

Abstract
Melanoma is generally resistant to chemotherapy, which may be related to defects in death receptor signaling and to defects in induction of apoptosis. Forkhead family transcription factors induce the expression of death receptor ligands such as Fas ligand (Fas-L) resulting in apoptosis. We therefore investigated whether a triple mutant form of forkhead transcription factor FKHRL1 (FKHRL1/TM) can enhance Fas-L mediated-apoptosis in melanoma cells.
Two melanoma cells A2058 or DM6 were tested for their sensitivity to agonistic anti-Fas antibody (CH-11); adenovirus expressing FKHRL1/TM (Ad-FKHRL1/TM) was assessed for its capability to induce activation of the caspase pathway; the role of Fas-L in the Ad-FKHRL1/TM mediated-cell death was also assessed in vitro. Ad-FKHRL1/TM antitumor activity in vivo was also evaluated in a mouse melanoma xenograft model. We found that DM6 melanoma cells were more resistant to Fas/Fas-L-mediated apoptosis induced by agonistic anti-Fas antibody than A2058 melanoma cells. Ectopic expression of FKHRL1/TM in melanoma cells upregulated Fas-L expression, decreased procaspase-8 levels, and significantly increased Fas/FasL-mediated cell death in both cells lines; this induced cell death was partially blocked by a Fas/Fas-L antagonist. Importantly, Ad-FKHRL1/TM treatment of subcutaneous melanoma xenografts in mice resulted in approximately 70% decrease in tumor size compared with controls. These data indicate that overexpression of FKHRL1/TM can induce the Fas-L pathway in melanoma cells. Ad-FKHRL1/TM therefore might represent a promising vector for melanoma treatment.
Keywords: FKHRL1/TM, Fas-L, cell death, apoptosis, melanoma
Introduction
Melanoma is the sixth most common fatal malignancy in the United States and is responsible for 4% of all cancer deaths and four out of every five skin cancer-related deaths.1 The incidence of this highly aggressive cancer is increasing worldwide, and its capability to metastasize and resist chemotherapy results in a poor prognosis.2,3 Melanoma, like other types of cancer, becomes resistant to apoptosis during tumor progression.3 Melanoma cells may be resistant to apoptosis because they lack, or have defective, death receptor signaling.4
The members of the forkhead box O (FoxO) family of transcription factors play important roles in many physiological and pathological processes. These factors have been postulated to be tumor suppressors because of their established roles in regulating cell-cycle arrest, apoptosis, DNA-damage repair, and scavenging of reactive oxygen species.5,6 Forkhead family members have been studied as potential gene therapy agents. For example, phosphatidylinositol 3-kinase (PI3K)/Akt inhibits apoptosis by phosphorylation of the FoxO family of transcription factors, therefore application of triple mutant forkhead human transcription factor like-1 (FKHRL1/TM - FoxO3) which cannot be phosphorylated by PI3K/Akt can overcome this problem; exogenous expression of FKHRL1/TM in Caov-3 ovarian cancer cells decreased cell viability in response to treatment with cisplatin.7 Similarly, overexpression of FKHRL1 and forkhead box O 1 (FKHR - FoxO1) in LAPC4 prostate carcinoma cells using adenoviral vectors induced extensive apoptosis.8 A recent study also showed that adenovirus encoding FoxO1 induced significant tumor suppression of glioma cells in vivo.9 We have also previously shown that an adenovirus expressing the transcription factor FKHRL1/TM (Ad-FKHRL1/TM) efficiently induces apoptosis in SK-MEL-2 and SK-MEL-28 melanoma cells in vitro.10
Part of the mechanism of induction of apoptosis by FKHRL1/TM involves effects on the expression of the death receptor ligand, Fas-ligand (Fas-L). Ionizing radiation can promote FKHRL1 transcriptional activity and stimulates expression of apoptosis-inducing proteins such as Fas-L.11 Other experiments using non-mutated FKHRL1 induced apoptosis in vascular smooth muscle cells (VSMC); this apoptosis was partially inhibited by treatment with a neutralizing antibody against Fas-L.12 Brunet et al. reported that FKHRL1-induced apoptosis was significantly reduced in cerebellar granule neurons treated with soluble Fas-Fc fusion protein that functions as a decoy for the newly synthesized Fas ligand.13 These studies all suggest that FKHRL1 induces apoptosis by a Fas ligand-dependent mechanism.
Reportedly some melanoma cell lines release cytochrome c from mitochondria and activate caspase-3 upon stimulation with the Fas agonist monoclonal antibody CH-11, however this apoptotic pathway was not activated in several other melanoma cell lines.14 Results from another study indicate that most melanoma cell lines express Fas, but lack expression of Fas-L, however all melanoma cells tested responded with increased apoptosis to conditional expression of Fas-L.15 Fas-L activation may therefore represent a promising approach for inducing apoptosis in otherwise resistant melanoma tumor cells.
The present study is a continuation of our previous report;10 we confirmed that Fas-L plays an important role in FKHRL1/TM-induced melanoma cell death. We show that ectopic expression of FKHRL1/TM induces the Fas-L pathway in melanoma cells and Ad-FKHRL1/TM has significant tumor suppression activity in a melanoma xenograft model in mice.
Results
Activation of Fas-L pathway in melanoma cells
Although activation of Fas-L by forkhead transcription factors has been widely documented in multiple cell types to induce apoptosis,11-13 this pathway has not been fully described in melanoma cells. Therefore, we first tested whether Fas-L pathway activation resulted in downstream signaling and apoptosis in DM6 and A2058 melanoma cells using agonistic anti-Fas antibody, CH-11.14,15 The CH-11 antibody recognizes the Fas cell surface antigen, initiating the Fas/FasL pathway. DM6 and A2058 melanoma cells were cultured in the absence or presence of CH-11 at a concentration of 1 µg/mL according to previous publications.14,15 After 72 h, downstream procaspase-8 levels were decreased in A2058 cells treated with CH-11 compared with untreated cells, suggesting cleavage to activate caspase-8 and initiation of apoptosis. A2058 cells showed a larger decrease of procaspase-8 levels than DM6 cells (Fig. 1A). In addition, a cell viability assay was performed to determine whether the decrease of procaspase-8 is related to melanoma cell survival. An MTT assay demonstrated that treatment of A2058 with CH-11 resulted in 70% cell survival, whereas DM6 cells were resistant to CH-11 (Fig. 1B). Induction of apoptosis by CH-11 was confirmed by annexin-V staining and flow cytometry analysis. Treatment of A2058 cells with CH-11 induced 22% apoptosis, whereas apoptosis was only 5% in untreated cells. No significant difference was observed in apoptosis levels in DM6 cells with CH-11 (8 and 10% apoptosis, respectively, Fig. 1C). These results indicate that CH-11 activates the Fas/Fas-L pathway in A2058 melanoma cells to induce apoptosis. DM6 cells were resistant to Fas/Fas-L pathway activation, and stimulation with CH-11 was unable to induce apoptosis which suggests that Fas/Fas-L pathway in DM6 cells is not functional.
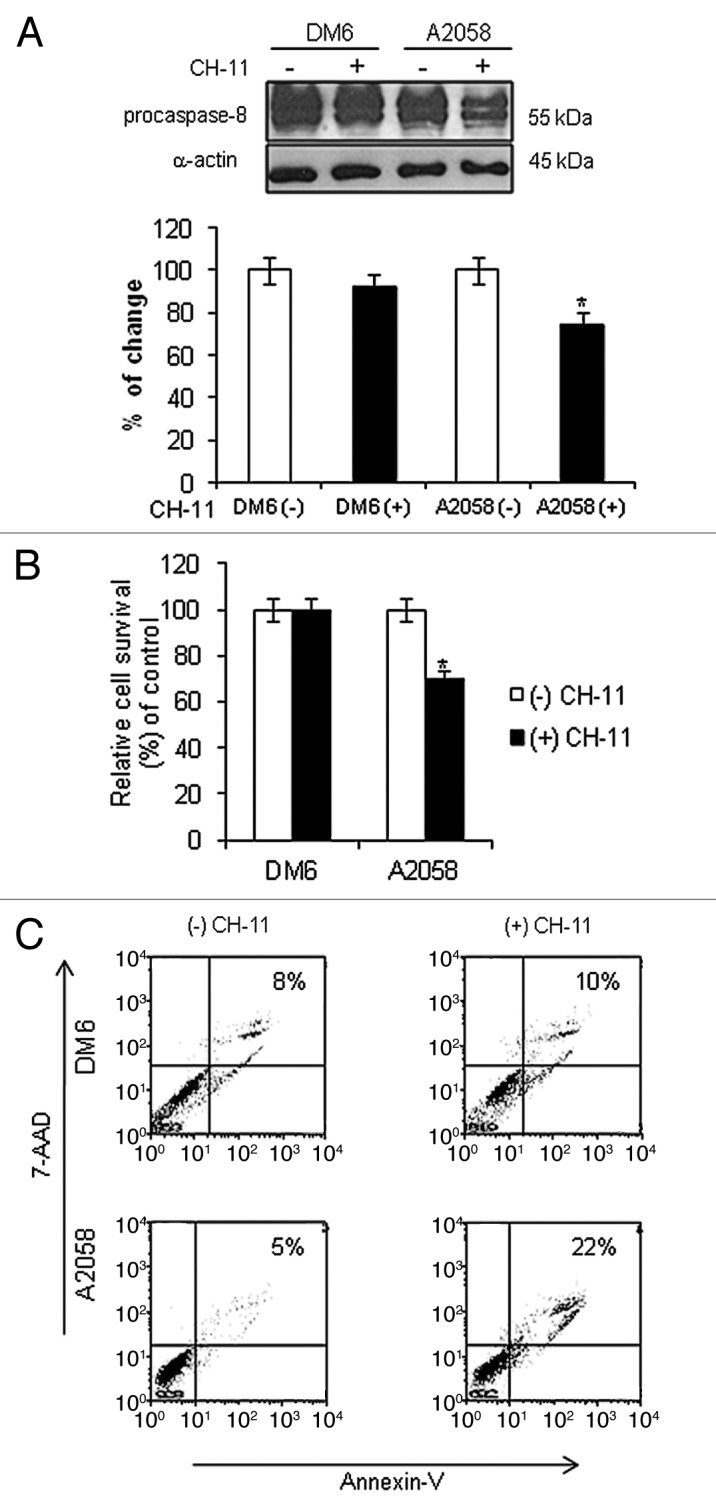
Figure 1. Activation of Fas/Fas-L pathway in melanoma cells. DM6 or A2058 melanoma cells were cultured in absence or presence of agonistic anti-Fas antibody, CH-11, at a concentration of 1 µg/mL for 72 h. (A) western blot and bar graphs of procaspase-8 expression after CH-11 treatment. Bars represent mean ± SEM expressed as percentage of change from three separate experiments, (*p < 0.05) decrease in the level of expression. (B) MTT assay was used to determine cell survival. Treatment of A2058 cells with CH-11 induced significant cytotoxicity compared with non-treated cells. Each bar represents the mean ± SD of three independent experiments (*p < 0.05). (C) Annexin V staining was used to determine the percentage of apoptosis. Cells were stained with annexin V-PE and 7-AAD 72 h after treatment. Cells positive for annexin V-PE and 7-AAD staining were analyzed by FACScan flow cytometer with FlowJo software. One representative experiment is shown from three performed.
Adenovirus-mediated FKHRL1/TM efficiently induces melanoma cell death by activation of the caspase pathway
We have previously shown that Ad-FKHRL1/TM efficiently induces apoptosis in SK-MEL-2 and SK-MEL-28 melanoma cells independent of p53.10 Therefore, we investigated whether Ad-FKHRL1/TM can also induce apoptosis in two other melanoma cell lines which are used to establish tumors in vivo, A2058 and DM6.16,17 Melanoma cell killing and FKHRL1/TM expression increased in a dose-dependent fashion in both cell lines (Fig. 2A and B). Melanoma cell killing activity was associated with expression of FKHRL1/TM and caspase-3 activation. A decrease in the proenzyme caspase-3 level was observed in both cells lines (Fig. 2B). An apoptosis-specific PARP cleavage fragment (85–90 kDa) was also present in both cell lines (Fig. 2B). Apoptosis was validated by annexin-V analysis. Annexin-V staining of A2058 and DM6 cell lines infected with Ad-FKHRL1/TM demonstrated 78 and 62% apoptotic cells, respectively, with minimal apoptosis detected in mock or Ad-LacZ infected-cells (Fig. 2C). Our previous report and these results indicate that FKHRL1/TM can efficiently kill a panel of melanoma cell lines by apoptosis, including the DM6 cells which showed resistance to agonistic anti-Fas antibody CH-11-mediated apoptosis.
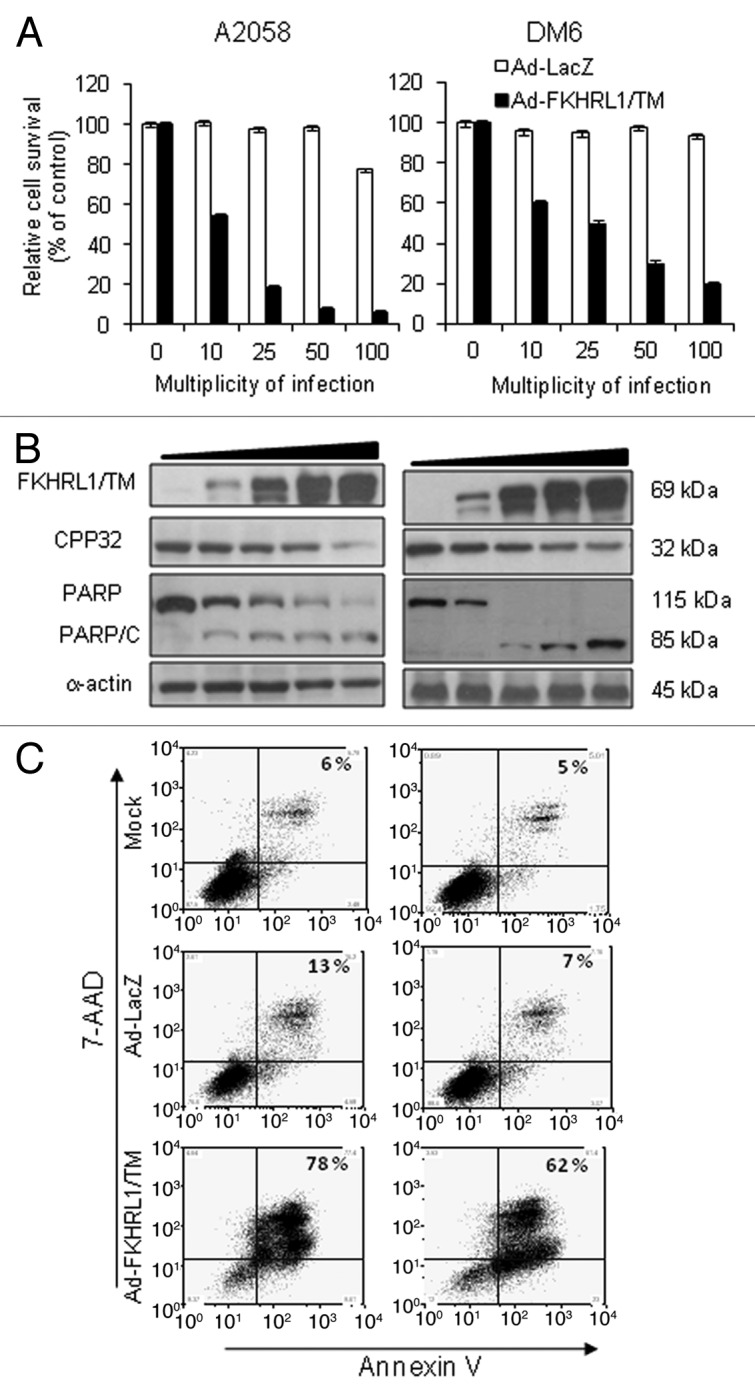
Figure 2. FKHRL1/TM induces cytotoxicity, caspase pathway activation, and apoptosis. A2058 and DM6 melanoma cell lines were infected at increasing doses (0–100 MOI) of either Ad-LacZ or Ad-FKHRL1/TM. At 72 h cells were analyzed by MTT, and western blot assays. (A) MTT assay was used to determine cell survival comparing Ad-LacZ with Ad-FKHRL1/TM treatments. Each point represents the mean ± SD of three independent experiments (p < 0.05). (B) Expression of FKHRL1/TM, proenzyme CPP32/caspase-3 (CPP32), PARP and cleaved components of PARP (PARP/C) were detected by western blot. α-Actin was used to demonstrate equal loading for each lane. (C) A2058 and DM6 melanoma cell lines were not infected (Mock) or infected with either Ad-LacZ or Ad-FKHRL1/TM at an MOI of 100. Three days post-infection the percentages of apoptosis were determined by annexin V staining, and analysis by FACScan flow cytometer with FlowJo software. Similar results were obtained in three independent experiments. A representative experiment is shown.
Ectopic expression of FKHRL1/TM enables Fas/Fas-L pathway activation in melanoma cells
Since we observed that FKHRL1/TM can activate the caspase pathway (Fig. 2B), and it has been previously reported that forkhead transcription factors can upregulate and activate the Fas/Fas-L mediated-extrinsic apoptotic pathway,11 we investigated whether FKHRL1/TM can induce activation of the Fas-L pathway in melanoma cells. DM6 and A2058 cells were not infected (Mock) or infected with Ad-LacZ (control vector) or Ad-FKHRL1/TM at an MOI of 100. Expression of procaspase-8 was analyzed after 24, 48 and 72 h by western blot, and expression of AdFKHRL1/TM was also confirmed by immunoblot. Procaspase-8 levels declined over time in both cell lines infected with Ad-FKHRL1/TM. Similar to our observations with CH-11 treatment, procaspase-8 decrease was greater in A2058 than in DM6 cells (Fig. 3A). No significant changes in procaspase-8 levels were observed in mock or Ad-LacZ infected-cells. In order to confirm that FKHRL1/TM does in fact upregulate Fas-L expression, protein extracts from the above experiment at 72 h after infection were used to detect Fas-L expression by western blot. We found that cells infected with Ad-FKHRL1/TM expressed significantly greater levels of Fas-L in comparison with mock or cells infected with Ad-LacZ (Fig. 3B). This result indicates that FKHRL1/TM can upregulate and activates the Fas-L pathway in melanoma cells, even in the DM6 cell line that was resistant to CH-11.

Figure 3. Activation of Fas/Fas-L pathway by FKHRL1/TM in melanoma cells. DM6 or A2058 cells were infected with either Ad-LacZ or Ad-FKHRL1/TM at an MOI of 100 or not infected (Mock); cells were harvested at 24, 48, and 72 h post-infection. (A) Western blot and bar graphs of procaspase-8 expression after adenovirus infection. Bars represent mean ± SEM expressed as percentage of change from three separate experiments, (*p < 0.05) decrease in the level of expression. (B) Western blot and bar graphs of Fas-L expression at 72 h after adenovirus infection. Bars represent mean ± SEM expressed as percentage of change from 3 separate experiments, (*p < 0.05) increase in the level of expression. α-Actin was used to demonstrate equal loading for each lane.
Inhibition of Fas-L partially blocks FKHRL1/TM-mediated cell death
We next assessed whether Fas/Fas-L antagonist, Kp7-6, could prevent FKHRL1/TM mediated-cell death. According to the manufacturer, Kp7-6 reduces FasL induced apoptosis by 58% in Jurkat cells at 1 mg/mL. The antagonists binds to both FasL (Kd = 11.2 µM) and Fas (Kd = 13.2 µM). DM6 and A2058 cells were cultured in the presence of increasing concentrations of Kp7-6 (0, 1, 2.5, 5, 10, and 20 µg/mL), followed by infection with Ad-LacZ or Ad-FKHRL1/TM. An MTT assay demonstrated that Fas/Fas-L antagonist inhibited FKHRL1/TM-mediated cytotoxicity in a dose-dependent manner. Cell survival was decreased with Ad-FKHRL1/TM infection compared with Ad-LacZ controls (20 and 40% viable cells in DM6 or A2058 cells, respectively). In contrast, in the presence of Kp7-6 at 10 µg/mL, the cell viability percentages were increased to 73 and 70% in Ad-FKHRL1/TM infected DM6 and A2058 cells at 72 h, respectively (Fig. 4A). These results confirm previous reports which suggest that Fas-L may be the main mediator of FKHRL1/TM induced-cell death.12 However, as there was not a complete cell death rescue with Kp7-6 treatment, this suggests that other cell death mechanisms such as autophagy18 may be involved.
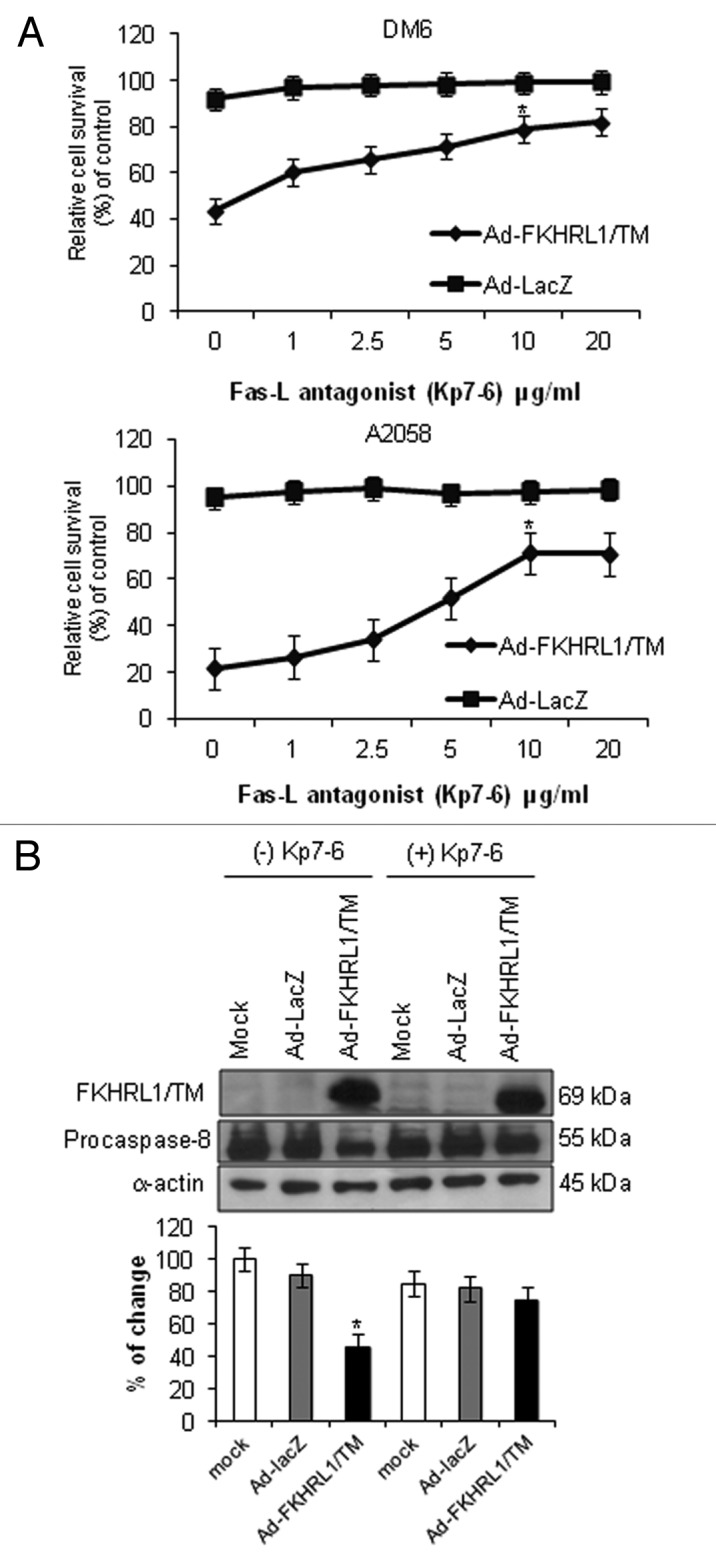
Figure 4. Inhibition of Fas-L-mediated cell death. (A) DM6 or A2058 cells were cultured with increasing concentrations of Fas/Fas-L antagonist Kp7-6 followed by infection with either Ad-LacZ or Ad-FKHRL1/TM at an MOI of 50. At 72 h an MTT assay was performed to determine cell survival. Ad-FKHRL1/TM alone was compared with Ad-FKHRL1/TM in presence of Kp7-6. Each point represents the mean ± SD of three independent experiments (*p < 0.05). (B) A2058 cells were cultured in absence or presence of Fas/Fas-L antagonist Kp7-6 at 10 µg/mL followed by mock infection or infection with either Ad-LacZ or Ad-FKHRL1/TM at an MOI of 100. Western blot and bar graphs of procaspase-8 expression after adenovirus infection. Bars represent mean ± SEM expressed as percentage of change from 3 separate experiments, (*p < 0.05) decrease in the level of expression. α-Actin was used to demonstrate equal loading for each lane. FKHRL1/TM expression was also confirmed.
The effectiveness of the Fas/Fas-L antagonist was evaluated by western blot analysis of procaspase-8 levels. A2058 cells were cultured in the absence or presence of Kp7-6 (10 µg/mL), followed by no infection (mock) or infection with either Ad-LacZ or Ad-FKHRL1/TM. Procaspase-8 expression was analyzed 72 h post infection by western blot of whole cell lysates. Kp7-6 inhibited the decrease of procaspase-8 expression in Ad-FKHRL1/TM infected cells, whereas procaspase-8 expression did not change significantly in mock or Ad-LacZ infected-cells (Fig. 4B). These data indicate that Fas-L antagonist Kp7-6 efficiently inhibits Fas-L pathway activation.
Transduction with Ad-FKHRL1/TM sensitizes melanoma cells to CH-11 mediated-apoptosis
We found that DM6 cells were resistant to agonistic anti-Fas antibody CH-11 mediated-cytotoxicity (Fig. 1). We therefore next inquired whether pretreatment with Ad-FKHRL1/TM could sensitize melanoma cells to CH-11 treatment. A2058 and DM6 cells were not infected (mock) or infected with either Ad-LacZ or Ad-FKHRL1/TM at an MOI of 10 (this MOI of concentration induces ~50% of cell viability in A2058 cells) (Fig. 2A), followed by treatment with CH-11 at 1 µg/mL. Annexin-V staining at 72 h showed that A2058 and DM6 cells infected with Ad-FKHRL1/TM alone induced 47 and 26% apoptosis, respectively, while Ad-FKHRL1/TM plus CH-11 induced 77% and 63% apoptosis, respectively (p < 0.05, Fig. 5A). In contrast Ad-LacZ infected cells plus CH-11 did not significantly increase apoptosis in comparison with CH-11 treatment alone (Fig. 5A). This result suggests that combination of Ad-FKHRL1/TM with CH-11 has at least additive apoptotic effect.
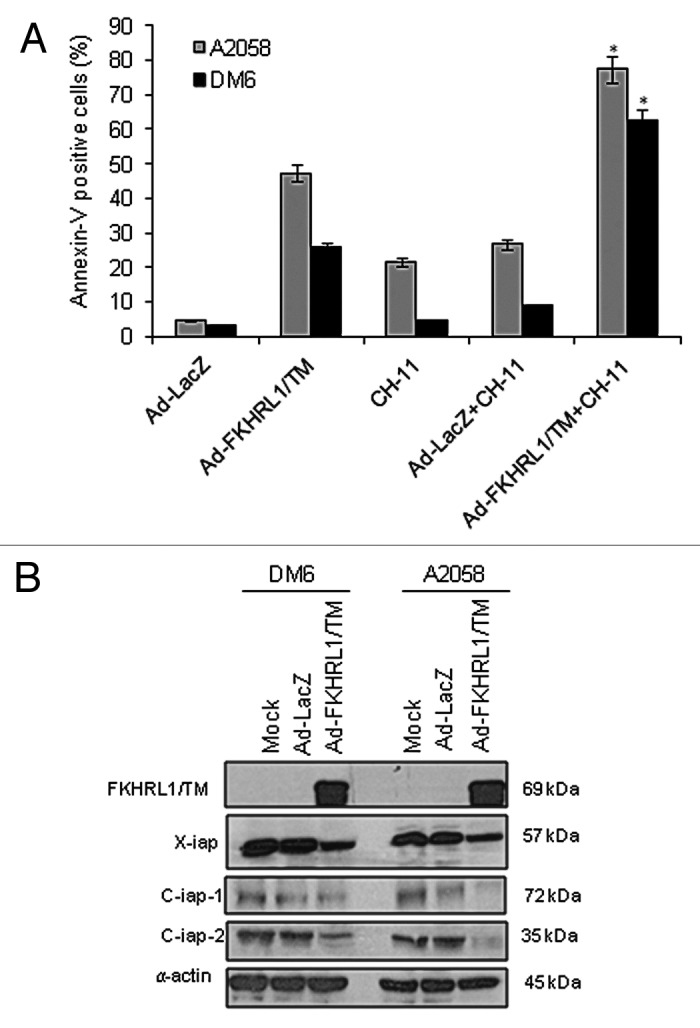
Figure 5. FKHRL1/TM sensitizes melanoma cells to CH-11 mediated-apoptosis and downregulates IAP family members. (A) A2058 or DM6 cells were infected with either Ad-LacZ or Ad-FKHRL1/TM at an MOI of 10 +/− treatment with CH-11 at 1 µg/mL. Annexin V staining was used to determine the percentage of apoptosis at 72 h after treatment by FACScan flow cytometer with FlowJo software. Ad-FKHRL1/TM infection alone was compared with Ad-FKHRL1/TM in combination with CH-11. Each bar represents the mean ± SD of three independent experiments (*p < 0.05). (B) A2058 or DM6 cells were no infected (mock) or infected with either Ad-LacZ or Ad-FKHRL1/TM at an MOI of 100 at 72 h post-infection expression of X-IAP, C-IAP-1, or C-IAP-2 were analyzed, a representative experiment is shown from three performed.
FKHRL1/TM downregulates X-IAP, c-IAP-1, and c-IAP-2 IAP family members
The inhibitor of apoptosis (IAP) gene family members (survivin, XIAP, c-IAP-1 or c-IAP-2) have been implicated in cancer developments.19 Other studies have shown that overexpression of FKHRL1/TM decreased survivin expression.20 Thus we analyzed the effect of FKHRL1/TM expression on protein levels of other IAP family members. To this end, A2058 and DM6 melanoma cells were uninfected (mock) or infected with Ad-LacZ or Ad-FKHRL1/TM, 3 d later expression of XIAP, c-IAP-1 or c-IAP-2 were analyzed. A western blot analysis showed that protein levels of XIAP, c-IAP-1, c-IAP-2 declined in both cell lines infected with Ad-FKHRL1/TM in comparison with mock or Ad-LacZ infected-cells (Fig. 5B). This result suggests that FKHRL1-induced downregulation of XIAP, c-IAP-1, and c-IAP-2 may be a mechanism that contribute to sensitization of melanoma cells to CH-11 treatment.
Ad-FKHRL1/TM suppresses tumor growth in vivo
Ad-FKHRL1/TM was further assessed for its antitumor activity in a melanoma mouse subcutaneous xenograft model. A2058 melanoma cells were injected subcutaneously into the flanks of nude mice. Six days later, mice containing palpable tumors were randomized and directly injected with Ad-FKHRL1/TM or Ad-GFP as a control. Intratumoral injections with adenovirus were performed every 3 d for a total of four treatments. The tumors of mice receiving FKHRL1/TM or GFP treatments were harvested 24 h after the final treatment following euthanization and subjected to histopathological analysis to assess whether transgene expression and apoptosis are possible mechanisms that induce tumor suppression in mice treated with Ad-FKHRL-1/TM. Immunohistochemical analysis revealed approximately 72% of tumor cells in mice injected with Ad-FKHRL-1/TM expressed FKHRL1/TM; no FKHRL1/TM expression was detected in tumors that of mice that received Ad-GFP (Fig. 6A). High expression of FKHRL1/TM was associated with a significant percentage of Fas-L (~60%) and cleaved caspase-3-positive cells (~70%), suggesting increased apoptosis in the tumors of mice treated with Ad-FKHRL1/TM. In contrast, neither Fas-L nor cleaved caspase-3 were detected in tumors of mice treated with Ad-GFP (Fig. 6A). About 75% of tumor cells in tumors of mice injected with Ad-GFP were positive for GFP expression (Fig. 6A). These results suggest that adenovirus has high transduction efficiency into the solid tumors and can produce a high therapeutic index.

Figure 6. Immunohistochemistry and antitumor effect of treatment with adenovirus expressing FKHRL1/TM. After subcutaneous administration of 5 × 106 A2058 cells, mice with palpable tumors were randomized to receive a local injection of Ad-GFP or Ad-FKHRL1/TM on days 0, 3, 6, and 9 (vertical arrows) at a concentration of 1 × 109 pfu (plaque forming units). (A) Immunohistochemistry of A2058 melanoma tumors at 24 h after last treatment with Ad-GFP or Ad-FKHRL1/TM. GFP expression was visualized by fluorescence microscopy. FKHRL1/TM, Fas-L, and cleaved caspase-3 expressions were detected by immunohistochemistry. (B) Tumor volume was plotted over time. Volume (V) was determined by V = (L × W 2)/2 of tumor measured using calipers. (C) On day 27, tumors were excised and photographed. The relative size of tumors is shown. (D) Tumors were also weighed on day 27. The results are expressed as the mean ± SEM for each treatment group. The differences in tumor size and weight in animals treated with Ad-FKHRL1/TM were statistically significant on day 27 compared with Ad-GFP control (*p < 0.05).
There was strong tumor suppression in six out of six mice (100%) treated with Ad-FKHRL1/TM by day 27 by observation of the palpable tumor. Tumor size reduction was approximately 70% in Ad-FKHRL1/TM-treated mice in comparison with control virus Ad-GFP-treated mice, this was statistically significant (p < 0.05; Fig. 6B). At 27 d after the first adenovirus injection, tumors were excised and weighed. The photographs reveal that tumors from the mice that were injected with Ad-GFP were significantly larger than tumors from mice injected with Ad-FKHRL1/TM (Fig. 6C). Those tumors treated with Ad-GFP injection weighed 1.2 ± 0.9 g, whereas tumors treated with Ad-FKHRL1/TM weighed significantly less at 0.3 ± 0.06 g (p < 0.05; Fig. 6D). These results indicate that adenovirus encoding a triple mutant form of FKHRL1 has potent antitumoral activity in vivo and FKHRL1/TM induced-tumor suppression was mediated, at least in part, through activation of caspase pathway.
Discussion
Although Fas-L treatment is a promising approach for the destruction of cancer cells, resistance to Fas/Fas-L induced apoptosis may occur through the downregulation of Fas expression. For example, in a 2008 study, 5 out of 13 melanoma cell lines had undetectable or low levels of cell surface Fas expression. In addition, resistance to recombinant Fas-L and absence or low levels of Fas expression were closely related.4 Another study reported that only 5 of 11 melanoma cell lines showed activation of Fas/Fas-L pathway upon treatment with agonistic monoclonal antibody CH-11, meaning the other remaining six melanoma cell lines were resistant to Fas/Fas-L.14 Eberle et al. reported that most of the melanoma cell lines that they analyzed expressed Fas but lacked expression of Fas-L. Overexpression of Fas enhanced sensitivity to Fas-L but was unable to trigger apoptosis by itself. In contrast, all melanoma cells responded with increased apoptosis to conditional expression of Fas-L.15 These studies suggest that melanoma cells have different levels of resistance to Fas/Fas-L pathway mediated-apoptosis and that targeted expression of Fas-L could be a promising strategy for melanoma therapy.
One alternative approach to manipulating expression of Fas-L in melanoma cells is by transferring Fas-L into the tumor using viral vectors, however it is possible that introduction of Fas-L alone may not be potent enough to suppress tumor growth. Currently, several investigators are exploring strategies to target different pathways to enhance the activation of the Fas/Fas-L pathway. We used an adenovirus expressing FKHRL1/TM because this transcription factor activates the Fas-L promoter, which then increases Fas-L expression and results in apoptosis induction.11 This vector not only induces apoptosis through Fas-L but also upregulates p27kip1, which is a cyclin dependent kinase inhibitor,10 and downregulates anti-apoptotic proteins such as survivin,20 Mcl-1, and Bcl-xL.10
In the present study we found that DM6 melanoma cells were resistant to CH-11 treatment, but ectopic expression of FKHRL1/TM decreased procaspase-8 levels over time, and Fas-L upregulation by FKHRL1/TM was confirmed. Although we did not test the status of Fas in A2058 or DM6 cells, this suggests that the Fas/Fas-L pathway is aberrant in DM6 and that overexpression of FKHRL1/TM induces Fas/Fas-L pathway. We demonstrated that it is likely that the Fas/Fas-L pathway is an important mechanism by which FKHRL1/TM induces cell death in melanoma cells, since application of Fas/Fas-L antagonist attenuated FKHRL1/TM induced-cell cytotoxicity. It is possible that other mechanisms are involved in the Ad-FKHRL1/TM mediated-cell death, for example it was reported that Ad expressing FKHRL1/TM downregulates survivin a member of inhibitor of apoptosis proteins (IAP) family.20 It was also reported that FoxO1 induces autophagy, a type II programmed cell death which was associated to tumor suppression activity.18 In our previous study we showed that overexpression of FKHRL1/TM induced lamin-B cleavage a substrate of caspase-6.10 We also found that pretreatment with Ad-FKHRL1/TM sensitized melanoma cells to CH-11 antibody, suggesting that the combination of Ad-FKHRL1/TM with CH-11 has at least an additive apoptotic effect. We found that FKHRL1/TM downregulated other AIP family members (Fig. 5B) suggesting that this may be an additional mechanism by which FKHRL1/TM sensitize melanoma cells to CH-11. Previous investigators have demonstrated that agonistic CH-1l induces cytosolic cytochrome c translocation and activates the apoptotic protein caspase-3.14 Whereas Ad-FKHRL1/TM may trigger Fas/FasL mediated-apoptosis in an autocrine manner. The combination of the two induces at least additive apoptotic effects. Further in vitro and in vivo studies are needed to fully characterize the mechanisms of these effects. The lack of direct comparison in vivo of Ad-FKHRL1/TM and agonistic anti-Fas antibody CH-11 with Ad-FKHRL1/TM alone is a limitation of this study.
Importantly, we have now validated the potency of Ad-FKHRL1/TM in vivo. Treatment of subcutaneous melanoma xenografts with Ad-FKHRL1/TM in a murine model produced approximately 70% decrease in tumor size compared with controls. Tumors that were treated with Ad-FKHRL-1/TM had high levels of FKHRL-1/TM expression and strongly induced Fas-L expression and caspase-3 activation. These results indicate that Ad-FKHRL1/TM can efficiently activate the apoptotic pathway and inhibit the growth of solid melanoma tumors in an in vivo model. Our previous and current data suggest that Ad-FKHRL1/TM has potential to be assessed in clinical settings. The clinical applications may include the use of Ad-FKHRL1/TM in combination of agents that target the Fas/Fas-L pathway in isolated limb perfusion treatment of in-transit metastatic disease, thus limiting systemic toxicity of adenovirus vector therapy. More investigation in this area is needed and is underway in our laboratory.
In summary, we confirmed that melanoma cells have different levels of resistance to agonistic anti-Fas antibody CH-11 treatment. Inactivation of the Fas/Fas-L pathway by resistance to Fas-L-induced apoptosis is a possible mechanism by which melanoma cells escape apoptosis. Pretreatment with Ad-FKHRL1/TM overcame this resistance and sensitized melanoma cells to CH-11 treatment, indicating that adenoviral mediated-gene transfer of FKHRL1/TM induces Fas/Fas-L pathway and increases the susceptibility of melanoma cells to Fas/Fas-L-targeted therapy. We also confirmed that the Fas/Fas-L pathway plays an important role in FKHRLI/TM mediated-cell death in melanoma cells. These experiments suggest that FKHRL1/TM-based viral gene therapy may be a promising way to circumvent some of the apoptosis-escape mechanisms developed by melanoma tumors. This adenoviral mediated-gene therapy may also provide clinicians with an alternative way to avoid the resistance of some tumor cells to Fas/Fas-L-targeted therapy by enhancing Fas/Fas-L pathway with FKHRL1/TM.
Materials and Methods
Cell culture conditions and reagents
Human melanoma cell line A2058 (ATCC no. CRL-11147) and human embryonic kidney cell line HEK-293 (ATCC no. CRL-1573), and the human melanoma DM6 cell line was kindly provided to us by Dr Douglas S. Tyler (Duke University Medical Center).21 A2058 cells were cultured in Dulbecco’s modified of Eagle’s medium (DMEM) (cat no. 21063029), DM6 cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM) (cat no. 12440), and HEK-293 cells were cultured in α minimal essential medium (α-MEM) (cat no. 32571). All media were supplemented with 10% heat-inactivated fetal bovine serum (FBS) (cat no. 12664025) and penicillin/streptomycin (100 U/mL) (cat no. 15140122). All cell culture reagents were obtained from Invitrogen. Cells were cultured in a 5% CO2 incubator at 37°C. An agonistic anti-Fas antibody, CH-11, (1 µg/mL, cat no. 05–201, Millipore) was used to activate the Fas/Fas-L pathway, and a Fas/Fas-L antagonist, Kp7-6, (0, 1, 2.5, 5, 10, and 20 µg/mL; cat no. 341291, Calbiochem) was used to inhibit the Fas/Fas-L pathway.
Adenoviral vectors
Three replication-defective recombinant adenoviral vectors were used. One was the Ad-CMV-LacZ (Ad-LacZ) vector containing the transgene nuclear-localized β-galactosidase under control of the CMV promoter as described by us previously.22 We constructed Ad-GFP expressing green fluorescent protein and Ad-FKHRL1/TM expressing forkhead human transcription factor like-1 triple mutant according to the Ad-Easy system (MP Biomedicals, AES1001A) as reported previously.10 All vectors were propagated in the HEK-293 cell line, and titers were determined using standard plaque assays. For infections, 1 × 106 cells were plated in 10-cm tissue culture plates. The following day, the media was removed, and the adenoviral vectors were added in 1 mL of DMEM at an indicated multiplicity of infection (MOI). Mock infection was performed by treatment of cells with vehicle only (media). After a 1-h incubation at 37°C 9 mL of fresh α-MEM with 5% FBS was added. Cells were harvested at specific time points for analysis.
Western blot analysis
Cells were harvested and lysed in RIPA buffer as described previously.10 Cell lysates were centrifuged, and protein concentration was determined by BCA Protein Assay kit (PIERCE, prod no. 23225). Equal amounts of cellular protein were electrophoresed in either 10% (Procaspase-8, FKHRL1, Fas-L, and PARP [poly (ADP-ribose) polymerase], Xiap, Ciap-1) or 12% proenzyme caspase-3/cysteine protease (CP) P32 (CPP32), Ciap-2 SDS-polyacrylamide gels and transferred to Hybond-PVDP membranes (Amersham). The membranes were first incubated with the following primary antibodies: rabbit-antihuman FKHRL1 (Cell Signaling, no. 9462), rabbit-antihuman caspase-8 and mouse antihuman Fas Ligand (PharMingen, cat no. 559932 and 65431A respectively), mouse-antihuman-caspase-3/CPP32 (BD Transduction Laboratories, cat no. 610322), mouse-antihuman poly (ADP-ribose) polymerase (PARP) (Calbiochem, cat no. 512739); mouse antihuman X-IAP (BD Transduction Laboratories cat no. 610763); mouse antihuman c-IAP-1 (PharMingen, cat no. 556533); rabbit antihuman c-IAP-2 (Santa Cruz Biotechnology, sc-7944), and rabbit-antihuman-α-actin (Sigma-Aldrich, A5060). Next, the membranes were incubated with anti-mouse immunoglobulin (Ig) or anti-rabbit Ig, peroxidase-linked, species-specific whole antibody (GE Healthcare, cat no. NA931V, and NA934V respectively). ECL reagents were used to detect the signals according to the manufacturer’s instructions (GE Healthcare, cat no. RPN2106V1). All films were scanned with an optical scanner (Epson Expression 1680) and quantified by measuring the density of each band using UNSCAN-IT software (Silk Scientific, Inc.). To correct for possible unequal loading, each band's density was normalized to its α-actin density. To allow for multiple comparisons between gels, each sample was compared with its respective mock (not infected) or not treated that was run on the same gel. Results are expressed in percentage of change, which was calculated by dividing the averages of each protein band.
MTT assay
Cell proliferation was assessed at 72 h after respective treatments by measuring the conversion of 3-(4, 5-dimethylthiazol-2-)-2, 5-diphenyltetrazolium bromide salt (MTT) to formazan, according to the manufacturer’s instructions (Sigma Aldrich, cat no. M2128). The supernatant from each plate was collected for measurement of absorbance at a wavelength of 570 nm. The results are expressed as the percentage of live cells.
Detection of apoptosis
The nonadherent cells were harvested, and adherent cells were trypsinized for 2 min then stopped with PBS/10% FCS. Nonadherent cells and adherent cells were pulled and stained with annexin V-PE and 7-AADaccording with the manufacturer’s instructions (BD PharMingen, cat no. 559763) and as reported previously.23 Cells were analyzed by FACScan flow cytometer (Becton Dickinson) and with FlowJo software (Tree Star, Inc.).
Immunohistochemistry
Tumors were excised 24 h after the fourth injection following euthanization, fixed in 10% formalin, embedded in paraffin blocks, and processed for histologic analysis and detection of FKHRL1/TM, Fas-L expression, and cleaved caspase-3. A rabbit-antihuman-FoxO3a (Cell Signaling, cat no. 9467) mAb at a 1:200 dilution was used to detect FKHRL1/TM expression, a rabbit-antihuman cleaved caspase-3 (Asp175)(5A1E) mAb at a 1:200 dilution (Cell Signaling, cat no. 9664) was used for detection of cleaved caspase-3, a mouse-antihuman Fas Ligand (65431A) mAb at a 1:200 dilution (PharMingen, cat no. 559932) was used to detection of Fas-L. The slides were then washed with PBS and incubated with the standard ultra-Sensitive ABC peroxidase staining kit (PIERCE, cat no. 32020) and detected with diaminobenzidine tetrahydrochloride (DAB) solution containing 0.006% H2O2. Hematoxylin was used as a counterstain. Tissue sections stained without primary antibodies were used as negative controls. Photographs were taken with ×20 magnification and analyzed with NIS-Elements BR 3.0 software (Nikon instruments, Inc.).
Melanoma xenograft study
Tumors were formed by injecting 5 × 106 A2058 melanoma cells into athymic BALB/c nu/nu male mice (6–8 weeks of age; Charles River Laboratories). The cells were injected subcutaneously into the bilateral flanks of the mice. Six days later, palpable tumors were randomized and directly injected with Ad-FKHRL1/TM (1 × 109 plaque forming units [pfu]) or Ad-GFP (1 × 109 pfu) (n = 6 for each group). Intratumoral injections were performed every 3 d, totaling four treatments. Each injection of purified virus was diluted in a total volume of 100 μl of 0.9% of NaCl solution. Tumors were measured every three days, and tumor volume was determined by externally measuring in two dimensions using a caliper. Volume (V) was determined by the following equation, where L is length and W is width of the tumor: V = (L × W 2)/2. Animal experiments were performed in accordance with institutional guidelines and approved by the University of Louisville Institutional Animal Care and Use Committee.
Statistical analysis
The results of the in vitro assays and tumor growth in mice from the two treatments groups were analyzed by unpaired Student’s t-test using a one-way ordinary parametric analysis of variance. A significance level of p < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by Lung Cancer Research Foundation (J.G.G.-G.), award numbers R01CA129975 (H.S.Z.) and R01CA90784 (K.M.M.) from the National Cancer Institute and GMB081410 (KM.M. and H.S.Z.) and G030983 (H.S.Z.) from the Kentucky Lung Cancer Research Program. We thank Melanie Scott for editing.
Glossary
Abbreviations:
- FKHRL1
forkhead human transcription factor like 1
- TM
triple mutant
- Ad
adenovirus
- PI3K
phosphatidylinositol 3-kinase
- VSMC
vascular smooth muscle cells
- CMV
cytomegalovirus
- GFP
green fluorescent protein
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/21349
References
- 1.Miller AJ, Mihm MC., Jr. Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 2.Losina E, Walensky RP, Geller A, Beddingfield FC, 3rd, Wolf LL, Gilchrest BA, et al. Visual screening for malignant melanoma: a cost-effectiveness analysis. Arch Dermatol. 2007;143:21–8. doi: 10.1001/archderm.143.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geserick P, Drewniok C, Hupe M, Haas TL, Diessenbacher P, Sprick MR, et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL- and CD95L-mediated apoptosis. Oncogene. 2008;27:3211–20. doi: 10.1038/sj.onc.1210985. [DOI] [PubMed] [Google Scholar]
- 4.Bullani RR, Wehrli P, Viard-Leveugle I, Rimoldi D, Cerottini JC, Saurat JH, et al. Frequent downregulation of Fas (CD95) expression and function in melanoma. Melanoma Res. 2002;12:263–70. doi: 10.1097/00008390-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–9. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008;18:421–9. doi: 10.1016/j.tcb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Arimoto-Ishida E, Ohmichi M, Mabuchi S, Takahashi T, Ohshima C, Hayakawa J, et al. Inhibition of phosphorylation of a forkhead transcription factor sensitizes human ovarian cancer cells to cisplatin. Endocrinology. 2004;145:2014–22. doi: 10.1210/en.2003-1199. [DOI] [PubMed] [Google Scholar]
- 8.Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277:47928–37. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- 9.Lau CJ, Koty Z, Nalbantoglu J. Differential response of glioma cells to FOXO1-directed therapy. Cancer Res. 2009;69:5433–40. doi: 10.1158/0008-5472.CAN-08-4540. [DOI] [PubMed] [Google Scholar]
- 10.Gomez-Gutierrez JG, Souza V, Hao HY, Montes de Oca-Luna R, Dong YB, Zhou HS, et al. Adenovirus-mediated gene transfer of FKHRL1 triple mutant efficiently induces apoptosis in melanoma cells. Cancer Biol Ther. 2006;5:875–83. doi: 10.4161/cbt.5.7.2911. [DOI] [PubMed] [Google Scholar]
- 11.Yang JY, Xia W, Hu MC. Ionizing radiation activates expression of FOXO3a, Fas ligand, and Bim, and induces cell apoptosis. Int J Oncol. 2006;29:643–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Suhara T, Kim HS, Kirshenbaum LA, Walsh K. Suppression of Akt signaling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol Cell Biol. 2002;22:680–91. doi: 10.1128/MCB.22.2.680-691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 14.Raisova M, Bektas M, Wieder T, Daniel P, Eberle J, Orfanos CE, et al. Resistance to CD95/Fas-induced and ceramide-mediated apoptosis of human melanoma cells is caused by a defective mitochondrial cytochrome c release. FEBS Lett. 2000;473:27–32. doi: 10.1016/S0014-5793(00)01491-5. [DOI] [PubMed] [Google Scholar]
- 15.Eberle J, Fecker LF, Hossini AM, Wieder T, Daniel PT, Orfanos CE, et al. CD95/Fas signaling in human melanoma cells: conditional expression of CD95L/FasL overcomes the intrinsic apoptosis resistance of malignant melanoma and inhibits growth and progression of human melanoma xenotransplants. Oncogene. 2003;22:9131–41. doi: 10.1038/sj.onc.1207228. [DOI] [PubMed] [Google Scholar]
- 16.Bauer TW, Gutierrez M, Dudrick DJ, Li J, Blair IA, Menon C, et al. A human melanoma xenograft in a nude rat responds to isolated limb perfusion with TNF plus melphalan. Surgery. 2003;133:420–8. doi: 10.1067/msy.2003.113. [DOI] [PubMed] [Google Scholar]
- 17.Canter RJ, Zhou R, Kesmodel SB, Zhang Y, Heitjan DF, Glickson JD, et al. Metaiodobenzylguanidine and hyperglycemia augment tumor response to isolated limb perfusion in a rodent model of human melanoma. Ann Surg Oncol. 2004;11:265–73. doi: 10.1245/ASO.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665–75. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 19.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27:6252–75. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- 20.Hilmi C, Larribere L, Deckert M, Rocchi S, Giuliano S, Bille K, et al. Involvement of FKHRL1 in melanoma cell survival and death. Pigment Cell Melanoma Res. 2008;21:139–46. doi: 10.1111/j.1755-148X.2008.00440.x. [DOI] [PubMed] [Google Scholar]
- 21.Ko SH, Ueno T, Yoshimoto Y, Yoo JS, Abdel-Wahab OI, Abdel-Wahab Z, et al. Optimizing a novel regional chemotherapeutic agent against melanoma: hyperthermia-induced enhancement of temozolomide cytotoxicity. Clin Cancer Res. 2006;12:289–97. doi: 10.1158/1078-0432.CCR-05-0210. [DOI] [PubMed] [Google Scholar]
- 22.Dong YB, Yang HL, Elliott MJ, McMasters KM. Adenovirus-mediated E2F-1 gene transfer sensitizes melanoma cells to apoptosis induced by topoisomerase II inhibitors. Cancer Res. 2002;62:1776–83. [PubMed] [Google Scholar]
- 23.Gomez-Gutierrez JG, Garcia-Garcia A, Hao H, Rao XM, Montes de Oca-Luna R, Zhou HS, et al. Adenovirus-mediated expression of truncated E2F-1 suppresses tumor growth in vitro and in vivo. Cancer. 2010;116:4420–32. doi: 10.1002/cncr.25322. [DOI] [PMC free article] [PubMed] [Google Scholar]