

Mitochondrial outer membrane tail-anchored proteins are a unique class of membrane proteins with unknown targeting mechanism. Using two high-throughput microscopy screens, we demonstrate that the inherent differences in membrane composition between organelle membranes is enough to determine membrane integration specificity in a living cell.
Abstract
Tail-anchored (TA) proteins have a single C-terminal transmembrane domain, making their biogenesis dependent on posttranslational translocation. Despite their importance, no dedicated insertion machinery has been uncovered for mitochondrial outer membrane (MOM) TA proteins. To decipher the molecular mechanisms guiding MOM TA protein insertion, we performed two independent systematic microscopic screens in which we visualized the localization of model MOM TA proteins on the background of mutants in all yeast genes. We could find no mutant in which insertion was completely blocked. However, both screens demonstrated that MOM TA proteins were partially localized to the endoplasmic reticulum (ER) in ∆spf1 cells. Spf1, an ER ATPase with unknown function, is the first protein shown to affect MOM TA protein insertion. We found that ER membranes in ∆spf1 cells become similar in their ergosterol content to mitochondrial membranes. Indeed, when we visualized MOM TA protein distribution in yeast strains with reduced ergosterol content, they phenocopied the loss of Spf1. We therefore suggest that the inherent differences in membrane composition between organelle membranes are sufficient to determine membrane integration specificity in a eukaryotic cell.
INTRODUCTION
Tail-anchored (TA) proteins are a group of proteins characterized by a single transmembrane domain (TMD) at their C-termini that tethers them to intracellular membranes with their N-termini facing the cytosol. The signal that allows them to be targeted to membranes of various cellular compartments is encoded within the TMD (Borgese et al., 2003b). All TA proteins are translated in the cytosol and inserted posttranslationally into their corresponding membranes (Borgese et al., 2007; Rabu et al., 2009). Despite the fact that TA proteins can be found on all organelles, their insertion into membranes seems to occur into either the mitochondrial outer membrane (MOM) or the endoplasmic reticulum (ER), which serve as the first station for TA proteins of all other compartments of the secretory pathway (Schuldiner et al., 2008, 2010).
Understanding the biogenesis of mitochondrial TA proteins is important, due to their essential roles in basic cellular functions and in the progression of diseases. For example, in yeast, TA proteins are required for mitochondrial fission (Fis1; Mozdy et al., 2000), for inheritance and motility (Gem1; Frederick et al., 2004) and for the functioning of the translocase of the outer membrane (TOM) complex (Tom5, Tom6, and Tom7; Walther and Rapaport, 2009). In mammals, Bcl2, and other BCL family members that are well-studied oncogenes are also mitochondrial TA proteins (Chipuk et al., 2010).
The structural characteristics that differentiate a mitochondrial targeting TA from an ER one are well characterized and include a relatively short TMD with moderate hydrophobicity and positively charged residues at its flanking regions (Horie et al., 2002; Beilharz et al., 2003; Borgese et al., 2003a, b; Habib et al., 2003). In fact, these exact features have been shown to reduce the affinity by which TA proteins bind to the chaperone Sgt2 (Wang et al., 2010). Sgt2 therefore associates with high-affinity only with ER-bound TA proteins and relays them to a dedicated and conserved insertion pathway: the Guided Entry of TA proteins (GET) complex in yeast (Schuldiner et al., 2005, 2008; Jonikas et al., 2009) and the Transmembrane domain Recognition Complex (TRC40) chaperone complex in mammals (Stefanovic and Hegde, 2007; Favaloro et al., 2008, 2010). However, it is still unknown whether dedicated protein machinery exists to recognize MOM TA proteins and selectively insert them into their target membranes or whether the mitochondrial insertion pathway occurs as a default for those proteins that do not bind Sgt2 (Rapaport, 2003; Setoguchi et al., 2006; Borgese et al., 2007; Kemper et al., 2008).
We, and others, have previously shown that mitochondria isolated from cells mutated in known outer membrane import components are not compromised in their TA insertion capacity. This implies that MOM TA proteins are inserted by other mechanisms (Setoguchi et al., 2006; Kemper et al., 2008). Moreover, recent evidence suggests that these proteins may not require any proteinaceous receptor component, as mitochondrial TA proteins insert in vitro into protease-treated mitochondrial membranes with the same efficiency as they insert into untreated ones (Kemper et al., 2008). Interestingly, in vitro insertion efficiency of MOM TA proteins into pure lipid vesicles is highly dependent on the composition of the receiving membrane, since efficiency decreases as ergosterol levels increase (Kemper et al., 2008). Because the MOM in yeast cells has the lowest sterol content among all membranes that face the cytosol (Zinser et al., 1991; Schneiter et al., 1999), such low ergosterol content could explain why MOM TA proteins do not require assistance for insertion. In support of this, we have previously shown that in the absence of the GET pathway, ER-destined TA proteins are wrongly inserted into mitochondria (Schuldiner et al., 2008). This would suggest that mitochondrial membranes in yeast cells serve as a “default” target for “unguided” TA protein insertion.
However, the ability to insert in vitro in the absence of protein machinery does not necessarily indicate lack of such machinery in vivo. Hence, in this study, we explored whether there are cellular factors influencing mitochondrial TA protein insertion in vivo. To identify such proteins, we took a novel approach in the study of MOM TA protein insertion and performed two high-throughput microscopy screens using the budding yeast Saccharomyces cerevisiae as a model system. We inspected the cellular localization of a model MOM TA protein (green fluorescent protein-Gem1 [GFP-Gem1]) on the background of strains harboring mutations in each yeast gene. This screen uncovered a single gene, SPF1, whose deletion causes mislocalization of GFP-Gem1 to the ER.
Spf1 is an ER localized P-type ATPase whose deletion causes a variety of severe phenotypes in relation to ER homeostasis but has no defined function (Cronin et al., 2000, 2002; Tipper and Harley, 2002; Vashist et al., 2002; Ando and Suzuki, 2005). We show here that in addition to the GFP-Gem1 ER mislocalization, the ∆spf1 strain exhibited a redistribution of additional MOM single-span proteins into ER membranes concomitant with changes in ergosterol distribution between ER and mitochondrial membranes. We thus uncovered a role for Spf1 in determining the lipid and protein composition of organelles’ membranes. Moreover, we suggest that the inherent unique lipid composition of the MOM, rather than dedicated insertion machinery, is sufficient to determine membrane-integration specificity in a living cell.
RESULTS
A screen for proteins affecting the localization of GFP-Gem1 reveals that Spf1 is required for correct membrane targeting of MOM TA proteins
To visually screen for factors influencing the insertion of yeast MOM TA proteins in vivo, we first created a yeast strain (based on the yeast S288C background; Brachmann et al., 1998) expressing a fluorescently tagged version of the model MOM TA protein, Gem1. The reporter was constructed by introducing a galactose-inducible promoter (GAL1p) driving an N-terminally fused GFP at the endogenous GEM1 coding region. This created an N-terminal GFP fusion protein (GFP-Gem1) whose expression is controlled by an inducible promoter. Indeed, when induced by growth in galactose-containing medium, GFP-Gem1 localized correctly to mitochondria, as could be seen by its colocalization with an mCherry protein fused to a mitochondrial targeting sequence (MTS; Figure 1A, top row).
FIGURE 1:
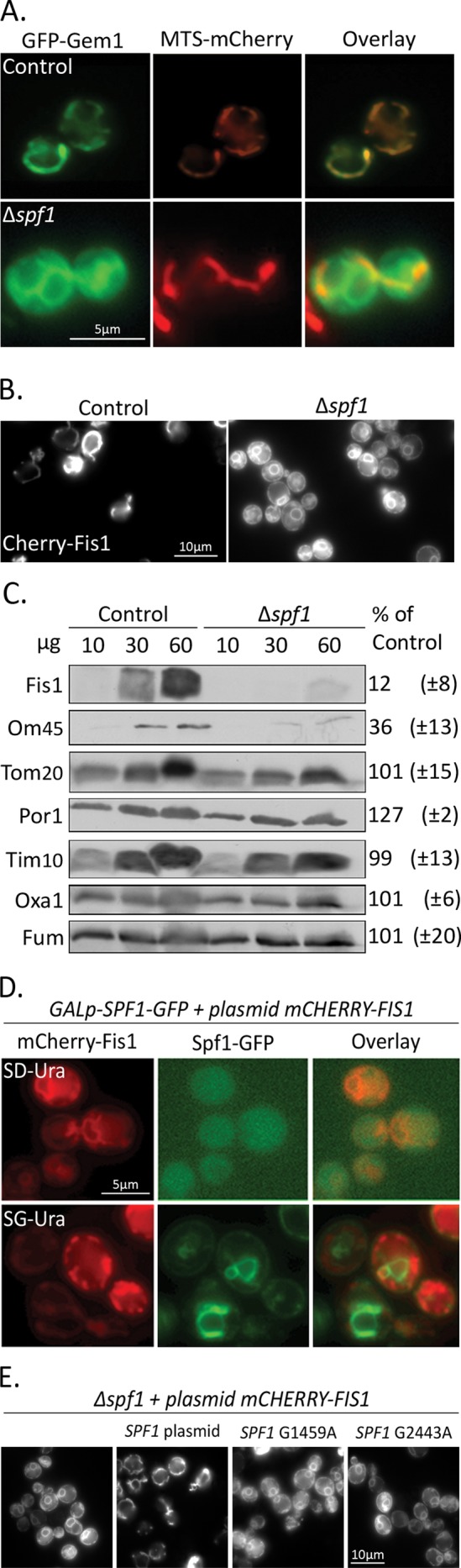
Deletion of SPF1 causes mislocalization of MOM TA proteins. (A) GFP-Gem1 was visualized in control or ∆spf1 cells expressing a mitochondria-targeted mCherry (MTS-mCherry). (B) mCherry-Fis1 was visualized in control or ∆spf1 cells. (C) Mitochondria were isolated from control and ∆spf1 cells grown to stationary phase at 30°C. The indicated amounts of mitochondrial proteins were analyzed by SDS–PAGE and immunodecorated with antibodies against various mitochondrial proteins. The intensity of the bands in three independent experiments was quantified and the average amount (± SD) of proteins in mutant mitochondria is expressed as percent of their level in the control organelle. (D) Cells expressing Spf1 under control of a galactose-inducible promoter were transformed with a plasmid encoding mCherry-Fis1. Cells were grown on synthetic selective medium containing either glucose (SD-Ura) or galactose (SG-Ura) and visualized using fluorescence microscopy. (E) mCherry-Fis1 was visualized in ∆spf1 cells either without any plasmid, with a plasmid containing full-length SPF1 gene, or with plasmids containing the full-length gene carrying one of two ATPase-dead mutations.
We used this strain to explore which cellular factors influence mitochondrial TA protein insertion in vivo. Using synthetic genetic array (SGA) methodology (Tong et al., 2001; Tong and Boone, 2006; Cohen and Schuldiner, 2011), we crossed this strain against two libraries that together encompass mutations in every yeast gene. The first library was the yeast deletion library (Giaever et al., 2002), which consists of ∼5000 strains, each harboring a deletion in one nonessential gene. The second was a decreased abundance by mRNA perturbation (DAmP) library, which consists of ∼1000 strains, each expressing a hypomorphic allele of an essential gene (Schuldiner et al., 2005; Breslow et al., 2008). This procedure allowed us to create a library of nearly 6000 haploid cells, each expressing the GFP-Gem1 fusion protein on the background of a mutation in a single gene (Supplemental Figure S1).
We then visualized the cellular localization of GFP-Gem1 in each of these mutated strains using a high-throughput microscopy setup (Cohen and Schuldiner, 2011). Microscopy was performed during post–logarithmic growth in the semifermentable carbon source galactose to induce maximal expression of GFP-Gem1. Under these conditions, the majority of the strains showed normal mitochondrial localization. Remarkably, solely in one strain, ∆spf1, we found a dramatic mislocalization of GFP-Gem1 but mitochondrial morphology remained unchanged (Figure 1A, bottom row).
Because only a single protein came up in our screen as influencing MOM TA protein distribution, we decided to verify our results by performing an additional screen based on a different query strain. For the new query strain, we constructed a fusion protein consisting of a GFP moiety fused to the Gem1 TMD (aa 626–662) with a Myc epitope tag as a linker. This protein was expressed from an exogenous locus (i.e., the endogenous coding region of GEM1 remained intact) under control of a constitutive promoter (TEF2p). The new query strain gave us three advantages: First, expression levels of the reporter were lower, therefore reducing the chances for mislocalization due to overexpression. Second, only the TMD of Gem1 was utilized, eliminating potential localization motifs in the N-terminal functional region. Third, and most importantly, our strain had normal mitochondrial morphology due to the normal expression of the endogenous GEM1.
Following the SGA procedure, we obtained a new library, which was screened in mid–logarithmic growth in media containing the fermentable carbon source glucose. Despite the differences in the fluorescent construct and the screening conditions, ∆spf1 was again identified as the only strain with altered MOM TA protein localization. Collectively, our microscopic screens suggested that aside from SPF1, there is no single gene whose loss dramatically compromises the specific targeting of a MOM TA protein.
Spf1 is a multiple-pass transmembrane protein that resides in the ER membranes and has been predicted to function as a P-type ATPase, although its substrates have not been identified (Cronin et al., 2000, 2002; Tipper and Harley, 2002; Vashist et al., 2002; Ando and Suzuki, 2005). Because Spf1 is not a mitochondrial protein, it has little chance of being part of the MOM TA protein insertion machinery. Therefore our findings suggest that its loss has an indirect effect on Gem1 biogenesis. To uncover whether the defect of ∆spf1 is specific only to Gem1 insertion, we transformed control and ∆spf1 cells with a plasmid encoding mCherry-Fis1 (another MOM TA protein; Mozdy et al., 2000). Indeed, mCherry-Fis1 mislocalized in the mutant cells in a manner similar to GFP-Gem1 (Figure 1B).
To confirm that the phenotype was not the result of the fluorophore tagging at the N-terminus of the TA proteins, we purified mitochondria from control and ∆spf1 cells during post–logarithmic growth. Using a specific antibody, we confirmed that endogenous Fis1 levels are dramatically reduced in mutant mitochondria (Figure 1C). Interestingly, we found that the levels of the signal-anchored protein OM45, which appears to follow the same insertion pathway as MOM TA proteins (Meineke et al., 2008; Walther and Rapaport, 2009), were also reduced in mutant mitochondria (Figure 1C). Indeed, OM45 tagged with a fluorophore on the background of ∆spf1 displayed the same phenotype as the MOM TA proteins and was also observed in nonmitochondrial membranes (Figure S2). Of note, staining mitochondria in the mutant cells with MTS-mCherry revealed normal mitochondrial morphology (Figure 1A) and the steady-state levels of other mitochondrial proteins were not altered in the ∆spf1 strain (Figure 1C). Hence, it seems that overall organelle biogenesis, structure, and dynamics were not altered in the mutant background.
To validate that the SPF1 deletion is causal for the MOM TA protein mislocalization phenotype, we replaced the endogenous promoter of SPF1 with an inducible one (GAL1p) and tagged the protein at its C-terminus with GFP to visualize its expression. By coexpressing the MOM TA protein mCherry-Fis1, we could see that in glucose-containing medium, cells did not express Spf1-GFP and mCherry-Fis1 was mislocalized (Figure 1D, top row). However, when cells were grown on galactose, Spf1-GFP expression was induced, causing a complete rescue back to the mitochondria-restricted localization of mCherry-Fis1 (Figure 1D, bottom row). As the genotype was unaltered between the two conditions and galactose as a carbon source does not cause redirection of TA protein insertion, we conclude that loss of Spf1 is the direct cause for MOM TA protein mislocalization in vivo.
To ascertain whether the role of Spf1 in correct MOM TA protein localization depends on its ATPase activity, we created two plasmids encoding for ATPase-dead Spf1 mutants (Suzuki and Shimma, 1999; Suzuki, 2001) and assayed their ability to rescue the ∆spf1 mislocalization phenotype. We found that neither of the mutants could rescue the phenotype, but a control plasmid carrying a wild-type copy of the gene could (Figure 1E). This observation suggests that the enzymatic activity of Spf1 is essential for its role in TA protein localization.
Deletion of SPF1 causes MOM TA proteins to accumulate in ER membranes
The new target membranes for MOM TA proteins in ∆spf1 cells displayed a pattern that is similar to that of the ER. To determine whether ER membranes do indeed become the target for MOM TA proteins when Spf1 is absent, we coexpressed an ER-localized red fluorescent protein (RFP; SS-RFP-HDEL) in ∆spf1 cells expressing GFP-Gem1. Under these conditions, substantial colocalization of the two fluorescent proteins was observed, indicating the nonmitochondrial membranes are, indeed, ER membranes (Figure 2A).
FIGURE 2:
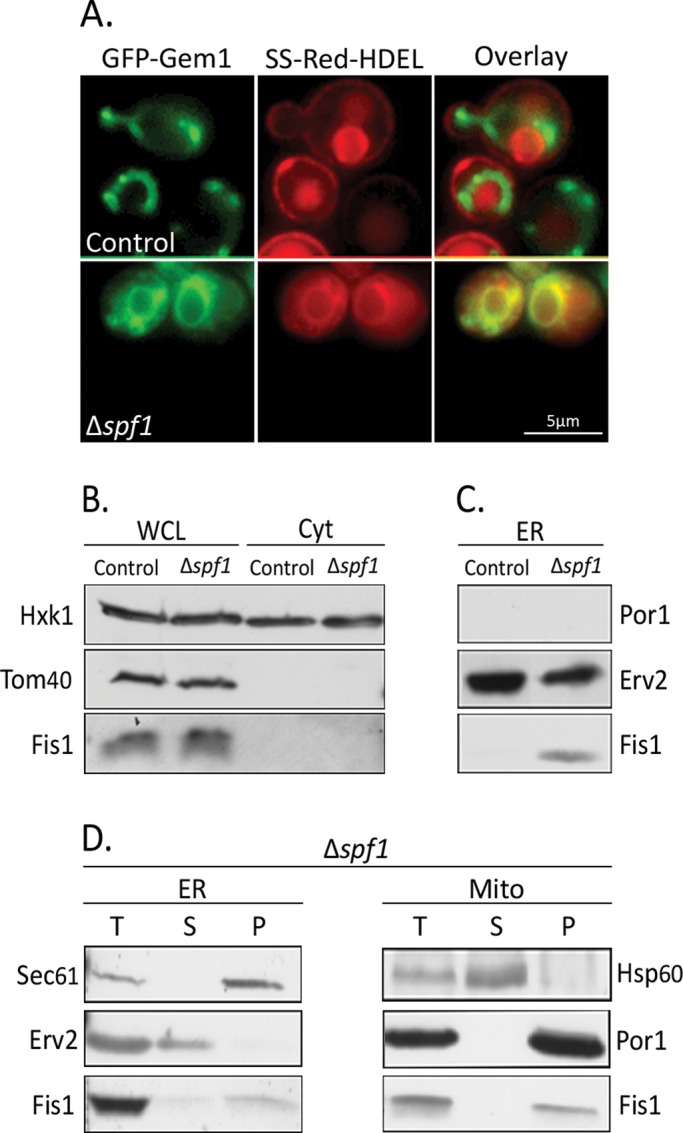
Loss of Spf1 results in insertion of MOM TA proteins into ER membranes. (A) GFP-Gem1 was visualized by fluorescence microscopy in control or ∆spf1 cells expressing an ER-localized RFP (SS-dsRED-HDEL). (B) Whole-cell lysates (WCL, 100 μg) and cytosol (Cyt, 50 μg) were isolated from control and ∆spf1 cells grown at 30°C to mid–log phase. Proteins were analyzed by SDS–PAGE and immunodecorated with antibodies against Hxk1 (cytosolic marker), Tom40 (MOM marker), and Fis1. (C) Mitochondrial and ER fractions were isolated from control and ∆spf1 cells grown at 30°C to mid–log phase. The indicated amounts of proteins were analyzed by SDS–PAGE and immunodecorated with antibodies against Erv2 (ER marker), Por1 (MOM marker), and Fis1. (D) Mitochondria and ER fractions were isolated from ∆spf1 (T = total) cells grown at 30°C to mid–log phase. All samples were subjected to carbonate extraction, in which membrane proteins are recovered in the pellet (P) and soluble proteins are found in the supernatant (S). Proteins were analyzed by SDS–PAGE and immunodecorated with the indicated antibodies.
To confirm this point, we performed Western analysis on endogenous Fis1 in either whole-cell lysates or fractionated samples. We found that in ∆spf1 cells, the overall Fis1 levels were not altered nor was the protein enriched in the cytosolic fraction (Figure 2B). However, when we analyzed ER-derived microsomes isolated from either control or ∆spf1 cells, we observed that Fis1 molecules could be detected in the ER fraction of ∆spf1, but not in the ER fraction of control cells (Figure 2C). The absence of the abundant mitochondrial marker protein Porin (Por1) in the ER fraction excludes the possibility that microsomes from the ∆spf1 cells were contaminated by mitochondrial proteins. Interestingly, in ∆spf1 strains, we did not observe any mislocalization of TA proteins destined to the secretory pathway, which suggests that the ∆spf1 mutation specifically affected localization of MOM TA proteins (Figure S3).
We then explored whether the MOM TA proteins truly insert into the ER membranes in the absence of Spf1 or whether they are merely attached to the cytosolic surface. Using carbonate extraction experiments, we could demonstrate that the MOM TA proteins of both the ER and mitochondrial membranes resisted this treatment as expected from bona fide membrane-embedded proteins (Figure 2D). Thus the phenotype we observed in ∆spf1 cells is indeed indicative of a change in membrane specificity.
Mitochondria from ∆spf1 strains do not display reduced insertion efficiency of Fis1
To better understand the effects of deleting SPF1 on MOM TA protein insertion, we first tested whether the absence of Spf1 directly affects the insertion efficiency of TA proteins into MOMs. To this end, we used an established in vitro insertion assay based on purified mitochondria and a Fis1 construct that contains a cysteine residue in its TMD (Fis1-TMC). Because the cysteine residue can be modified only if it is exposed due to improper insertion of the TMD, this assay allowed us to assess the fraction of correctly inserted Fis1 molecules into MOMs (Kemper et al., 2008). To our surprise, we found that mitochondria isolated from ∆spf1 cells could import Fis1 to the same extent and at the same rate as mitochondria isolated from control cells (Figure 3, see both at 15 and at 30 min). It therefore appears that the absence of Spf1 does not affect the import capacity or fidelity of mitochondria.
FIGURE 3:
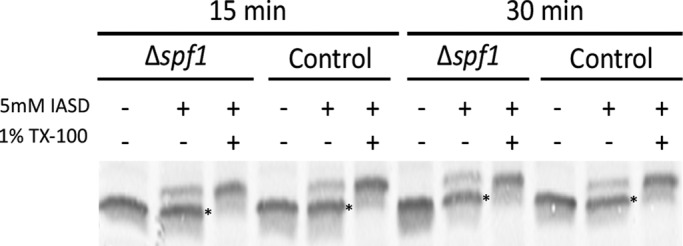
Fis1 is imported with equal efficiency into ∆spf1 and control mitochondria. Radiolabeled Fis1 carrying a cysteine in its transmembrane domain (Fis1-TMC; see Kemper et al., 2008) was incubated at 25°C for 15 or 30 min with mitochondria isolated from either control or ∆spf1 cells. Mitochondria were reisolated by centrifugation and resuspended in labeling buffer containing the labeling reagent 4-acetamido-4′-[(iodoacetyl)amino]stilbene-2,2′-disulfonic acid in the presence or absence of the detergent Triton (TX-100). Mitochondrial proteins were analyzed by SDS–PAGE and autoradiography. Bands corresponding to the protected (correctly inserted) protein are indicated with asterisks.
Mislocalization of MOM TA proteins in ∆spf1 cells is not caused by an overactive GET pathway
The aforementioned results suggest that neither efficiency of insertion into MOM nor overall Fis1 levels were changed in ∆spf1 cells. Therefore the change in distribution observed in vivo is most likely a result of overinsertion into the ER membranes. Such an increase in ER insertion could, in theory, stem from up-regulation or loss of specificity of the GET pathway, which is responsible for insertion of secretory pathway TA proteins into the ER membranes (Schuldiner et al., 2008; Jonikas et al., 2009). However, we found that Get3 steady-state levels were not altered in ∆spf1 cells (Figure 4A). Moreover, overexpression of GET3 in control cells did not cause mislocalization of mCherry-Fis1 into ER membranes (Figure 4B). Most importantly, deletion of GET3 on the background of ∆spf1 did not suppress the mislocalization of MOM TA proteins to the ER (Figure 4C). We conclude that the insertion of MOM TA proteins into ER membranes in ∆spf1 cells is independent of the GET machinery.
FIGURE 4:

The mislocalization of MOM TA proteins in ∆spf1 cells is not caused by the GET pathway. (A) A plasmid encoding mCherry-Fis1 was introduced into control, ∆spf1, and control overexpressing Get-3 hemagglutinin (Get3HA↑) cells. Whole-cell lysates were analyzed by SDS–PAGE and immunodecoration with antibodies against Get3 and Hxk1 (as a loading control). The bands corresponding to Get3 and Get3-HA are indicated. (B) All three cell types were grown at 30°C to mid–log phase and analyzed by fluorescence microscopy. Arrowheads indicate ER structures stained in ∆spf1 cells. (C) A plasmid encoding GFP-Gem1 was introduced into either ∆spf1 or ∆spf∆get3 cells. Transformed cells were grown at 30°C to mid–log phase and analyzed by fluorescence microscopy.
Reduced ergosterol levels in ∆spf1 cells are sufficient to cause ER localization of MOM TA proteins
We next tested whether deletion of SPF1 caused other cellular changes that could account for the difference in MOM TA protein targeting. Even though Spf1 has no known cellular function, it has been previously shown that its deletion causes severe misregulation of cellular Ca2+ homeostasis that activates the calcineurin pathway (Cronin et al., 2002). However, we found that TA protein insertion into the MOM was not affected by extracellular calcium levels, the pH of the medium, or constitutive activation of the calcineurin pathway (Figure S4). Along the same line, deleting the major Golgi membrane Ca2+ P-type ATPase PMR1 did not cause any mislocalization of mCherry-Fis1 (unpublished data). It therefore appears that altered Ca2+ levels are not the direct cause for mislocalization of MOM TA proteins.
We therefore wondered what other effects ∆spf1 could have on organelle membranes. We have previously shown in vitro that translocation of both MOM TA proteins and signal-anchored proteins is influenced by ergosterol levels (Kemper et al., 2008; Merklinger et al., 2012). We therefore monitored ergosterol levels in mitochondria and ER-derived microsomes from both control and ∆spf1 cells. In agreement with previous reports (Zinser et al., 1991; Schneiter et al., 1999), we found that mitochondrial ergosterol levels in control cells were lower than those in the ER (p value = 0.04). Remarkably, in ∆spf1 cells, ergosterol content of both compartments did not show any significant difference (Figure 5A). These results suggest that the equal sterol content in the membranes of both compartments may underlie the nonspecific targeting of MOM TA proteins.
FIGURE 5:
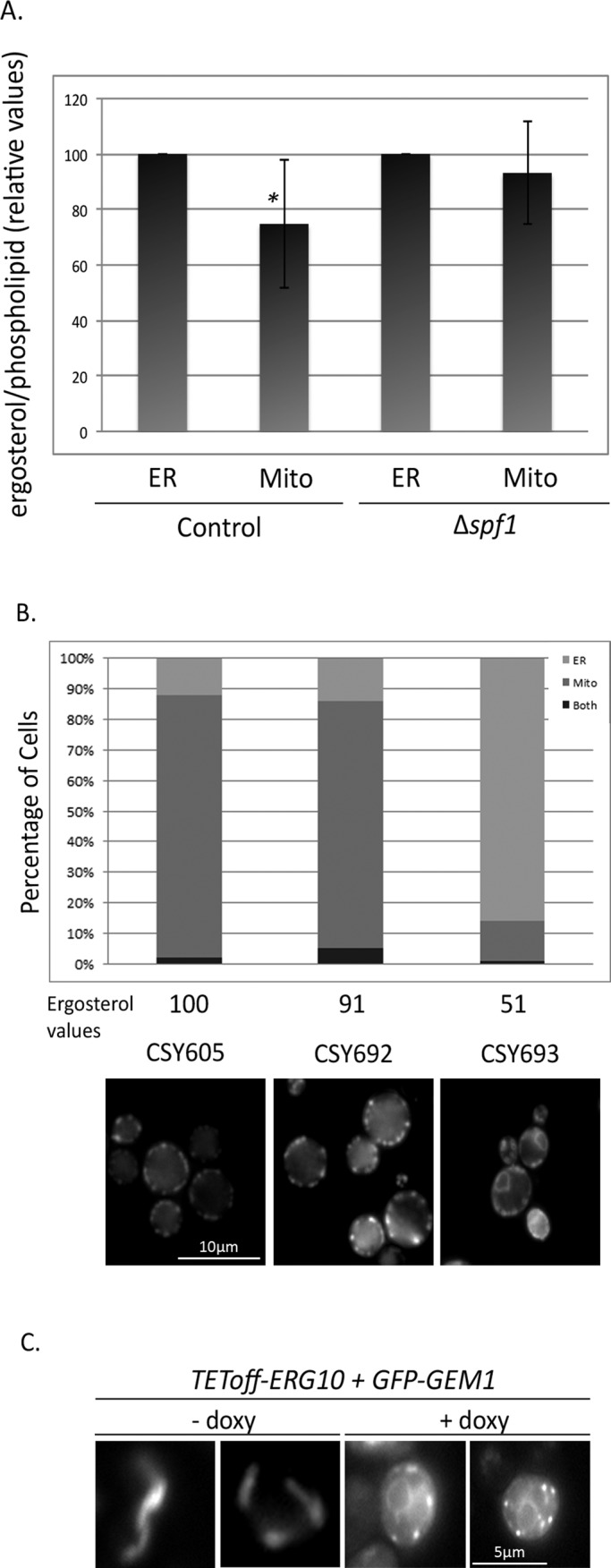
Reduced ergosterol levels in ∆spf1 cells are sufficient to cause ER localization of MOM TA proteins. (A) Lipids were extracted from mitochondria and microsomes isolated from control and ∆spf1 cells. Equal amounts of lipids (according to phospholipid determination) were analyzed for their ergosterol levels by gas chromatography/mass spectrometry. The results represent six independent experiments. The asterisk represents statistical significance (p < 0.05). (B) mCherry-Fis1–expressing strains engineered to have reduced REV (REV as measured by Babiskin and Smolke [2011]) denoted as “Ergosterol values”) were grown for 36 h and analyzed by fluorescence microscopy. Cells (n = 100) were monitored and scored as having either mitochondrial or ER signal, depending on the most abundant signal. (C) Strains with a TEToff-repressible promoter driving ERG10 and expressing a galactose-inducible GFP-GEM1 were visualized following 5 h of doxycycline-mediated repression of ERG10 expression, which was followed by an additional 6 h in both doxycycline and galactose to induce expression of GFP-GEM1.
To test whether ergosterol levels are a major determinant of membrane targeting specificity in vivo, we used two approaches. First, we utilized a set of engineered strains harboring various alleles of the essential ERG9 gene. These strains express an increasingly enhanced RNA degradation signal, causing correlated reduction in the levels of ERG9 that in turn cause an associated reduction in cellular amounts of ergosterol (Babiskin and Smolke, 2011). We used these strains to test the effects of dialing down sterols on MOM TA protein insertion. When we visualized the localization of Fis1 on the entire spectrum of mutants, we saw that only in the strain with the lowest relative ergosterol values (REV; CSY693; Babiskin and Smolke, 2011) was there a major shift in the distribution of the protein to ER membranes (representative strains are shown in Figure 5B). Second, we created a strain expressing GFP-Gem1 under control of a galactose-inducible promoter (GAL1p-GFP-GEM1) on the background of a strain carrying a repressible promoter (TEToff) driving expression of the first enzyme in sterol biosynthesis, the essential ERG10 gene (Mnaimneh et al., 2004). Turning on expression of GFP-GEM1 after having shut down expression of ERG10 caused relocalization of the MOM TA protein to ER membranes (Figure 5C). Taken together, these experiments support the notion that severe reduction in ergosterol levels is sufficient to cause a shift in membrane targeting. Based on our findings, we propose that Spf1 is required to maintain the normal lipid composition of intracellular compartments and that in its absence, mistargeting of MOM TA proteins may occur by spontaneous insertion into the ER.
DISCUSSION
TA proteins form a unique group of membrane proteins that completely rely on posttranslational import for their insertion into either mitochondrial or ER membranes. This is a result of their single TMD existing at the most C-terminal region, the TA leaving the ribosome only upon completion of translation (Borgese et al., 2003b). Recently, the cellular machinery that inserts most of the ER-bound TA proteins has been discovered in both yeast and mammals (Stefanovic and Hegde, 2007; Schuldiner et al., 2008; Jonikas et al., 2009; Favaloro et al., 2010). However, dedicated machinery for the biogenesis of mitochondrial TA proteins has not been identified.
By systematically screening for genes that take part in the biogenesis of the MOM TA protein Gem1, we could not find a single deletion strain that displayed accumulation of the protein in a cytosolic form or in which the protein could not be detected. These would be the phenotypes expected from mutants of dedicated insertion machinery, in which a preinserted form either accumulates in the cytosol or is rapidly degraded. Genetic screens have their limitations and, at least hypothetically, our results do not necessarily imply that no such factors exist. Our inability to find such a factor could potentially stem from a redundancy of the protein required for insertion (a single mutation would not demonstrate a phenotype robust enough to be detected). Another theoretical explanation is that if such machinery is essential (although none of the mitochondrial TA proteins is an essential one, and the dedicated ER machinery is also not essential), then the hypomorphic alleles of essential genes used in the screen might not have been compromised enough to induce a detectable phenotype. Finally, the mutant might not have been represented in our library, or we might not have screened under conditions that require such protein machinery (for example, GET complex–dependent insertion of ER TA proteins seems to be required only following the diauxic shift; Schuldiner et al., 2008). To rule out these concerns, increase our coverage, and verify our results, we performed our screen twice with two different types of reporter proteins (either the TMD of Gem1 alone or the entire protein). Moreover, to minimize loss of strains due to the handling history of the library, we carried out the two screens with yeast deletion libraries taken from different sources. Finally, the screens were performed under two very different growth conditions, either on a semifermentable carbon source (galactose) in non–logarithmic growth phase or on a fully fermentable carbon source (glucose) during mid–logarithmic growth. Taking into consideration our extensive screens and the fact that previous work in vitro did not identify a proteinaceous component essential for TA protein insertion into the MOM (Setoguchi et al., 2006; Kemper et al., 2008), we suggest that MOM TA protein insertion in yeast most probably does not require dedicated proteins. It may be that in higher eukaryotes, in which many more mitochondrial TA proteins exist (Kalbfleisch et al., 2007), a dedicated system might have evolved, but it has eluded identification to date (Setoguchi et al., 2006).
Our screens did uncover a single protein, Spf1, that affects insertion of MOM TA proteins that are not subunits of the TOM complex, namely Gem1 and Fis1. It is currently unclear whether Spf1 plays a similarly important role in the biogenesis of Tom5, Tom6, and Tom7. The latter proteins may follow a different import pathway as several MOM proteins such as Mim1, Mas37/Sam37 and the Tom receptors were reported to be involved in their biogenesis (Dietmeier et al., 1997; Stojanovski et al., 2007; Thornton et al., 2010). However, cofactors for correct biogenesis do not exclude a universal mechanism for the membrane insertion process. It could be that the small TOM components simply require additional proteins for downstream steps, such as assembly into the TOM complex. Although triple deletion in Neurospora crassa of TOM5, TOM6, and TOM7 and double deletion in yeast of TOM5 and additional TOM components were reported to be lethal (Dietmeier et al., 1997; Sherman et al., 2005), the lack of obvious growth and morphology phenotypes in ∆spf1 cells does not contradict a potential requirement for functional Spf1 in the biogenesis of these proteins. Under logarithmic growth conditions, ∆spf1 mitochondria still display about half the normal Fis1 levels. Hence, these cells most likely still harbor adequate amounts of any TA protein that is affected in this background. It is anticipated that even reduced mitochondrial amounts of small TOM proteins are sufficient to maintain normal function of the TOM complex, which in turn can support normal growth and mitochondrial morphology.
Spf1 is the first yeast protein ever shown to affect insertion of MOM TA proteins. However, since the exact biochemical role of Spf1 is not yet known, it is currently impossible to determine the precise mechanism by which its deletion affects insertion. The fact that Spf1 resides in ER membranes suggests that its effect is indirect. Because ∆spf1 cells display loss of differential sterol levels between MOM and ER membranes, and reduced ergosterol levels caused mistargeting in vivo, our findings strongly support the notion that Spf1, either directly or indirectly, regulates ergosterol levels and this in turn affects MOM TA protein insertion.
It is well documented that the ergosterol content of cellular membranes is tightly regulated, and even minor changes can result in major effects on cellular processes (Stuven et al., 2003). Ergosterol levels in the ER are usually higher than those in mitochondria, and it was shown that ergosterol increases the rigidity of a membrane. The ER membrane should therefore consistently be an unfavorable target for spontaneous insertion of MOM TA proteins. In support of the role of sterols in insertion of TA proteins into membranes, it has been shown that insertion of MOM TA proteins into liposomes is inhibited by increased levels of sterols in the membrane (Kemper et al., 2008). This is also the case for mitochondrial signal-anchored proteins, such as OM45 (Merklinger et al., 2012). Conversely, when ER-derived microsomes were depleted of sterols, they became more rapidly targeted by the ER TA protein cytB5 in a manner that did not require protein factors nor energy (Brambillasca et al., 2005). Taken together, the inability to identify a proteinaceous component as being required for MOM TA protein insertion and the idea that sterol levels have a major role in determining insertion specificity strongly suggest that the lipid composition of a membrane can be the determinant guiding MOM TA protein distribution in yeast.
The hypothesis that sterol levels themselves are responsible for insertion specificity raises the question of why none of the mutants in the biosynthetic pathway of ergosterol came up in our screen as affecting MOM TA proteins insertion. One possible answer is that the majority of proteins in this pathway are essential, and it may be that their DAmP alleles did not cause a reduction in sterol levels considerable enough to cause mislocalization that could pass our threshold of detection. In support of this assumption, only when we visualized Fis1 insertion on a highly compromised ERG9 mutant (Babiskin and Smolke, 2011) or Gem1 insertion on a strongly repressed ERG10 strain did we find that they phenocopied the mislocalization observed in ∆spf1 cells.
The idea that sterol levels have such a strong regulatory role in protein insertion raises new questions of how TA protein insertion efficiency, membrane specificity, and regulation of targeting can be achieved. One way to achieve both efficiency and specificity would be to target the mRNA encoding TA proteins before translation to mitochondrial membranes. Indeed, it has been shown that the mRNA of mitochondrial proteins is often localized to the surface of mitochondria (Marc et al., 2002; Haim et al., 2007; Saint-Georges et al., 2008; Eliyahu et al., 2010). Another pathway to achieve specificity would be selective degradation of proteins inserted into the wrong target membranes; however, there is no evidence in support of this. Finally, regulation of insertion could simply be dependent on the membrane composition of different organelles, which would imply that a regulatory network of proteins exists to maintain accurate levels of sterols in each membrane. Future investigations can address the mechanism of such a network.
Organization of eukaryotic cells into intracellular organelles allows the creation of distinct biochemical environments. By controlling the characteristics (e.g., fluidity, thickness, and charge) of their membranes, cells can create unique environments into which proteins can insert or bind. Indeed, understanding how the composition of boundary membranes controls the shape, size, and activity of the respective organelles is still a major question in cell biology. Our work demonstrates an important aspect of how organelle identity and function can be affected by changes in the lipid composition of its membranes.
MATERIALS AND METHODS
Media and growth conditions
Yeast cells were grown on regular yeast media (either synthetic media with dextrose [SD] or yeast extract, peptone, and dextrose [YEPD]) plates supplemented with hygromycin (300 μg/ml; AG Scientific, San Diego, CA), G418 (200 μg/ml; Calbiochem, San Diego, CA), or nourseothricin (200 μg/ml; Werner BioAgents, Jena, Germany) when needed for selection. In cases in which G418 was required in an SD-based medium, yeast nitrogen base without ammonium sulfate (Conda Pronadisa, Madrid, Spain) was added and supplemented with monosodium glutamate (Sigma-Aldrich, St. Louis, MO). When necessary, dextrose was replaced by galactose (2%; Amresco, Solon, OH) or raffinose (2%; Amresco) as a carbon source. For the TETOff experiments, doxycycline (Sigma-Aldrich) was used at 15 μg/ml in YEP-based medium.
SGA
SGA was performed as previously described (Tong et al., 2001, 2004; Tong and Boone, 2006; Cohen and Schuldiner, 2011). For more details, see the Supplemental Material.
Fluorescence microscopy
High-throughput microscopy.
High-throughput screens were performed with a previously described system (Cohen and Schuldiner, 2011). For more details, see the Supplemental Material.
Manual microscopy.
The microscopy for follow-up analysis was performed using an Olympus IX71 microscope controlled by DeltaVision SoftWoRx 3.5.1 software with either 60× or 100× oil-immersion lenses. Images were captured by a Photometrics (Tucson, AZ) Coolsnap HQ camera with excitation at 490/20 nm and emission at 528/38 nm (GFP) or excitation at 555/28 nm and emission at 617/73 nm (mCherry). Images were transferred to Adobe Photoshop CS2 for slight contrast and brightness adjustments (San Jose, CA).
Purification of mitochondria and microsomes
Mitochondria were isolated from yeast cells grown on lactate-containing medium by differential centrifugation as previously described (Daum et al., 1982). For highly pure mitochondria, a Percoll gradient purification was performed. Isolated mitochondria were layered on top of a self-forming gradient (25% Percoll in an SEM buffer [250 mM sucrose, 1 mM EDTA, and 10 mM MOPS, pH 7.2]) and centrifuged (80,000 × g, 45 min, 4°C). The mitochondrial fraction from the lower third of the gradient was collected, resuspended in 30 ml SEM buffer, and reisolated by centrifugation (15,000 × g, 15 min, 4°C).
Microsomes were isolated from yeast cells by differential centrifugation. After the first mitochondrial pellet was obtained, the supernatant was centrifuged again (15,000 × g, 15 min, 4°C) to avoid contamination with mitochondrial elements. The postmitochondrial fraction was subjected to centrifugation (100,000 × g, 1 h, 4°C), and the pelleted microsomes were resuspended in SEM buffer.
Isolation and measurement of lipids
Mitochondrial and microsomal lipids were extracted and subjected to phosphate analysis as previously described (Brugger et al., 2000). Mass spectrometry analysis was performed in positive-ion mode on a quadrupole time-of-flight mass spectrometer (QStar Elite; Applied Biosystems, Bedford, MA). Ergosterol quantification was performed as previously described (Ejsing et al., 2009). Prior to extraction, stigmasta-5,7,22-trienol was added as a standard. Each sample was normalized by total phospholipid content. For each strain, ergosterol/phospholipid content in microsomes was denoted as 100%, and values for mitochondria from the same strain are given as percent relative to microsomes.
In vitro protein import and membrane insertion assay
Radiolabeled precursor proteins were synthesized in rabbit reticulocyte lysate in the presence of [35S]methionine (Perkin Elmer-Cetus, Waltham, MA) after in vitro transcription by SP6 polymerase from pGEM4 vectors (Promega, Madison, WI) encoding Fis1. In vitro import experiments were performed as previously reported (Kemper et al., 2008).
Supplementary Material
Acknowledgments
We gratefully acknowledge A. Azem for helpful discussions. We thank B. Schwappach, J. Nunnari, C. Loewen, J. Audhya, and T. Edlind for plasmids; K. Rehn, J. Johänning, and E. Kracker for technical support; and T. Sachsenheimer and D. Wistuba for help with the ergosterol determination. We also thank A. Futerman for reagents, M. Breker for setting up the ScanR microcopy system, and Y. Cohen and T. Ast for assistance. We thank I. Yofe, T. Ast, O. Schuldiner, and Y. Cohen for critical reading of the manuscript. This work was supported by: the Deutsche Forschungsgemeinschaft (TP A1 SFB/TRR 83 to B.B.; RA 1048/4-1 and RA 1048/5-1 to D.R.); the CellNetworks Cluster of Excellence (ECX81 to B.B.); the Feinberg Foundation Visiting Faculty Program (D.R.); the Marie Curie Re-integration grant (program FP7 of the EU 239224 to the Schuldiner laboratory); an EMBO short-term fellowship (I.F.); and the Israeli Science Foundation (ISF) Legacy Heritage fund (grant # 1995/08 to Y.H.). The robotic setup was purchased through a career development award from the Human Frontiers Science Program (CDA0006/2008-C), as well as the above-mentioned ISF grant, the generous donation of the J&R Foundation, and the estate of Lela London.
Abbreviations used:
- DAmP
decreased abundance by mRNA perturbation
- ER
endoplasmic reticulum
- GFP-Gem1
green fluorescent protein-Gem1
- MOM
mitochondrial outer membrane
- MTS
mitochondrial targeting sequence
- REV
relative ergosterol values
- RFP
red fluorescent protein
- SGA
synthetic genetic array
- TA
tail-anchored
- TMD
transmembrane domain
- TOM
translocase of the outer membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-12-0994) on August 23, 2012.
*These authors contributed equally to this work.
REFERENCES
- Ando A, Suzuki C. Cooperative function of the CHD5-like protein Mdm39p with a P-type ATPase Spf1p in the maintenance of ER homeostasis in Saccharomyces cerevisiae. Mol Genet Genomics. 2005;273:497–506. doi: 10.1007/s00438-005-1153-6. [DOI] [PubMed] [Google Scholar]
- Babiskin AH, Smolke CD. A synthetic library of RNA control modules for predictable tuning of gene expression in yeast. Mol Syst Biol. 2011;7:471. doi: 10.1038/msb.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz T, Egan B, Silver PA, Hofmann K, Lithgow T. Bipartite signals mediate subcellular targeting of tail-anchored membrane proteins in Saccharomyces cerevisiae. J Biol Chem. 2003;278:8219–8223. doi: 10.1074/jbc.M212725200. [DOI] [PubMed] [Google Scholar]
- Borgese N, Brambillasca S, Colombo S. How tails guide tail-anchored proteins to their destinations. Curr Opin Cell Biol. 2007;19:368–375. doi: 10.1016/j.ceb.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Borgese N, Brambillasca S, Soffientini P, Yabal M, Makarow M. Biogenesis of tail-anchored proteins. Biochem Soc Trans. 2003a;31:1238–1242. doi: 10.1042/bst0311238. [DOI] [PubMed] [Google Scholar]
- Borgese N, Colombo S, Pedrazzini E. The tale of tail-anchored proteins: coming from the cytosol and looking for a membrane. J Cell Biol. 2003b;161:1013–1019. doi: 10.1083/jcb.200303069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Brambillasca S, Yabal M, Soffientini P, Stefanovic S, Makarow M, Hegde RS, Borgese N. Transmembrane topogenesis of a tail-anchored protein is modulated by membrane lipid composition. EMBO J. 2005;24:2533–2542. doi: 10.1038/sj.emboj.7600730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, Newman HW, Braun S, Madhani HD, Krogan NJ, Weissman JS. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B, Sandhoff R, Wegehingel S, Gorgas K, Malsam J, Helms JB, Lehmann WD, Nickel W, Wieland FT. Evidence for segregation of sphingomyelin and cholesterol during formation of COPI-coated vesicles. J Cell Biol. 2000;151:507–518. doi: 10.1083/jcb.151.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Y, Schuldiner M. Cell biology in the 21st century—the revolution of high throughput microscopy methods. In: Emili A, Cagney G, editors. In: Network Biology: Methods and Applications (Methods in Molecular Biology) Totowa, NJ: Humana Press; 2011. [Google Scholar]
- Cronin SR, Khoury A, Ferry DK, Hampton RY. Regulation of HMG-CoA reductase degradation requires the P-type ATPase Cod1p/Spf1p. J Cell Biol. 2000;148:915–924. doi: 10.1083/jcb.148.5.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SR, Rao R, Hampton RY. Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J Cell Biol. 2002;157:1017–1028. doi: 10.1083/jcb.200203052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G, Gasser SM, Schatz G. Import of proteins into mitochondria. Energy-dependent, two-step processing of the intermembrane space enzyme cytochrome b2 by isolated yeast mitochondria. J Biol Chem. 1982;257:13075–13080. [PubMed] [Google Scholar]
- Dietmeier K, Honlinger A, Bomer U, Dekker PJ, Eckerskorn C, Lottspeich F, Kubrich M, Pfanner N. Tom5 functionally links mitochondrial preprotein receptors to the general import pore. Nature. 1997;388:195–200. doi: 10.1038/40663. [DOI] [PubMed] [Google Scholar]
- Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, Simons K, Shevchenko A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci USA. 2009;106:2136–2141. doi: 10.1073/pnas.0811700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliyahu E, Pnueli L, Melamed D, Scherrer T, Gerber AP, Pines O, Rapaport D, Arava Y. Tom20 mediates localization of mRNAs to mitochondria in a translation-dependent manner. Mol Cell Biol. 2010;30:284–294. doi: 10.1128/MCB.00651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro V, Spasic M, Schwappach B, Dobberstein B. Distinct targeting pathways for the membrane insertion of tail-anchored (TA) proteins. J Cell Sci. 2008;121:1832–1840. doi: 10.1242/jcs.020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro V, Vilardi F, Schlecht R, Mayer MP, Dobberstein B. Asna1/TRC40-mediated membrane insertion of tail-anchored proteins. J Cell Sci. 2010;123:1522–1530. doi: 10.1242/jcs.055970. [DOI] [PubMed] [Google Scholar]
- Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol. 2004;167:87–98. doi: 10.1083/jcb.200405100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Habib SJ, Vasiljev A, Neupert W, Rapaport D. Multiple functions of tail-anchor domains of mitochondrial outer membrane proteins. FEBS Lett. 2003;555:511–515. doi: 10.1016/s0014-5793(03)01325-5. [DOI] [PubMed] [Google Scholar]
- Haim L, Zipor G, Aronov S, Gerst JE. A genomic integration method to visualize localization of endogenous mRNAs in living yeast. Nat Methods. 2007;4:409–412. doi: 10.1038/nmeth1040. [DOI] [PubMed] [Google Scholar]
- Horie C, Suzuki H, Sakaguchi M, Mihara K. Characterization of signal that directs C-tail–anchored proteins to mammalian mitochondrial outer membrane. Mol Biol Cell. 2002;13:1615–1625. doi: 10.1091/mbc.01-12-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbfleisch T, Cambon A, Wattenberg BW. A bioinformatics approach to identifying tail-anchored proteins in the human genome. Traffic. 2007;8:1687–1694. doi: 10.1111/j.1600-0854.2007.00661.x. [DOI] [PubMed] [Google Scholar]
- Kemper C, Habib SJ, Engl G, Heckmeyer P, Dimmer KS, Rapaport D. Integration of tail-anchored proteins into the mitochondrial outer membrane does not require any known import components. J Cell Sci. 2008;121:1990–1998. doi: 10.1242/jcs.024034. [DOI] [PubMed] [Google Scholar]
- Marc P, Margeot A, Devaux F, Blugeon C, Corral-Debrinski M, Jacq C. Genome-wide analysis of mRNAs targeted to yeast mitochondria. EMBO Rep. 2002;3:159–164. doi: 10.1093/embo-reports/kvf025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meineke B, Engl G, Kemper C, Vasiljev-Neumeyer A, Paulitschke H, Rapaport D. The outer membrane form of the mitochondrial protein Mcr1 follows a TOM-independent membrane insertion pathway. FEBS Lett. 2008;582:855–860. doi: 10.1016/j.febslet.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Merklinger E, Gofman Y, Kedrov A, Driessen AJ, Ben-Tal N, Shai Y, Rapaport D. Membrane integration of a mitochondrial signal-anchored protein does not require additional proteinaceous factors. Biochem J. 2012;442:381–389. doi: 10.1042/BJ20111363. [DOI] [PubMed] [Google Scholar]
- Mnaimneh S, et al. Exploration of essential gene functions via titratable promoter alleles. Cell. 2004;118:31–44. doi: 10.1016/j.cell.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabu C, Schmid V, Schwappach B, High S. Biogenesis of tail-anchored proteins: the beginning for the end? J Cell Sci. 2009;122:3605–3612. doi: 10.1242/jcs.041210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport D. Finding the right organelle. Targeting signals in mitochondrial outer-membrane proteins. EMBO Rep. 2003;4:948–952. doi: 10.1038/sj.embor.embor937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Georges Y, Garcia M, Delaveau T, Jourdren L, Le Crom S, Lemoine S, Tanty V, Devaux F, Jacq C. Yeast mitochondrial biogenesis: a role for the PUF RNA-binding protein Puf3p in mRNA localization. PLoS One. 2008;3:e2293. doi: 10.1371/journal.pone.0002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiter R, et al. Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J Cell Biol. 1999;146:741–754. doi: 10.1083/jcb.146.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, Weissman JS. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell. 2008;134:634–645. doi: 10.1016/j.cell.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Schwappach B, Weissman JS. In: Encyclopedia of Life Sciences. Hoboken, NJ: John Wiley & Sons. http://onlinelibrary.wiley.com/doi/10.1002/9780470015902.a0021876/full; 2010. Membrane insertion of tail anchored proteins. [Google Scholar]
- Setoguchi K, Otera H, Mihara K. Cytosolic factor- and TOM-independent import of C-tail-anchored mitochondrial outer membrane proteins. EMBO J. 2006;25:5635–5647. doi: 10.1038/sj.emboj.7601438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman EL, Go NE, Nargang FE. Functions of the small proteins in the TOM complex of Neurospora crassa. Mol Biol Cell. 2005;16:4172–4182. doi: 10.1091/mbc.E05-03-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Hegde RS. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell. 2007;128:1147–1159. doi: 10.1016/j.cell.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Stojanovski D, Guiard B, Kozjak-Pavlovic V, Pfanner N, Meisinger C. Alternative function for the mitochondrial SAM complex in biogenesis of α-helical TOM proteins. J Cell Biol. 2007;179:881–893. doi: 10.1083/jcb.200706043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuven E, Porat A, Shimron F, Fass E, Kaloyanova D, Brugger B, Wieland FT, Elazar Z, Helms JB. Intra-Golgi protein transport depends on a cholesterol balance in the lipid membrane. J Biol Chem. 2003;278:53112–53122. doi: 10.1074/jbc.M300402200. [DOI] [PubMed] [Google Scholar]
- Suzuki C. Immunochemical and mutational analyses of P-type ATPase Spf1p involved in the yeast secretory pathway. Biosci Biotechnol Biochem. 2001;65:2405–2411. doi: 10.1271/bbb.65.2405. [DOI] [PubMed] [Google Scholar]
- Suzuki C, Shimma YI. P-type ATPase spf1 mutants show a novel resistance mechanism for the killer toxin SMKT. Mol Microbiol. 1999;32:813–823. doi: 10.1046/j.1365-2958.1999.01400.x. [DOI] [PubMed] [Google Scholar]
- Thornton N, Stroud DA, Milenkovic D, Guiard B, Pfanner N, Becker T. Two modular forms of the mitochondrial sorting and assembly machinery are involved in biogenesis of α-helical outer membrane proteins. J Mol Biol. 2010;396:540–549. doi: 10.1016/j.jmb.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Tipper DJ, Harley CA. Yeast genes controlling responses to topogenic signals in a model transmembrane protein. Mol Biol Cell. 2002;13:1158–1174. doi: 10.1091/mbc.01-10-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Vashist S, Frank CG, Jakob CA, Ng DT. Two distinctly localized P-type ATPases collaborate to maintain organelle homeostasis required for glycoprotein processing and quality control. Mol Biol Cell. 2002;13:3955–3966. doi: 10.1091/mbc.02-06-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther DM, Rapaport D. Biogenesis of mitochondrial outer membrane proteins. Biochim Biophys Acta. 2009;1793:42–51. doi: 10.1016/j.bbamcr.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Wang F, Brown EC, Mak G, Zhuang J, Denic V. A chaperone cascade sorts proteins for posttranslational membrane insertion into the endoplasmic reticulum. Mol Cell. 2010;40:159–171. doi: 10.1016/j.molcel.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991;173:2026–2034. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.