies have shown that even increased systemic concentrations of cytokines or secretion at sites distant from the tumor were still not effective in the treatment of tumor.26,34,37 In our study, we detected the TRAIL concentration of tumor homogenate to determine the secretion of TRAIL-MSCs. The level of TRAIL in TRAIL-MSCs and cotreatment groups were more than 4-fold than those in control and cisplatin groups, which contributed to suppress the tumor growth. However, the TRAIL concentration in blood serum wasn’t increased obviously in TRAIL-MSCs and cotreatment groups. These data confirmed that high local secretion of TRAIL within the tumor was essential in the treatment of tumor.
As a new standard treatment for HCC, sorafenib inhibits several Tyrosine protein kinases, Raf kinases and serine/threonine kinases. Several previous studies have shown sorafenib overcomes TRAIL resistance and inhibition of Mcl-1 and STAT3 represents the major mechanism in HCC.14,15,43 Therefore sorafenib not only inhibits tumor angiogenesis and induces tumor cell apoptosis, but sensitizes HCC cells to TRAIL. We hypothesize that combined use of sorafenib and TRAIL-MSCs may produce a favorable response in patients with advanced HCC and is worthy of further study.
In summary, we explored the therapeutic potential of combination chemotherapy and TRAIL-MSCs. MSCs can be genetically modified using lentiviral vectors (pLenti6.3-TNFSF10-IRES-hrGFP) and express TRAIL efficiently. In addition to restrict the volume and relative intensity (photons/sec) of tumors, cotreatment with cisplatin and TRAIL-MSCs also inhibited tumor angiogenesis and restricted the blood supply. Based on this therapeutic strategy, the chemotherapeutic drugs and genes carried by MSCs might be replaced rationally according to the biological characteristics of different tumors.
Materials and methods
Cell culture
An immortalized human bone marrow-derived MSCs lines UE7T-13 cells (JCRB 1154, JCRB Cell Bank),44-47 the life span of which was prolonged by infecting retrovirus encoding human papillomavirus E7 and human telomerase reverse transcriptase (hTERT) was used in the present study. HCC cells line MHCC97-H (kindly provided by Prof. Zhao-You Tang, Liver Cancer Institute, Fudan University)48 and MSCs were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and penicillin (100 U/ml)/streptomycin (100 μg/ml) and were maintained at 37°C in a humidified atmosphere with 5% CO2.
Reagents
For fluorescence-activated cell sorting (FACS) analysis, Mouse IgG1 Isotype Control-PE (IC002P), IgG2B Isotype Control-PE (IC0041P), TRAIL R1 (DR4)-PE (FAB347P), TRAIL R2 (DR5)-PE (FAB6311P), TRAIL R3 (DcR1)-PE (FAB6302P) and TRAIL R4 (DcR2)-PE (FAB633P) were purchased from R&D Systems. Soluble recombinant human TRAIL was purchased from PeproTech. Cisplatin was obtained from Sigma. Human TRAIL enzyme-linked immunosorbent assay (ELISA) Kit (BMS2004) was purchased from Bender MedSystems. Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Laboratories. All other chemicals were commercial products of the highest purity available.
Construction of lentiviral vectors and cell transduction
TRAIL gene cDNA (TNFSF10) was amplicated by polymerase chain reaction (PCR) and cloned into the pLenti6.3 vector. The reconstructed Lentiviral Vectors (pLenti6.3-TNFSF10-IRES-hrGFP) was prepared, purified and their functional titers were determined as described previously.35,49 For lentiviral transduction of MSCs, MSCs (1.5 × 106 cells) were plated on a T-25 culture flask and allowed to attach overnight. The medium was replaced with 5 ml fresh lentiviral supernatant with a multiplicity of infection (MOI) of 10 and 8 μg/ml polybrene (Sigma) to assist the uptake of viral particles. MSCs transduced by pLenti6.3-TNFSF10-IRES-hrGFP were collected 72 h later. The number of GFP-positive cells was determined and purified by fluorescence-activated cell sorting analysis (FACS).
The report gene vector pLenti6.3-Firefly luciferase-IRES-RFP (mkate2) were constructed and stored in our laboratory. MHCC97-H cells were infected with fresh lentiviral supernatant with a MOI of 30 and 8 μg/ml polybrene mentioned above. The number of RFP-positive cells was determined and purified by FACS.
Cell viability assay
Cell viability assay was performed as Miyoshi N as described.50 In brief, MHCC97-H cells (20,000 cells per well) and TRAIL-MSCs (5,000 cells per well) were seeded in a 96-well plate. After an overnight incubation, MHCC97-H cells were treated with 100 μl medium containing cisplatin (0, 2.5 and 5.0 μg/ml) and TRAIL (0, 50, 100, 200 ng/ml) for 24 h, and TRAIL-MSCs were treated with 100 μl medium containing cisplatin (0, 5, 10, 20, 40, 80 μg/ml) for 24 h. Then, 10 μl CCK-8 solution was added to each well. After incubation at 37°C for 2 h in a humidified CO2 incubator, absorbance of each well was monitored with a microplate reader (ELX-800, Bio-Tek) using 450 nm as the primary wavelength (630 nm as the reference wavelength). The concentrations required to inhibit growth by 50% (IC50) were calculated from survival curves using the Bliss method.51
FACS analysis of TRAIL receptors
MHCC97-H cells and MSCs (controls or treated with cisplatin at 5.0 μg/ml for 24 h) were detached non-enzymatically with citric saline buffer (0.135 M potassium chloride, 0.015 M sodium citrate) for 5–8 min at 37°C. FACS analysis of TRAIL receptors was performed according to the manufacturer's protocol (R&D Systems). In brief, Cells were centrifuged and washed three times in an isotonic PBS buffer (supplemented with 0.5% BSA), and were incubated with IgG1 Isotype, IgG2B Isotype and TRAIL R1-R4 antibodies (10 μL/100,000 cells) respectively for 40 min at 4°C. Excess unbound antibodies were removed by washing twice with PBS before submission for FACS. The levels of receptor expression were quantified by the PE mean fluorescence intensity index (ratio of PE mean fluorescence intensity of cells incubated with anti-receptor antibodies and the background PE mean fluorescence intensity in cells incubated with IgG isotype control) and the percentages of cells gated positive for receptor expression.
Differentiation potential of TRAIL-MSCs
To evaluate the effect of TRAIL gene on the multilineage potential of MSCs, TRAIL-MSCs were induced to differentiate into osteogenic and adipogenic lineages. TRAIL-MSCs (6 × 103 cells per well) were seeded in a 24-well plate pretreated with collagen type I. After an overnight incubation, the differentiation potential of TRAIL-MSCs was assayed as follows.
For osteogenic differentiation, TRAIL-MSCs were cultured in DMEM medium supplemented with 0.1 μM dexamethasone, 0.2 mM vitamin C and 10 mM β-glycerophosphate for 2 weeks with medium changes every three days. At the end of this period, alizarin red staining was used to visualize matrix mineralization. For staining, cell layers were first fixed by 70% methanol for 60 min and then subjected to alizarin red solution for 30 min.
For adipogenic differentiation, TRAIL-MSCs were cultured in an adipogenic culture medium that included 1 μM dexamethasone, 100 μM indomethacin, 0.5 mM 3-isobutyl-1-methylxanthine and 10 μg/ml insulin for 2 weeks with medium changes every 3 d. Cells were then rinsed twice with PBS, fixed with 4% formalin for 40 min, washed with distilled water and covered with a 0.3% oil red O solution in 60% isopropanol for 50 min.
ELISA for expression of human TRAIL in vitro
To detect the expression of TRAIL, TRAIL-MSCs (6 × 103 cells per well) were seeded in a 24-well plate within 1 ml culture. The culture supernatant was collected at 0, 24, 48 and 72 h. The TRAIL concentrations were detected by ELISA according to the manufacturer's protocol (Bender MedSystems).
HCC model in Balb/c nude mice
Animal experimental procedures were performed in the Experimental Animal Center of Sun Yat-sen University in accordance with the institutional guidelines. A HCC model was established according to the methods described by Yu J et al.52 In brief, a suspension of MHCC97-H cells (1 × 107 cells in 0.2 ml PBS) was injected subcutaneously into the neck of the female mice. Ten days later, when a small vascularized tumor (5–6 mm diameter) had developed, 24 mice were randomized into four groups. The four experimental groups were as follows: untreated control group, cisplatin treatment group, TRAIL-MSCs treatment group, and cisplatin combined TRAIL-MSCs group. Cisplatin (1.5 mg/kg) was given intraperitoneally in 100 μl PBS every 3 d. The dosage of cisplatin is based on dosages commonly used in murine models of HCC.53,54 In TRAIL-MSCs treatment group, TRAIL-MSCs (1 × 106 cells in 0.2 ml PBS) were injected into the tail vein. The mice treated with both cisplatin and TRAIL-MSCs were administered by using the same schedule for the single treatment. All control mice received an equal volume of PBS. After three weeks of treatment, the mice were sacrificed. Tumor diameter was measured with a caliber ruler every week. Tumor volume (mm3) was estimated by measuring the longest (a) and shortest (b) diameter of the tumor and calculating volume as: volume = a × b2 × π/6.
Bioluminescence imaging
At the end of treatment period, BLI of the live mice was performed using Xenogen IVIS-Spectrum system (Caliper; Xenogen) according to the procedure described by Love Z et al.55 Briefly, at 10 min before each scan, each mouse was given an intraperitoneal injection of 4.5 mg D-luciferin in 150 μl of sterile PBS. And then the mice were anesthetized with isoflurane administrated by use of an EZ-Anesthesia system (Euthanex) and were restrained in a horizontal position after induction. After acquiring the photographic images of each mouse, the optical images were displayed and analyzed as described previously.56 Regions were manually drawn around the bodies of the mice to assess relative signal intensity emitted. Optical signal was expressed as photon intensity, in units of photons/second (p/s) within the region of interest (ROI).
In vivo migration assay
TRAIL-MSCs homing to the tumor sites in vivo were determined by detecting the GFP-positive cells. The mice were sacrificed within 6 h after BLI. The tumors were resected and frozen immediately in liquid nitrogen before 10 μm frozen sections were obtained. Migration of GFP-positive cells (green fluorescence) was assessed by direct visualization using a fluorescence and phase-contrast microscope (Nikon ECLIPSE 80i).
Immunohistochemistry on tumors
The degree of angiogenesis was determined by calculating the intratumoral microvessel density (MVD). Immunohistochemical staining of paraffin slides was performed as described previously.57,58 The following antibodies were used at 37°C in humidity chamber: rat anti-mouse CD34 monoclonal antibody (ab8158, Abcam) at a dilution of 1:50 for 1 h as a primary antibody, subsequently with HRP-conjugated anti-rat IgG (PV-6004) for 30 min as a secondary antibody. Immunocomplexes were visualized with diaminobenzidine/hydrogen peroxidase. The slides were counterstained with hematoxylin. The stained slides were examined at low-power magnification (×40 and ×100 total magnification) to identify the areas with the most intense neovascularization (hot-spots) of the tumor. In each section, five hotspots were selected. Microvessel counts of these hot-spots were performed using a high-power field (×200). In all samples, the averages of the number of microvessels in each tumor were calculated from the five vascular hotspots, which were referred to as the density counts.
Expression of human TRAIL in vivo
After administration of treatment, blood was collected from the mice on days 7, 14, 21 and serum was stored at -70°C until use. The frozen tumors (0.1 g) were homogenized in 0.9 ml cold PBS using a glass-glass homogenizer on ice. The homogenate was further homogenized with an ultrasonic homogenizer for 1 min on ice and then centrifuged at 10,000 g for 15 min at 4°C.59 The supernatant was collected and stored at -70°C for subsequent analysis. TRAIL in serum and in tumor homogenate was determined using a commercial TRAIL ELISA kit.
Statistical analysis
All data were expressed as mean ± SD and analyzed by 2-tailed Student’s t-tests using SPSS 13.0 software (SPSS, Inc.). A result with a p value of < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by Joint Funds of the National Natural Science Foundation of China and the Government of Guangdong Province (U1032002), National Natural Science Foundation of China (81071206, 81172193), Team Project of Natural Science Foundation of Guangdong Province (05200177) and Sci-Tech Project Foundation of Guangdong Province (2009B030801026).
Glossary
Abbreviations:
- HCC
hepatocellular carcinoma
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- MSCs
mesenchymal stem cells
- TRAIL-MSCs
TRAIL gene modified MSCs
- FACS
fluorescence-activated cell sorting
- ELISA
enzyme-linked immunosorbent assay
- BLI
bioluminescence imaging
- MVD
microvessel density
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/21347
References
- 1.Hertl M, Cosimi AB. Liver transplantation for malignancy. Oncologist. 2005;10:269–81. doi: 10.1634/theoncologist.10-4-269. [DOI] [PubMed] [Google Scholar]
- 2.Roberts LR. Sorafenib in liver cancer--just the beginning. N Engl J Med. 2008;359:420–2. doi: 10.1056/NEJMe0802241. [DOI] [PubMed] [Google Scholar]
- 3.Aravalli RN, Steer CJ, Cressman EN. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48:2047–63. doi: 10.1002/hep.22580. [DOI] [PubMed] [Google Scholar]
- 4.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. SHARP Investigators Study Group Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 6.Pinter M, Hucke F, Graziadei I, Vogel W, Maieron A, Königsberg R, et al. Advanced-stage hepatocellular carcinoma: transarterial chemoembolization versus sorafenib. Radiology. 2012;263:590–9. doi: 10.1148/radiol.12111550. [DOI] [PubMed] [Google Scholar]
- 7.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 8.Pollack IF, Erff M, Ashkenazi A. Direct stimulation of apoptotic signaling by soluble Apo2l/tumor necrosis factor-related apoptosis-inducing ligand leads to selective killing of glioma cells. Clin Cancer Res. 2001;7:1362–9. [PubMed] [Google Scholar]
- 9.Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med. 2002;8:808–15. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 10.Ray S, Almasan A. Apoptosis induction in prostate cancer cells and xenografts by combined treatment with Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand and CPT-11. Cancer Res. 2003;63:4713–23. [PubMed] [Google Scholar]
- 11.Naka T, Sugamura K, Hylander BL, Widmer MB, Rustum YM, Repasky EA. Effects of tumor necrosis factor-related apoptosis-inducing ligand alone and in combination with chemotherapeutic agents on patients’ colon tumors grown in SCID mice. Cancer Res. 2002;62:5800–6. [PubMed] [Google Scholar]
- 12.Zhang B, Shan H, Li D, Li ZR, Zhu KS, Jiang ZB, et al. Cisplatin sensitizes human hepatocellular carcinoma cells, but not hepatocytes and mesenchymal stem cells, to TRAIL within a therapeutic window partially depending on the upregulation of DR5. Oncol Rep. 2011;25:461–8. doi: 10.3892/or.2010.1084. [DOI] [PubMed] [Google Scholar]
- 13.Chen KF, Yeh PY, Hsu C, Hsu CH, Lu YS, Hsieh HP, et al. Bortezomib overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells in part through the inhibition of the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem. 2009;284:11121–33. doi: 10.1074/jbc.M806268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen KF, Tai WT, Liu TH, Huang HP, Lin YC, Shiau CW, et al. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res. 2010;16:5189–99. doi: 10.1158/1078-0432.CCR-09-3389. [DOI] [PubMed] [Google Scholar]
- 15.Rosato RR, Almenara JA, Coe S, Grant S. The multikinase inhibitor sorafenib potentiates TRAIL lethality in human leukemia cells in association with Mcl-1 and cFLIPL down-regulation. Cancer Res. 2007;67:9490–500. doi: 10.1158/0008-5472.CAN-07-0598. [DOI] [PubMed] [Google Scholar]
- 16.Yamanaka T, Shiraki K, Sugimoto K, Ito T, Fujikawa K, Ito M, et al. Chemotherapeutic agents augment TRAIL-induced apoptosis in human hepatocellular carcinoma cell lines. Hepatology. 2000;32:482–90. doi: 10.1053/jhep.2000.16266. [DOI] [PubMed] [Google Scholar]
- 17.Xu ZW, Kleeff J, Friess H, Büchler MW, Solioz M. Synergistic cytotoxic effect of TRAIL and gemcitabine in pancreatic cancer cells. Anticancer Res. 2003;23(1A):251–8. [PubMed] [Google Scholar]
- 18.Jane EP, Premkumar DR, Pollack IF. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-kappaB signaling pathway. Mol Cancer Ther. 2011;10:198–208. doi: 10.1158/1535-7163.MCT-10-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding L, Yuan C, Wei F, Wang G, Zhang J, Bellail AC, et al. Cisplatin restores TRAIL apoptotic pathway in glioblastoma-derived stem cells through up-regulation of DR5 and down-regulation of c-FLIP. Cancer Invest. 2011;29:511–20. doi: 10.3109/07357907.2011.605412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang M, Liu Z, Xiang Y, Ma H, Liu S, Liu Y, et al. Synergistic antitumor effect of AAV-mediated TRAIL expression combined with cisplatin on head and neck squamous cell carcinoma. BMC Cancer. 2011;11:54. doi: 10.1186/1471-2407-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shareef MM, Cui N, Burikhanov R, Gupta S, Satishkumar S, Shajahan S, et al. Role of tumor necrosis factor-alpha and TRAIL in high-dose radiation-induced bystander signaling in lung adenocarcinoma. Cancer Res. 2007;67:11811–20. doi: 10.1158/0008-5472.CAN-07-0722. [DOI] [PubMed] [Google Scholar]
- 22.Hori T, Kondo T, Kanamori M, Tabuchi Y, Ogawa R, Zhao QL, et al. Ionizing radiation enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through up-regulations of death receptor 4 (DR4) and death receptor 5 (DR5) in human osteosarcoma cells. J Orthop Res. 2010;28:739–45. doi: 10.1002/jor.21056. [DOI] [PubMed] [Google Scholar]
- 23.Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J, et al. Preclinical studies to predict the disposition of Apo2L/TRAIL in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001;299:31–8. [PubMed] [Google Scholar]
- 24.Kelley SK, Ashkenazi A. Targeting death receptors in cancer with Apo2L/TRAIL. Curr Opin Pharmacol. 2004;4:333–9. doi: 10.1016/j.coph.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Ozawa K, Sato K, Oh I, Ozaki K, Uchibori R, Obara Y, et al. Cell and gene therapy using mesenchymal stem cells (MSCs) J Autoimmun. 2008;30:121–7. doi: 10.1016/j.jaut.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Eliopoulos N, Francois M, Boivin MN, Martineau D, Galipeau J. Neo-organoid of marrow mesenchymal stromal cells secreting interleukin-12 for breast cancer therapy. Cancer Res. 2008;68:4810–8. doi: 10.1158/0008-5472.CAN-08-0160. [DOI] [PubMed] [Google Scholar]
- 27.Duan X, Guan H, Cao Y, Kleinerman ES. Murine bone marrow-derived mesenchymal stem cells as vehicles for interleukin-12 gene delivery into Ewing sarcoma tumors. Cancer. 2009;115:13–22. doi: 10.1002/cncr.24013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao Y, Yao A, Zhang W, Lu S, Yu Y, Deng L, et al. Human mesenchymal stem cells overexpressing pigment epithelium-derived factor inhibit hepatocellular carcinoma in nude mice. Oncogene. 2010;29:2784–94. doi: 10.1038/onc.2010.38. [DOI] [PubMed] [Google Scholar]
- 29.Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E, et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res. 2010;70:3718–29. doi: 10.1158/0008-5472.CAN-09-1865. [DOI] [PubMed] [Google Scholar]
- 30.Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009;69:4134–42. doi: 10.1158/0008-5472.CAN-08-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohr A, Lyons M, Deedigan L, Harte T, Shaw G, Howard L, et al. Mesenchymal stem cells expressing TRAIL lead to tumour growth inhibition in an experimental lung cancer model. J Cell Mol Med. 2008;12(6B):2628–43. doi: 10.1111/j.1582-4934.2008.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi SA, Hwang SK, Wang KC, Cho BK, Phi JH, Lee JY, et al. Therapeutic efficacy and safety of TRAIL-producing human adipose tissue-derived mesenchymal stem cells against experimental brainstem glioma. Neuro Oncol. 2011;13:61–9. doi: 10.1093/neuonc/noq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SM, Lim JY, Park SI, Jeong CH, Oh JH, Jeong M, et al. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008;68:9614–23. doi: 10.1158/0008-5472.CAN-08-0451. [DOI] [PubMed] [Google Scholar]
- 34.Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 35.Menon LG, Kelly K, Yang HW, Kim SK, Black PM, Carroll RS. Human bone marrow-derived mesenchymal stromal cells expressing S-TRAIL as a cellular delivery vehicle for human glioma therapy. Stem Cells. 2009;27:2320–30. doi: 10.1002/stem.136. [DOI] [PubMed] [Google Scholar]
- 36.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 37.Kidd S, Caldwell L, Dietrich M, Samudio I, Spaeth EL, Watson K, et al. Mesenchymal stromal cells alone or expressing interferon-beta suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment. Cytotherapy. 2010;12:615–25. doi: 10.3109/14653241003631815. [DOI] [PubMed] [Google Scholar]
- 38.Dwyer RM, Khan S, Barry FP, O’Brien T, Kerin MJ. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res Ther. 2010;1:25. doi: 10.1186/scrt25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zielske SP, Livant DL, Lawrence TS. Radiation increases invasion of gene-modified mesenchymal stem cells into tumors. Int J Radiat Oncol Biol Phys. 2009;75:843–53. doi: 10.1016/j.ijrobp.2008.06.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Secchiero P, Melloni E, Corallini F, Beltrami AP, Alviano F, Milani D, et al. Tumor necrosis factor-related apoptosis-inducing ligand promotes migration of human bone marrow multipotent stromal cells. Stem Cells. 2008;26:2955–63. doi: 10.1634/stemcells.2008-0512. [DOI] [PubMed] [Google Scholar]
- 41.Uchibori R, Okada T, Ito T, Urabe M, Mizukami H, Kume A, et al. Retroviral vector-producing mesenchymal stem cells for targeted suicide cancer gene therapy. J Gene Med. 2009;11:373–81. doi: 10.1002/jgm.1313. [DOI] [PubMed] [Google Scholar]
- 42.Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. 2009;109:259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- 43.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–40. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishihira S, Okubo N, Takahashi N, Ishisaki A, Sugiyama Y, Chosa N. High-cell density-induced VCAM1 expression inhibits the migratory ability of mesenchymal stem cells. Cell Biol Int. 2011;35:475–81. doi: 10.1042/CBI20100372. [DOI] [PubMed] [Google Scholar]
- 45.Ishii K, Yoshida Y, Akechi Y, Sakabe T, Nishio R, Ikeda R, et al. Hepatic differentiation of human bone marrow-derived mesenchymal stem cells by tetracycline-regulated hepatocyte nuclear factor 3beta. Hepatology. 2008;48:597–606. doi: 10.1002/hep.22362. [DOI] [PubMed] [Google Scholar]
- 46.Shimomura T, Yoshida Y, Sakabe T, Ishii K, Gonda K, Murai R, et al. Hepatic differentiation of human bone marrow-derived UE7T-13 cells: Effects of cytokines and CCN family gene expression. Hepatol Res. 2007;37:1068–79. doi: 10.1111/j.1872-034X.2007.00162.x. [DOI] [PubMed] [Google Scholar]
- 47.Mori T, Kiyono T, Imabayashi H, Takeda Y, Tsuchiya K, Miyoshi S, et al. Combination of hTERT and bmi-1, E6, or E7 induces prolongation of the life span of bone marrow stromal cells from an elderly donor without affecting their neurogenic potential. Mol Cell Biol. 2005;25:5183–95. doi: 10.1128/MCB.25.12.5183-5195.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology. 2011;54:879–89. doi: 10.1002/hep.24450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Damme A, Thorrez L, Ma L, Vandenburgh H, Eyckmans J, Dell’Accio F, et al. Efficient lentiviral transduction and improved engraftment of human bone marrow mesenchymal cells. Stem Cells. 2006;24:896–907. doi: 10.1634/stemcells.2003-0106. [DOI] [PubMed] [Google Scholar]
- 50.Miyoshi N, Oubrahim H, Chock PB, Stadtman ER. Age-dependent cell death and the role of ATP in hydrogen peroxide-induced apoptosis and necrosis. Proc Natl Acad Sci U S A. 2006;103:1727–31. doi: 10.1073/pnas.0510346103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi Z, Liang YJ, Chen ZS, Wang XW, Wang XH, Ding Y, et al. Reversal of MDR1/P-glycoprotein-mediated multidrug resistance by vector-based RNA interference in vitro and in vivo. Cancer Biol Ther. 2006;5:39–47. doi: 10.4161/cbt.5.1.2236. [DOI] [PubMed] [Google Scholar]
- 52.Yu J, Qiao L, Zimmermann L, Ebert MP, Zhang H, Lin W, et al. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology. 2006;43:134–43. doi: 10.1002/hep.20994. [DOI] [PubMed] [Google Scholar]
- 53.Hsieh JL, Wu CL, Lee CH, Shiau AL. Hepatitis B virus X protein sensitizes hepatocellular carcinoma cells to cytolysis induced by E1B-deleted adenovirus through the disruption of p53 function. Clin Cancer Res. 2003;9:338–45. [PubMed] [Google Scholar]
- 54.Huynh H, Soo KC, Chow PK, Panasci L, Tran E. Xenografts of human hepatocellular carcinoma: a useful model for testing drugs. Clin Cancer Res. 2006;12:4306–14. doi: 10.1158/1078-0432.CCR-05-2568. [DOI] [PubMed] [Google Scholar]
- 55.Love Z, Wang F, Dennis J, Awadallah A, Salem N, Lin Y, et al. Imaging of mesenchymal stem cell transplant by bioluminescence and PET. J Nucl Med. 2007;48:2011–20. doi: 10.2967/jnumed.107.043166. [DOI] [PubMed] [Google Scholar]
- 56.Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–23. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luetzkendorf J, Mueller LP, Mueller T, Caysa H, Nerger K, Schmoll HJ. Growth inhibition of colorectal carcinoma by lentiviral TRAIL-transgenic human mesenchymal stem cells requires their substantial intratumoral presence. J Cell Mol Med. 2010;14:2292–304. doi: 10.1111/j.1582-4934.2009.00794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maeda K, Chung YS, Ogawa Y, Takatsuka S, Kang SM, Ogawa M, et al. Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer. 1996;77:858–63. doi: 10.1002/(SICI)1097-0142(19960301)77:5<858::AID-CNCR8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 59.Iwasaki A, Kuwahara M, Yoshinaga Y, Shirakusa T. Basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) levels, as prognostic indicators in NSCLC. Eur J Cardiothorac Surg. 2004;25:443–8. doi: 10.1016/j.ejcts.2003.11.031. [DOI] [PubMed] [Google Scholar]