

Abstract
Polo-like kinase 1 (PLK1) is a serine/threonine protein kinase and plays a critical role in mitosis. PLK1 has also been regarded as a valuable target for cancer treatment, and several PLK1 inhibitors are currently undergoing clinical investigations. In this study, our data show that the expression level of PLK1 is upregulated in human pancreatic cancer cells. Molecular modeling studies indicate that DMTC inhibits PLK1 activity through competitive displacement of ATP from its binding pocket. Our data further show that DMTC suppresses the proliferation of pancreatic cancer cells and induces the formation of multinucleated cells, ultimately resulting in apoptosis. In addition, combination index analysis demonstrates that DMTC acts synergistically with the chemotherapeutic drug gemcitabine in inhibiting the proliferation of pancreatic cancer cells. These results thus suggest a potential of using PLK1 inhibitors for the treatment of pancreatic cancer.
Keywords: PLK1, PLK1 inhibitor, cell proliferation and apoptosis, gemcitabine, pancreatic cancer
Introduction
Polo-like kinase 1 (PLK1) is a conserved serine/threonine protein kinase and is essential for cell division.1,2 Previous studies suggest that PLK1 plays pivotal roles in mitotic progress, functioning in processes from mitosis entry, centrosome maturation, spindle formation to chromosome segregation in anaphase as well as division completion.3-6 Recent studies also demonstrate that PLK1 regulates DNA damage checkpoint and is responsible for maintaining genomic stability during DNA replication.7-9 The multiple functions of PLK1 are potentially due to its specific localizations to mitotic structures and its enzymatic activity to phosphorylate a variety of substrates. As a consequence, the intact structure and catalytic activity of PLK1 are both necessary for its cellular functions.10-12
Pancreatic cancer is an aggressive disease. Early diagnosis of pancreatic cancer is difficult at present, which often leads to delay in effective treatment. Moreover, the poor response of pancreatic cancer to conventional chemotherapy adds to a bad prognosis.13,14 Recent studies had discovered some potentially useful biomarkers in pancreatic cancer, but more specific molecules involved in the pathogenesis still need to be developed.15-20 Improved understanding about the mechanisms of this disease and detailed knowledge for its diagnosis and treatment are in urgent need.
PLK1 is found to be an oncogene for its overexpression in various human cancers.21,22 Analyses about clinical data suggest that the expression level of PLK1 positively correlates with tumor progression and that the patients with high PLK1expression suffer from a significantly poorer rate of survival.21,23,24 In this study, we examined the expression level of PLK1 and found the expression is upregulated in pancreatic cancer cell lines. After demonstrating its important regulatory function in cell proliferation and its strong association with tumorigenesis, we propose PLK1 as an attractive anticancer drug target, and add PLK1-targeted small molecule inhibitors as potential agents for patients with cancer.25-29 DMTC is a PLK1 inhibitor, and competes with ATP to bind with PLK1.30 Our data showed that DMTC reduced pancreatic cancer cell proliferation and led to apoptosis, which implied its potential activity in cancer treatment. When the cells were treated with appropriate proportion of DMTC and gemcitabine, the cell proliferation level was reduced remarkably. The results suggested the potential usage of drug combination for clinical treatment.
Results
Expression levels of PLK1 is upregulated in human pancreatic cancer cells
It is reported that PLK1 is overexpressed in pancreatic tumor samples.31 To confirm this result, we tested the expression levels of PLK1 in human pancreatic cancer cell lines AsPC-1, BxPC-3, CFPAC-1, EPP85 and PANC-1. First, we extracted the total RNAs from these cells and examined the amount of PLK1 mRNA by RT-PCR (Fig. 1A). Using GAPDH as the internal standard, the relative PLK1 mRNA levels of these cancer cells were shown in Figure 1B compared with the normal pancreatic cells. PLK1 mRNA showed much higher levels in 4 of the 5 tested cancer cell lines, whereas the level of AsPC-1 was moderately increased. This result indicates that the expression levels of PLK1 mRNA is upregulated in pancreatic cells.
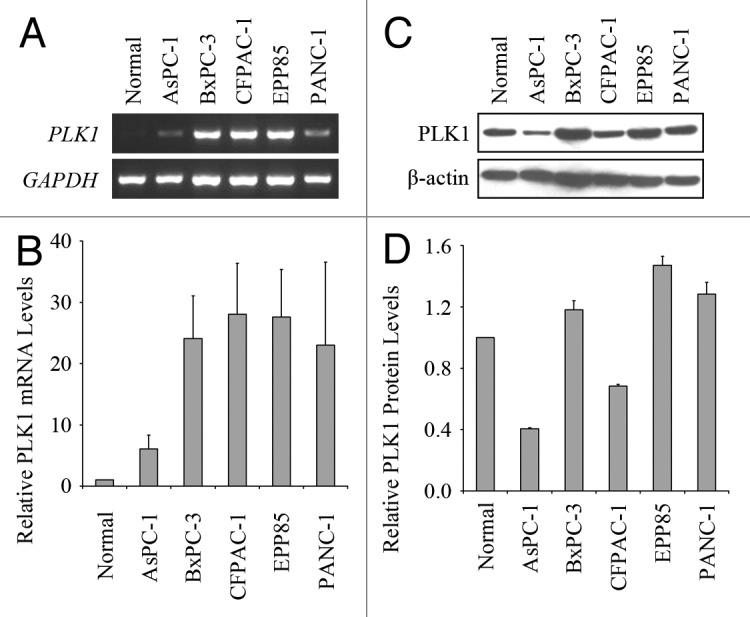
Figure 1. Expression levels of PLK1 mRNA and protein in human pancreatic cancer cells. (A) RT-PCR analysis of PLK1 mRNA expression in normal human pancreatic cells and five pancreatic cancer cell lines. (B) Experiments were performed as in (A) and the relative PLK1 mRNA levels were then quantified using GAPDH as the internal standard. (C) Western blotting analysis of PLK1 protein expression in normal and tumor-derived pancreatic cells. (D) Experiments were performed as in (C) and the relative PLK1 protein levels were then quantified using β-actin as the internal standard. Values, averages of two independent experiments; bars, SD.
Then we analyzed the protein levels of PLK1 in the cell lysates by western blotting (Fig. 1C). After quantification using β-actin as the internal standard, we found that PLK1 protein level in each cell line did not show such an obvious increase as its mRNA level (Fig. 1D). Moreover, two of these cell lines had lower expression levels of PLK1 protein than normal pancreatic cells. This result suggests that these cancer cells have differences in their mechanisms regulating the protein levels of PLK1. According to these results, EPP85 cells have the highest expression levels of PLK1 mRNA and protein among these pancreatic cancer cells, as a result, we chose this cell line to perform the following experiments.
Molecular modeling of the interaction between DMTC and PLK1
DMTC is a thiophenecarboxamide compound that acts as a potent and ATP-competitive inhibitor of PLK1 with excellent selectivity over IKK-2 and IKK-3.30 To obtain mechanistic insight into the inhibitory mechanism of DMTC, we simulated the interaction between DMTC and PLK1 by molecular modeling. DMTC was docked onto the 2.1 Å coordinates obtained from the crystal structure of PLK1 kinase domain,32 and the lowest-energy interaction model was shown in Figure 2A. In this model, the PLK1 kinase domain displayed the classical kinase fold forming a cleft between two lobes, and the DMTC binding pocket resided in this cleft, the same place where the ATP/ADP binding site was located. A detailed analysis of the molecular interaction presented the amino acid residues of PLK1 which are involved in the interaction with DMTC, including L59-G62, C67, K82 and L132-S137 (Fig. 2B), many of which are crucial for ATP/ADP binding and PLK1 activity, such as K8233 and S137.34 These data show that DMTC could inhibit PLK1 activity by competitively displacing ATP from its binding pocket and also further confirm our previous conclusion.30

Figure 2. Molecular modeling of the interaction between DMTC and PLK1. (A) Schematic models showing PLK1 kinase domain with ADP (left panel) or DMTC (right panel). These models were created by molecular docking as described in Section 2.5. (B) Details of important interactions between DMTC and PLK1. DMTC is color-coded with ivory showing carbon, gray depicting hydrogen, red representing oxygen, blue standing for nitrogen and yellow symbolizing sulfur. Amino acid residues of PLK1 involved in the interaction with DMTC are labeled.
Inhibition of PLK1 activity with DMTC prevents the proliferation of human pancreatic cancer cells
After clarifying of the inhibitory mechanism of DMTC, we examined the therapeutic potential of this inhibitor in pancreatic cancer cells. EPP85 cells were treated with gradient concentrations of DMTC, and the percentage of cell proliferation was then measured by SRB assay. We found that the prevention of EPP85 cell proliferation by DMTC was concentration-dependent, and the IC50 value, which stands for the drug concentration needed for 50% inhibition of cell proliferation, was determined to be 20 nM (Fig. 3A). Phase contrast microscopic analysis of cell morphology showed that while EPP85 cells normally proliferated upon DMSO treatment as the control, their proliferation was obviously impaired at the presence of DMTC (Fig. 3B).

Figure 3. Inhibition of PLK1 activity with DMTC prevents the proliferation of human pancreatic cancer cells. (A) EPP85 cells were treated with varying concentrations of DMTC for 72 h, and the percentage of cell proliferation was measured by SRB-based in vitro cell proliferation assay. The drug concentration needed for 50% inhibition of cell proliferation (IC50) is shown at the upper side. (B) Phase contrast images of EPP85 cells treated with 20 nM DMTC or equal volume of the DMSO control for 0, 24, 48 or 72 h. (C) EPP85 cells were treated for 72 h with 20 nM DMTC, 50 nM DMTC or DMSO as the control. After 2 weeks of culture, the colonies were shown with crystal violet staining. (D) Experiments were performed as in (C), and the number of colonies in each well was then quantified. Values, averages of three independent experiments; bars, SD; **p < 0.01.
To further confirm this effect, we performed the colony formation assay with EPP85 cells. After 2 weeks of culture, DMSO-treated cells formed colonies which originated from a single cell (Fig. 3C). In contrast, DMTC-treated cells formed much fewer (20 nM treatment) or no (50 nM treatment) colonies. As shown in Figure 3D, the number of colonies was significantly reduced upon DMTC treatment in a concentration-dependent manner. Taken together, these results demonstrate that this PLK1 inhibitor effectively prevents the proliferation of pancreatic cancer cells.
DMTC treatment results in the accumulation of cells with abnormal nuclei and triggers apoptosis in pancreatic cancer cells
To investigate the mechanism underlying the anti-proliferative activity of DMTC in pancreatic cancer cells, we examined the morphology of microtubules and DNA in DMTC-treated EPP85 cells by immunofluorescence microscopy. As shown in Figure 4A, after a 24 h treatment with 20 nM DMTC, many cells were seen containing multiple nuclei which were smaller than normal nuclei in the control cells. In addition, this abnormality caused by DMTC exhibited a time-dependent trait (Fig. 4B). Of 24 h DMTC-treated cells, 47.8% had abnormal nuclei, while after a 48 h treatment, the percentage of abnormal cells reached 68.0%. In contrast, less than 5% of cells with abnormal nuclei were detected in their respective control groups.

Figure 4. DMTC causes the accumulation of cells with abnormal nuclei and triggers apoptosis in pancreatic cancer cells. (A) Immunofluorescence images of microtubules (red) and DNA (blue) in EPP85 cells treated for 24 h with 20 nM DMTC or the DMSO control. (B) Cells were treated with 20 nM DMTC or DMSO (control) for 0, 24 or 48 h, and the percentage of cells with abnormal nuclei was then quantified by immunofluorescence staining of microtubules and DNA as in (A). Values, averages of three independent experiments; bars, SD; **p < 0.01. (C) Quantitation of apoptosis by annexin V/PI staining in EPP85 cells treated for 72 h with 20 nM DMTC or DMSO (control).
To test whether DMTC would induce apoptotic cell death after a longer period of treatment, we performed the annexin V/PI staining assay, which shows the exposure of phosphatidylserine to the outer plasma membrane in apoptotic cells. As shown in Figure 4C, after a 72 h treatment, the DMTC-treated EPP85 cells had a higher percentage of apoptotic cells (shown in the lower right box of each panel) than the level in the control cells. This result indicates that DMTC could lead to apoptosis in pancreatic cancer cells.
Combined effects of DMTC and gemcitabine in inhibiting EPP85 cell proliferation
Gemcitabine, a nucleoside analog, has been used as an anti-pancreatic cancer drug in the clinic.35 As DMTC could effectively inhibit the proliferation of pancreatic cancer cells, we wanted to know whether the combination of DMTC and gemcitabine would lead to a synergistic effect in preventing cell proliferation. We treated EPP85 cells with varying concentrations of DMTC (Fig. 3A) or gemcitabine (Fig. 5A) alone and in combination at a fixed ratio of 1:10 (Fig. 5B) for 72 h. At the end of this period, the inhibition of cell proliferation was measured by the SRB assay for each condition. Treatment interaction effects of DMTC and gemcitabine were then determined by calculating the CI values for each fraction affected using the CalcuSyn program, which is based on the median-effect principle of Chou and Talalay.36 As shown in Figure 5C, the CI values were less than 1 when the fraction affected was lower than 0.75, while the CI values were more than 1 when the fraction affected was higher than 0.75. This result indicates that DMTC acts synergistically with gemcitabine when lower concentrations of drugs are used to treat cells; however, when cells are treated with very high concentration of DMTC and gemcitabine, these two drugs show antagonistic instead of synergistic interaction. This result also suggests the optimal concentration range of these two drugs for their synergistic interaction.
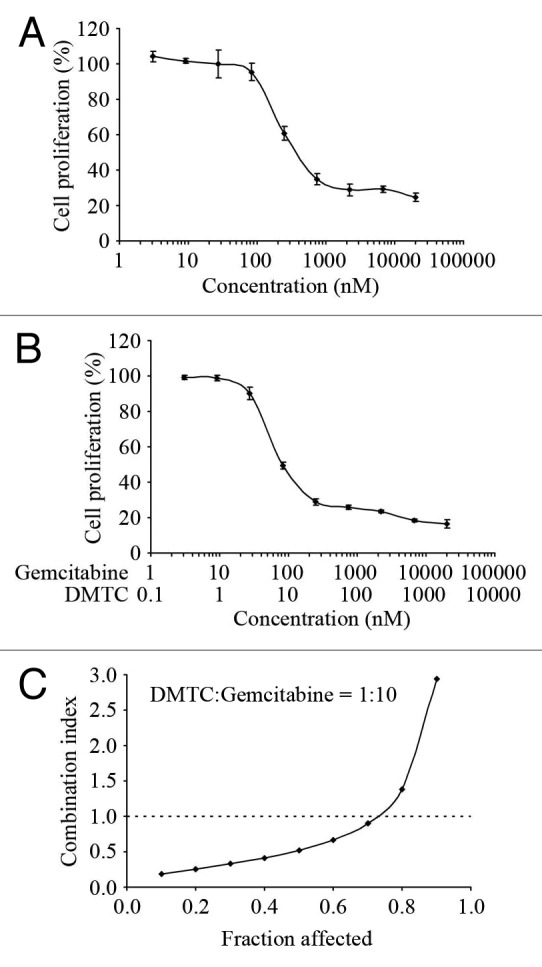
Figure 5. Treatment interaction effects of DMTC and gemcitabine in inhibiting EPP85 cell proliferation. (A) Cells were treated with varying concentrations of gemcitabine alone for 72 h, and the percentage of cell proliferation was measured by SRB-based in vitro cell proliferation assay. (B) Cells were treated with varying concentrations of DMTC and gemcitabine in combination at a fixed ratio of 1:10 for 72 h, and the percentage of cell proliferation was measured as in (A). Values, averages of three independent experiments; bars, SD (C) Treatment interaction effects of DMTC and gemcitabine were determined by calculating the CI values for each fraction affected using the CalcuSyn program. CI < 1, synergistic interactions; CI = 1, additive interactions; CI > 1, antagonistic interactions.
Discussion
At present, many chemotherapy drugs are being used as major treatment options for patients with cancer, but their severe side effects sometimes increase patients suffering from the disease. As a consequence, developing new drugs that are able to target proliferation cells specifically is both rewarding and challenging.37,38 It is suggested that PLK1 regulates mitotic progress by acting at DNA damage checkpoint, the defect of which is associated with various mitotic failures.39 Overexpression of PLK1 has been found in human tumors and clinical studies have revealed the correlation between decreased survival rate and increased PLK1 expression level. Therefore PLK1 is seen as a novel diagnostic marker and a drug target, with several PLK1 inhibitors underway for clinical studies.40
Pancreatic cancer is a disease with high lethality, owing to the difficulty in its diagnosis and the resistance that many patients displayed to conventional treatment. Pancreatic cancer is now the fourth most common cause of cancer related death in the world. In this study, we found that the expression level of PLK1 was upregulated in pancreatic cancer cell lines and that PLK1 overexpression correlated with tumor aggressiveness and poor prognosis. Concerning the key roles of PLK1 in cell proliferation and its specific activity during mitosis progress, inhibitors of PLK1 were expected to work as effective agents in treating pancreatic cancer.
DMTC is an ATP-competitive inhibitor of PLK1 and our molecular modeling simulated the association of DMTC and PLK1. The model indicated that DMTC bound the same pocket as ATP, which also inhibited PLK1 kinase activity. ATP binding pocket is conservative in PLKs and other kinases,41,42 so it is possible that this ATP-competitive inhibitor would target other kinases besides PLK1. The remarkable effects of DMTC in reducing cell proliferation might also be due to its ability to inhibit more potential targets.
When pancreatic cancer cells were treated with DMTC, the cell proliferation was inhibited and the reduction displayed dosage dependent manner. As a positive regulator of mitosis, PLK1 is an important modulator of cell division. It is reasonable that PLK1 inhibitors prevent cell proliferation; however, the detailed mechanisms still need to be better understood.
After treating with DMTC, pancreatic cancer cells exhibited multi nuclei and induced apoptosis. PLK1 is phosphorylated by Aurora A kinase and the active protein could phosphorylate cyclin B-Cdk1, which is responsible for mitotic entry.43 PLK1 activation is also essential for RhoA accumulation, which functions as an upstream regulator of contractile ring formation. As a result, PLK1 inactivation is detrimental for cleavage furrow ingression during anaphase and results in cytokinesis failure.4,44,45 Inactivation of PLK1 would prevent both mitosis entry and completion of cell division. When the PLK1 activation was inhibited by DMTC, cells displayed multinucleation and became apoptotic, due to failure of cytokinesis and DNA separation.
Patients with pancreatic cancer are often resistant to conventional chemotherapy and the amount of drugs for clinical treatment is limited. To develop new synergistic drug combinations thus seems imperative.46,47 Gemcitabine is a kind of pyramidine antimetabolite that perturbs with DNA synthesis, which is used clinically as an anti-pancreatic cancer drug.35 Concerning that DMTC prevents cell proliferation of pancreatic cancer cell line and induces apoptosis, we further discuss the potential effect of drug combination. The result suggested DMTC acted synergistically with Gemcitabine at lower concentration, and the two drugs showed antagonistic effect at high concentration. Gemcitabine inhibits DNA duplication by activating DNA damage checkpoints, which further prevents cell divisions. The activated checkpoints allow time for DNA repair and prevent genotoxic stress. DNA damage would lead to different possible results, and they are known as checkpoint recovery, checkpoint adaptation and cell apoptosis. PLK1 was found to regulate several important components of the DNA damage checkpoint complex to silence the checkpoint signal and involved in all the three processes.48-50 We suggest that Gemcitabine induced DNA damage and stimulated checkpoint activation, DMTC silenced PLK1 and maintained the integrity of checkpoint. As a consequence, incorrectly duplicated DNA failed to enter mitosis and ended up in apoptosis. When the cells were treated with more doses of DMTC, PLK1 inactivation would lead to checkpoint adaptation and chromosome separation. When using drugs at high concentrations, the drugs displayed antagonistic effect. The pharmacological studies indicate the potent usage of the two drugs and suggest optimal combination concentration.
Materials and methods
Materials
5-(5,6-Dimethoxybenzimidazol-1-yl)-3-(4-methanesulfonyl-benzyloxy)-thiophene-2-carboxamide (DMTC) was purchased from Calbiochem. 2'-Deoxy-2', 2'-difluorocytidine Hydrochloride (known as gemcitabine) was obtained from Toronto Research Chemicals. Sulforhodamine B (SRB), crystal violet, 4', 6-diamidino-2-phenylindole (DAPI) and mouse monoclonal antibodies against α-tubulin and β-actin were from Sigma-Aldrich. The rabbit monoclonal antibody against PLK1 was purchased from Cell Signaling Technology. Horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies were obtained from Amersham Biosciences. Rhodamine-conjugated anti-mouse secondary antibody was from Jackson ImmunoResearch Laboratories.
Cell culture
Human pancreatic cancer cell lines BxPC-3, EPP85, PANC-1 and the normal human pancreatic cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (FBS). Human pancreatic cancer cell line AsPC-1 was cultured in RPMI 1640 medium supplemented with 10% FBS. Human pancreatic cancer cell line CFPAC-1 was cultured in Iscove’s modified Dulbecco’s medium supplemented with 10% FBS. All cells were grown at 37°C in a humidified atmosphere with 5% CO2.
RT-PCR
Total RNAs were isolated using the TRIzol reagent following standard protocols (Invitrogen). Two micrograms of total RNA were used to synthesize the cDNA with M-MLV reverse transcriptase (Promega). Forward primer 5'-AAGTGGGTGGACTATTCGG-3' and reverse primer 5'-CAGGAGACTCAGGCGGTAT-3' were used to examine the expression of the human PLK1 gene by PCR. Primers used for the GAPDH gene were 5'-ATCACTGCCACCCAGAAGAC-3' (forward) and 5'-ATGAGGTCCACCACCCTGTT-3' (reverse). A total of 30 cycles were used to amplify PLK1, whereas 25 cycles were used to amplify the internal standard GAPDH.
Western blotting
Proteins were resolved by sodium dodecyl sulfate PAGE and transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked in Tris-buffered saline containing 0.05% Tween 20 and 5% fat-free dry milk, and incubated first with primary antibodies and then with horseradish peroxidase-conjugated secondary antibodies. Specific proteins were visualized with enhanced chemiluminescence detection reagent according to the manufacturer’s instructions (Pierce Biotechnology).
Molecular modeling
DMTC was docked onto the 2.1 Å coordinates obtained from the crystal structure of PLK1 kinase domain,32 using standard docking method.51
In vitro cell proliferation assay
Cells were seeded in 96-well plates at a density of 1 × 104 cells per well and they were treated with gradient concentrations of drugs the following day. After 72 h of drug treatment, the SRB assay was performed as described previously.52 The percentage of cell proliferation as a function of drug concentration was plotted to determine the IC50 value, which stands for the drug concentration needed to prevent cell proliferation by 50%.
Colony formation assay
Cells grown in 6-well plates at a density of 200 cells per well were treated with DMTC for 72 h. After 2 weeks of culture, the colonies were fixed with methanol and stained with 0.1% crystal violet. The number of colonies in each well was then counted.
Immunofluorescence microscopy
Cells grown on glass coverslips were fixed with cold (-20°C) methanol for 5 min and then washed with phosphate-buffered saline (PBS) for 5 min. Nonspecific sites were blocked by incubating with 2% bovine serum albumin in PBS for 30 min. Cells were incubated with mouse monoclonal anti-α-tubulin antibody for 2 h and then rhodamine-conjugated anti-mouse secondary antibody for 2 h followed by staining with DAPI for 10 min. Coverslips were mounted with 90% glycerol in PBS and examined with an Olympus fluorescence microscope.
Annexin V/propidium iodide (PI) staining assay
The annexin V/PI staining assay was performed by using the Alexa Fluor 488 annexin V/PI apoptosis assay kit following the manufacturer’s protocol (Molecular Probes). Briefly, cells were washed with PBS and then resuspended in the binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Cells were incubated with Alexa Fluor 488-conjugated annexin V and PI for 15 min at room temperature in the dark. The binding buffer was then added and cells were analyzed by flow cytometry.
Analysis of combined drug effects
Cells were treated with a range of DMTC or gemcitabine concentrations alone and in combination at a fixed ratio of 1:10 for 72 h. At the end of this period, the inhibition of cell proliferation was measured for each condition. Treatment interaction effects of DMTC and gemcitabine were then determined by calculating the combination index (CI) values for each fraction affected using the commercially available CalcuSyn program (Biosoft), which is based on the median-effect principle of Chou and Talalay.36 The CalcuSyn program automatically analyzes a data set using both the mutually exclusive assumption (similar mechanisms of action of both drugs) and the mutually nonexclusive assumption (dissimilar mechanisms of action of both drugs). The CI equation determines the additive effect of drug combinations, such that synergism is defined as a more than expected additive effect, and antagonism is defined as a less than expected additive effect. Thus, CI values less than 1 correspond to synergistic drug interactions, CI values equal to 1 correspond to additive interactions and CI values greater than 1 correspond to antagonistic interactions.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (90913021) and the 111 Project (B08011).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/21412
References
- 1.Sunkel CE, Glover DM. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci. 1988;89:25–38. doi: 10.1242/jcs.89.1.25. [DOI] [PubMed] [Google Scholar]
- 2.Llamazares S, Moreira A, Tavares A, Girdham C, Spruce BA, Gonzalez C, et al. polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991;5(12A):2153–65. doi: 10.1101/gad.5.12a.2153. [DOI] [PubMed] [Google Scholar]
- 3.Kang YH, Park JE, Yu LR, Soung NK, Yun SM, Bang JK, et al. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol Cell. 2006;24:409–22. doi: 10.1016/j.molcel.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, et al. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci USA. 2007;104:4383–8. doi: 10.1073/pnas.0701140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petronczki M, Glotzer M, Kraut N, Peters JM. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell. 2007;12:713–25. doi: 10.1016/j.devcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Moshe Y, Boulaire J, Pagano M, Hershko A. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci USA. 2004;101:7937–42. doi: 10.1073/pnas.0402442101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000;2:672–6. doi: 10.1038/35023629. [DOI] [PubMed] [Google Scholar]
- 8.Tsou MF, Wang WJ, George KA, Uryu K, Stearns T, Jallepalli PV. Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Dev Cell. 2009;17:344–54. doi: 10.1016/j.devcel.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yim H, Erikson RL. Polo-like kinase 1 depletion induces DNA damage in early S prior to caspase activation. Mol Cell Biol. 2009;29:2609–21. doi: 10.1128/MCB.01277-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299:1228–31. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 11.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, et al. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003;115:83–95. doi: 10.1016/S0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 12.Casenghi M, Meraldi P, Weinhart U, Duncan PI, Körner R, Nigg EA. Polo-like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation. Dev Cell. 2003;5:113–25. doi: 10.1016/S1534-5807(03)00193-X. [DOI] [PubMed] [Google Scholar]
- 13.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 14.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–49. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 15.Sun XD, Shi XJ, Sun XO, Luo YG, Wu XJ, Yao CF, et al. Dimethylenastron suppresses human pancreatic cancer cell migration and invasion in vitro via allosteric inhibition of mitotic kinesin Eg5. Acta Pharmacol Sin. 2011;32:1543–8. doi: 10.1038/aps.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M, Wang X, Yang Y, Li D, Ren H, Zhu Q, et al. Ectopic expression of the microtubule-dependent motor protein Eg5 promotes pancreatic tumourigenesis. J Pathol. 2010;221:221–8. doi: 10.1002/path.2706. [DOI] [PubMed] [Google Scholar]
- 17.Dangi-Garimella S, Krantz SB, Barron MR, Shields MA, Heiferman MJ, Grippo PJ, et al. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011;71:1019–28. doi: 10.1158/0008-5472.CAN-10-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim ST, Lim H, Jang KT, Lim T, Lee J, Choi YL, et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol Cancer Ther. 2011;10:1993–9. doi: 10.1158/1535-7163.MCT-11-0269. [DOI] [PubMed] [Google Scholar]
- 19.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 20.Liu M, Yu H, Huo L, Liu J, Li M, Zhou J. Validating the mitotic kinesin Eg5 as a therapeutic target in pancreatic cancer cells and tumor xenografts using a specific inhibitor. Biochem Pharmacol. 2008;76:169–78. doi: 10.1016/j.bcp.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–91. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 22.Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24:267–76. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 23.Knecht R, Oberhauser C, Strebhardt K. PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int J Cancer. 2000;89:535–6. doi: 10.1002/1097-0215(20001120)89:6<535::AID-IJC12>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 24.Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:2794–7. [PubMed] [Google Scholar]
- 25.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010;9:643–60. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 26.Schmit TL, Zhong W, Setaluri V, Spiegelman VS, Ahmad N. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small-molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. J Invest Dermatol. 2009;129:2843–53. doi: 10.1038/jid.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA. 2003;100:5789–94. doi: 10.1073/pnas.1031523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y. Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res. 2005;65:2698–704. doi: 10.1158/0008-5472.CAN-04-2131. [DOI] [PubMed] [Google Scholar]
- 29.Evans RP, Dueck G, Sidhu R, Ghosh S, Toman I, Loree J, et al. Expression, adverse prognostic significance and therapeutic small molecule inhibition of Polo-like kinase 1 in multiple myeloma. Leuk Res. 2011;35:1637–43. doi: 10.1016/j.leukres.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 30.Bamborough P, Christopher JA, Cutler GJ, Dickson MC, Mellor GW, Morey JV, et al. 5-(1H-Benzimidazol-1-yl)-3-alkoxy-2-thiophenecarbonitriles as potent, selective, inhibitors of IKK-epsilon kinase. Bioorg Med Chem Lett. 2006;16:6236–40. doi: 10.1016/j.bmcl.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 31.Gray PJ, Jr., Bearss DJ, Han H, Nagle R, Tsao MS, Dean N, et al. Identification of human polo-like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol Cancer Ther. 2004;3:641–6. [PubMed] [Google Scholar]
- 32.Kothe M, Kohls D, Low S, Coli R, Cheng AC, Jacques SL, et al. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry. 2007;46:5960–71. doi: 10.1021/bi602474j. [DOI] [PubMed] [Google Scholar]
- 33.McInnes C, Mezna M, Fischer PM. Progress in the discovery of polo-like kinase inhibitors. Curr Top Med Chem. 2005;5:181–97. doi: 10.2174/1568026053507660. [DOI] [PubMed] [Google Scholar]
- 34.Jang YJ, Ma S, Terada Y, Erikson RL. Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase. J Biol Chem. 2002;277:44115–20. doi: 10.1074/jbc.M202172200. [DOI] [PubMed] [Google Scholar]
- 35.FDA expands access to gemcitabine for pancreatic cancer. Am J Health Syst Pharm. 1995;52:931. doi: 10.1093/ajhp/52.9.931. [DOI] [PubMed] [Google Scholar]
- 36.Chou TC, Talalay P. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem. 1981;115:207–16. doi: 10.1111/j.1432-1033.1981.tb06218.x. [DOI] [PubMed] [Google Scholar]
- 37.Murugan RN, Park JE, Kim EH, Shin SY, Cheong C, Lee KS, et al. Plk1-targeted small molecule inhibitors: molecular basis for their potency and specificity. Mol Cells. 2011;32:209–20. doi: 10.1007/s10059-011-0126-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun X, Li D, Yang Y, Ren Y, Li J, Wang Z, et al. Microtubule-binding protein CLIP-170 is a mediator of paclitaxel sensitivity. J Pathol. 2012;226:666–73. doi: 10.1002/path.3026. [DOI] [PubMed] [Google Scholar]
- 39.Barr FA, Silljé HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–40. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 40.Beria I, Ballinari D, Bertrand JA, Borghi D, Bossi RT, Brasca MG, et al. Identification of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as a new class of orally and selective Polo-like kinase 1 inhibitors. J Med Chem. 2010;53:3532–51. doi: 10.1021/jm901713n. [DOI] [PubMed] [Google Scholar]
- 41.Lowery DM, Lim D, Yaffe MB. Structure and function of Polo-like kinases. Oncogene. 2005;24:248–59. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 42.Sun L, Li D, Dong X, Yu H, Dong JT, Zhang C, et al. Small-molecule inhibition of Aurora kinases triggers spindle checkpoint-independent apoptosis in cancer cells. Biochem Pharmacol. 2008;75:1027–34. doi: 10.1016/j.bcp.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Cogswell JP, Brown CE, Bisi JE, Neill SD. Dominant-negative polo-like kinase 1 induces mitotic catastrophe independent of cdc25C function. Cell Growth Differ. 2000;11:615–23. [PubMed] [Google Scholar]
- 44.Seong YS, Kamijo K, Lee JS, Fernandez E, Kuriyama R, Miki T, et al. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem. 2002;277:32282–93. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- 45.Sun L, Gao J, Dong X, Liu M, Li D, Shi X, et al. EB1 promotes Aurora-B kinase activity through blocking its inactivation by protein phosphatase 2A. Proc Natl Acad Sci USA. 2008;105:7153–8. doi: 10.1073/pnas.0710018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu M, Aneja R, Wang H, Sun L, Dong X, Huo L, et al. Modulation of multidrug resistance in cancer cells by the E3 ubiquitin ligase seven-in-absentia homologue 1. J Pathol. 2008;214:508–14. doi: 10.1002/path.2312. [DOI] [PubMed] [Google Scholar]
- 47.Liu M, Li D, Aneja R, Joshi HC, Xie S, Zhang C, et al. PO(2)-dependent differential regulation of multidrug resistance 1 gene expression by the c-Jun NH2-terminal kinase pathway. J Biol Chem. 2007;282:17581–6. doi: 10.1074/jbc.M702206200. [DOI] [PubMed] [Google Scholar]
- 48.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19:238–45. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Yoo HY, Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell. 2004;117:575–88. doi: 10.1016/S0092-8674(04)00417-9. [DOI] [PubMed] [Google Scholar]
- 50.van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong SE, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 2010;8:e1000287. doi: 10.1371/journal.pbio.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kick EK, Roe DC, Skillman AG, Liu G, Ewing TJ, Sun Y, et al. Structure-based design and combinatorial chemistry yield low nanomolar inhibitors of cathepsin D. Chem Biol. 1997;4:297–307. doi: 10.1016/S1074-5521(97)90073-9. [DOI] [PubMed] [Google Scholar]
- 52.Zhou J, Liu M, Aneja R, Chandra R, Joshi HC. Enhancement of paclitaxel-induced microtubule stabilization, mitotic arrest, and apoptosis by the microtubule-targeting agent EM012. Biochem Pharmacol. 2004;68:2435–41. doi: 10.1016/j.bcp.2004.08.032. [DOI] [PubMed] [Google Scholar]