

The septin-associated kinase Gin4 is required for the localization and activation of Bnr1, and the septin Shs1 is essential for Bnr1 activation. The loss of Gin4 or Shs1 phenocopies the loss of Bnr1; these defects are suppressed by constitutive activation of Bnr1. The data reveal novel regulatory links between the actin and septin cytoskeletons.
Abstract
Formin-family proteins promote the assembly of linear actin filaments and are required to generate cellular actin structures, such as actin stress fibers and the cytokinetic actomyosin contractile ring. Many formin proteins are regulated by an autoinhibition mechanism involving intramolecular binding of a Diaphanous inhibitory domain and a Diaphanous autoregulatory domain. However, the activation mechanism for these Diaphanous-related formins (DRFs) is not completely understood. Although small GTPases play an important role in relieving autoinhibition, other factors likely contribute. Here we describe a requirement for the septin Shs1 and the septin-associated kinase Gin4 for the localization and in vivo activity of the budding yeast DRF Bnr1. In budding yeast strains in which the other formin, Bni1, is conditionally inactivated, the loss of Gin4 or Shs1 results in the loss of actin cables and cell death, similar to the loss of Bnr1. The defects in these strains can be suppressed by constitutive activation of Bnr1. Gin4 is involved in both the localization and activation of Bnr1, whereas the septin Shs1 is required for Bnr1 activation but not its localization. Gin4 promotes the activity of Bnr1 independently of the Gin4 kinase activity, and Gin4 lacking its kinase domain binds to the critical localization region of Bnr1. These data reveal novel regulatory links between the actin and septin cytoskeletons.
INTRODUCTION
The formin family of proteins plays a major role in regulating the actin cytoskeleton and is required for diverse cellular processes including cytokinesis, polarized growth, filopodia assembly, and cell–cell adhesion. Formins promote the assembly of linear actin filaments through barbed-end binding of their formin homology 1 and 2 (FH1 and FH2) domains (Faix and Grosse, 2006; Goode and Eck, 2007). Diaphanous-related formins (DRFs) are typically regulated by autoinhibition, by which their N-terminal Diaphanous inhibitory domain (DID) binds their C-terminal Diaphanous autoregulatory domain (DAD; Li and Higgs, 2003; Higgs, 2005). Current models suggest that autoinhibition can be relieved by binding of Rho-family GTPases at the Rho-binding domain (RBD), which is adjacent to the DID domain (Li and Higgs, 2005; Seth et al., 2006). However, in vitro and in vivo data suggest that Rho-GTPases are not sufficient for the complete activation of formins (Li and Higgs, 2003; Martin et al., 2007). Other components that cooperate with Rho to activate formins have been identified in vivo. For example, the fission yeast formin For3 requires both Cdc42 and Bud6 for complete activity in vivo (Martin et al., 2007); mDia2 is activated by Rho kinase in combination with RhoA (Staus et al., 2011). In contrast, some formins are autoinhibited but are activated by proteins other than Rho-GTPases; for example, Daam1 is activated, at least in part, by the binding of Dishevelled to the DAD domain (Liu et al., 2008). Therefore knowledge of how DRFs are regulated is incomplete.
Formins not only are important for regulating actin assembly in cells, but they are also implicated in other aspects of cytoskeletal dynamics (Chesarone et al., 2010; Mao, 2011). Some formins bind and stabilize microtubules and are required for centrosome positioning in polarized cells (Bartolini et al., 2008; Bartolini and Gundersen, 2011; Ang et al., 2010). In budding yeast, genetic experiments suggest that formins are required for the normal assembly of the septin scaffold at the bud neck (Kadota et al., 2004; Gladfelter et al., 2005). Septins are a highly conserved family of GTPases required for cytokinesis and other processes in fungal and animal cells (Weirich et al., 2008). In budding yeast, septins create a scaffold at the bud neck for the recruitment of an assortment of proteins involved in cytokinesis and polarized growth (Faty et al., 2002; McMurray and Thorner, 2009). In addition to their scaffolding functions, septins establish membrane diffusion barriers and thus define specific membrane domains (Mostowy and Cossart, 2012). Although increasing evidence suggests intimate associations between the actin and septin cytoskeletons, the molecular basis for these interactions and their functional role is not well understood.
The budding yeast Saccharomyces cerevisiae is a useful experimental system in which to study formins and formin–septin interactions. Unlike animal cells, which contain numerous formin isoforms, budding yeast has only two formins, Bni1 and Bnr1 (Goode and Eck, 2007; Campellone and Welch, 2010; Chesarone et al., 2010). Our lab and others developed conditional strains in which formin function can be rapidly inactivated (within ∼5 min). Mutant analysis showed that budding yeast formins are required for actin cable assembly and, therefore, the control of polarized morphogenesis (Evangelista et al., 2002; Pruyne et al., 2002; Sagot et al., 2002a, b). Budding yeast formins are also required for spindle positioning (via their role in actin cable assembly; Lee et al., 1999) and the assembly of the cytokinetic actomyosin ring (Tolliday et al., 2002; Pruyne et al., 2004a; Yoshida et al., 2006). Although individually Bni1 and Bnr1 are not essential, deletion of both formins is lethal (Kohno et al., 1996; Imamura et al., 1997; Ozaki-Kuroda et al., 2001). Two pieces of evidence suggest that Bnr1 is regulated by autoinhibition. First, the lethality of a rho3Δ rho4Δ double-deletion strain can be rescued by deletion of the RBD of Bnr1 (Dong et al., 2003). Second, deletion of the DAD domain of Bnr1 generates an actin cable defect, consistent with the generation of excess actin filaments (Chesarone et al., 2009). Although the RBD is clearly required to inhibit Bnr1, the identification of the relevant Rho-type GTPase is less clear. There is evidence to suggest that Bnr1 interacts with Rho4-GTP (Kamei et al., 1998); however, there is no obvious growth or actin assembly defect in a rho4Δ bni1Δ strain (Dong et al., 2003; our unpublished results). Taken together, these results suggest that Bnr1 is likely autoinhibited, but the relevant Rho-GTPase has not been identified, and it is not known whether other regulators are required. Therefore we investigated other proteins that are required for the assembly of actin cables by Bnr1.
Bnr1 localizes to the bud neck at the onset of bud emergence and remains there until cytokinesis (Kikyo et al., 1999; Buttery et al., 2007). This bud neck localization is entirely dependent on septins (Pruyne et al., 2004a; Gao et al., 2010). Recent results showed that Bnr1 has two distinct localization signals that both require an intact septin cytoskeleton: the L1 domain is required for the spatial restriction of Bnr1 to the mother side of the bud neck (mother-side bias), and the L2 domain is required for the normal cell cycle timing of Bnr1 localization to the bud neck (i.e., its recruitment at bud emergence and loss at septin ring splitting; Gao et al., 2010). However, the binding partners of these domains are unknown; it is also unclear whether Bnr1 interacts directly with septins or indirectly with other components of the septin scaffold, such as the septin-associated kinases Elm1 and Gin4. In general, the relationship between the localization of Bnr1 and its function in the assembly of actin cables is unclear.
Here we present evidence that the septin-associated microtubule-affinity regulating kinase (MARK)/Par1-family kinase Gin4 is required for the localization of Bnr1 to the bud neck, as well as for its ability to promote actin assembly in yeast. A structure–function analysis suggests that Gin4 recruits Bnr1 to the bud neck in a manner that requires the Bnr1 L2 domain. The recruitment of Bnr1 is independent of the Gin4 N-terminal kinase domain, and the Gin4 C-terminus binds the minimal localization domain (N-terminus) of Bnr1. We also define an unexpected role for the septin Shs1 in regulating Bnr1. There are five septins in budding yeast: Cdc3, Cdc10, Cdc11, Cdc12, and Shs1. Shs1 is nonessential; its deletion results in defects in the septin cytoskeleton (Carroll et al., 1998; Garcia et al., 2011). Unlike Gin4, Shs1 is not required for Bnr1 localization. However, in common with the loss of Gin4, the deletion of Shs1 causes a striking defect in Bnr1-dependent actin cable assembly. Taken together, these data define novel regulatory links between the septin and actin cytoskeletons. We speculate that Gin4 and Shs1 may activate Bnr1 by a novel regulatory mechanism.
RESULTS
Partial requirement for the septin-associated proteins Gin4 and Elm1 for the bud neck localization of full-length Bnr1
Bnr1 localizes to the bud neck at the onset of bud emergence with a bias toward the mother side of the bud neck. Bnr1 remains at the bud neck until mitotic exit, when it is dispersed at the time of septin ring splitting (Kikyo et al., 1999; Buttery et al., 2007; Gao et al., 2010). The bud neck localization of Bnr1 is dependent on an intact septin scaffold (Pruyne et al., 2004a). To identify candidate bud neck receptors for Bnr1, we examined strains lacking specific septins (Shs1) or septin-associated proteins (Gin4, Elm1, and Nap1) for defects in Bnr1 localization (Carroll et al., 1998; Longtine et al., 1998a; Gladfelter et al., 2004). Shs1 is the nonessential septin in budding yeast; deletion of Shs1 results in defects in the septin cytoskeleton (Carroll et al., 1998; Garcia et al., 2011). Gin4 and Elm1 are septin-associated kinases that, when deleted, result in varying degrees of septin defects, which are exacerbated at higher temperatures (Longtine et al., 1998a; Bouquin et al., 2000; Gladfelter et al., 2004). Deletion of Nap1, an upstream activator of Gin4 (Altman and Kellogg, 1997), shows aberrant septin staining similar to a gin4Δ strain (Gladfelter et al., 2004).
We compared endogenous level Bnr1–green fluorescent protein (GFP) localization to that of endogenous level Myo1–cyan fluorescent protein (CFP), the type II myosin in budding yeast, which also requires septins for its localization the bud neck. Myo1 specifically depends on Shs1, Bni5, and Iqg1 for localization (Fang et al., 2010). We imaged fixed cells at 24°C (Supplemental Figure S1) or after a temperature shift of 4 h at 37°C. We found that both Elm1 and Gin4 are required for the normal localization of Bnr1. In strains lacking Elm1 or Gin4, the fraction of Myo1-CFP–positive cells with Bnr1-GFP at the bud neck was diminished; this phenotype was more prominent at 37°C (Figure 1, A and B, and Supplemental Figure S1, A and B). By contrast, the localization of Myo1 was unaffected in these strains and conditions (Figure 1, A and B, and Supplemental Figure S1, A and B). The requirement of Elm1 and Gin4 for Bnr1-GFP localization was even more obvious when we measured the background-corrected fluorescence intensity of Bnr1-GFP at the bud neck relative to Myo1-CFP at the bud neck in gin4∆ and elm1∆ strains (Figure 1C). In wild-type cells, the Bnr1-GFP–to–Myo1-CFP fluorescence intensity ratio had a mean ratio of 0.501 ± 0.041 (mean ± SE; n = 83). The gin4∆ and elm1∆ strains exhibited a significantly decreased mean of the fluorescence intensity ratio compared with the wild-type strain (p < 0.0001). The gin4∆ strains showed a mean of 0.036 ± 0.028 (n = 104); elm1Δ strains showed a mean of 0.007 ± 0.029 (n = 100); the differences in the mean fluorescence intensity ratios of the gin4∆ and elm1∆ strains were not statistically significant (p = 0.4787). The negative values observed in the gin4∆ and elm1∆ strains are due to loss of Bnr1 at the neck, with the concomitant increase in the fluorescence intensity of Bnr1 in the cytoplasm (where we measured the background fluorescence intensity). The mean, background-corrected fluorescence intensity for Myo1 was 117.9, 100, and 108 arbitrary units (AU) for wild type, gin4Δ, and elm1Δ, respectively. The mean, background-corrected fluorescence intensity for Bnr1 was 48.8, 1.1, and 2.5 AU for wild type, gin4Δ, and elm1Δ, respectively. Therefore these experiments demonstrate that Bnr1 localization is dependent, in part, on the septin-associated kinases Gin4 and Elm1.
FIGURE 1:
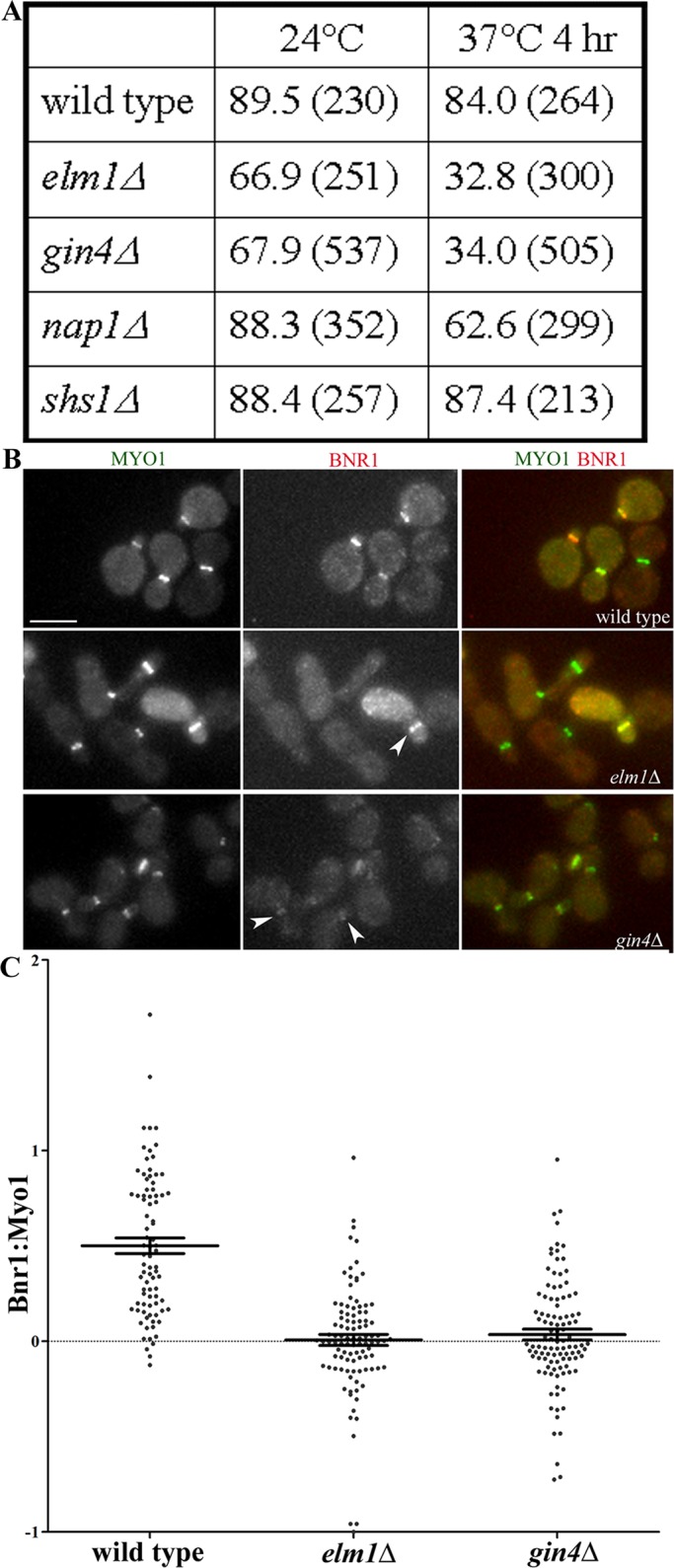
Partial requirement for Gin4 and Elm1 for the localization of full-length Bnr1. (A) The absence of Gin4 or Elm1 disrupts the localization of Bnr1-GFP to the bud neck, unlike the absence of Nap1 and Shs1. Shown is the mean percentage of Myo1-CFP–positive cells with Bnr1-GFP localization at the bud neck for the number of cells counted (indicated in parentheses). Experiments were performed twice with >100 cells per sample. (B) Representative images of Bnr1-GFP and Myo1-CFP in the indicated strains. Cells were fixed after 4 h at 37°C and imaged in three dimensions with five 0.5-μm stacks using the same exposure conditions for all strains. Scale bar, 5 μm. Shown are representative, normalized, maximum-projection images. (C) The ratio of the fluorescence intensity of Bnr1 (YFP) to Myo1 (CFP) was measured (see Materials and Methods for details) in the cells imaged in B. Each point represents the ratio of the background-corrected fluorescence intensity of Bnr1 to Myo1 for one cell; bars, mean ± SE.
A gin4Δ elm1Δ double-deletion strain showed no further decrease in the percentage of cells with Bnr1 localization than either single deletion (Supplemental Figure S1C). This is consistent with published genetic data suggesting that Elm1 and Gin4 collaborate to promote septin function (Gladfelter et al., 2004). The Nap1 protein is believed to be an upstream activator of Gin4 (Altman and Kellogg, 1997); nap1Δ shows aberrant septin staining similar to a gin4Δ strain (Gladfelter et al., 2004). However, in a nap1Δ strain, we observed only a small decrease in Bnr1 localization to the bud neck at either temperature tested (Figure 1A and Supplemental Figure S1, A and B). This suggests that the partial requirement of Gin4 and Elm1 for the localization of Bnr1 is not likely to be solely due to abnormal septins. Moreover, deletion of the nonessential septin Shs1 did not affect Bnr1 localization at any temperature tested, although it is required for normal Myo1 localization (Figure 1A and Supplemental Figure S1, A and B). The steady-state levels of Bnr1-GFP protein were modestly reduced in gin4Δ or elm1Δ strains by comparison with wild-type, nap1Δ, or shs1Δ strains (Supplemental Figure S1B). However, the experiments described later using Bnr1-truncation constructs whose levels are not affected by these gene deletions demonstrate that Gin4 and Elm1 have a specific role in targeting Bnr1 to the bud neck. Taken together, our data suggest that Bnr1 localization to the bud neck specifically involves Elm1 and Gin4.
The Bnr1 L2 domain is required for Gin4-dependent localization
Previously published work defined two separate localization signals within the Bnr1 N-terminus (Gao et al., 2010). Amino acids 1–466, or “L1,” is required for the normal concentration of Bnr1 on the mother side of the bud neck. Amino acids 466–733, or “L2,” is required for the proper timing of bud neck recruitment (at the onset of bud emergence) and dissociation (at the start of cytokinesis/septin splitting; Gao et al., 2010). We independently confirmed and extended these findings to define the domain that mediates Gin4-dependent Bnr1 recruitment.
We generated a series of Bnr1 truncations expressed from the BNR1 promoter (Figure 2A). Bnr1 constructs with the first 660 or 758 amino acids localized with a similar pattern and apparent fluorescence intensity as full-length Bnr1-GFP (Figure 2B). A construct with only the first 596 amino acids of Bnr1 localized to the bud neck but with a less robust signal than full-length Bnr1-GFP (Figure 2B). Although there was variation in the steady-state protein expression of these truncation constructs, there was no correlation between the expression level and the robustness of the signal of Bnr1 at the bud neck (Supplemental Figure S2A).
FIGURE 2:

The Gin4 effect on Bnr1 localization is mediated by the L2 domain of Bnr1. (A) Domain architecture of Bnr1. DAD, Diaphanous autoregulatory domain; DD, dimerization domain; DID, Diaphanous inhibitory domain; FH1, formin homology 1; FH2, formin homology 2; GBD, GTPase-binding domain; IH, interdomain helix. The truncations used in the experiments in C are indicated. (B) Endogenous-level expression of Bnr1-758–GFP or Bnr1-660–GFP defines a minimal localization signal for Bnr1. Images are maximum-intensity projections with normalized fluorescence intensity. (C) C-terminally GFP-tagged Bnr1 constructs containing either L1 or L2 were expressed from the inducible GAL1,10 promoter. Bnr1 L2 (596–758) requires Gin4 for localization to the bud neck. Cells were fixed after 1 h (L2) or 4 h (L1 and ΔL1) of induction with medium containing 2% galactose. (D) Endogenous-level Bnr1 constructs confirm a requirement for Bnr1-L2 for Gin4-dependent localization to the bud neck. Note that images were chosen to represent the fluorescence intensity of the Bnr1 constructs rather than the percentage of Bnr1 localized to the bud neck. (C, D) Number in the upper left indicates the average percentage of cells with Bnr1 localized to the bud neck (n > 100 cells; three independent experiments). (B–D) Cells were imaged in three dimensions with five 0.5-μm stacks using the same exposure conditions for all experiments. Images are normalized, maximum-projection images; scale bar, 5 μm. (E) Fluorescence intensity of full-length 3GFP-Bnr1, 3GFP-Bnr1-ΔL1, or 3GFP-Bnr1-ΔL2 measured from the images obtained for bnr1Δ strains as in D. Each point represents the background-subtracted fluorescence intensity of a single cell (in arbitrary units); bars, mean ± SE.
The localization of the Bnr1 minimal localization domain (Bnr1-660–GFP) was strikingly compromised in the absence of Gin4. In gin4Δ strains at room temperature, only 47% of cells showed Bnr1-660–GFP localization at the bud neck. In those cells that did show bud neck signal of Bnr1-660–GFP, the fluorescence intensity appeared to be markedly reduced (Supplemental Figure S2B). These findings are not due to differences in protein expression because the steady-state expression of Bnr1-660–GFP was comparable in the wild-type and gin4Δ strains (Supplemental Figure S2C). Thus we have determined the minimal domain of Bnr1 sufficient for localization at endogenous levels and confirmed that Gin4 is partially required for its recruitment to the bud neck. Similar results were obtained in elm1Δ strains (unpublished data).
We next examined the role of the L1 and L2 domains in the Gin4-dependent localization of Bnr1. When overexpressed from the GAL promoter, the L1 and L2 constructs gave the expected patterns of localization. L1 concentrated at the mother side of the bud neck but failed to dissociate from the bud neck after the septin rings split during mitotic exit; L2 failed to display the mother-side bias but exhibited a cell cycle–regulated localization that mirrored the full-length protein (Figure 2C). Of great interest, L2 localization was entirely dependent upon the presence of Gin4, whereas L1 localized independently of Gin4. Moreover, an N-terminal deletion lacking L1 (Bnr1-ΔL1(596-C)–GFP) also exhibited an absolute requirement for Gin4 for its localization (Figure 2C). The expression of these constructs was similar in all of the strains examined (Supplemental Figure S2D).
The specific requirement of the L2 domain for Gin4-dependent localization was confirmed using domain deletion constructs expressed from the native promoter. For these experiments full-length Bnr1 and ΔL1 (466–C) and ΔL2 (466–733) derivatives were visualized with an N-terminal triple GFP tag (Figure 2D). Using maximum-projection images, which were obtained with the same imaging conditions for all the strains, we compared the fluorescence intensity at the bud neck of the different truncation constructs in the bnr1Δ strains (Figure 2E). After subtracting the background fluorescence in the cytoplasm, we observed that 3GFP-Bnr1 full-length (mean ± SE, 974.5 ± 53.7 AU) and 3GFP-Bnr1-ΔL1 displayed similar mean fluorescence intensities (795.8 ± 42.5 AU). In contrast, 3GFP-Bnr1-ΔL2 showed a clear decrease in the signal at the bud neck (297.4 ± 14.9 AU). These results suggest that the Gin4/Bnr1 L2 mechanism for Bnr1 localization is more important than the L1 domain–dependent mechanism. In contrast with 3GFP-Bnr1-ΔL2 or the full-length control, 3GFP-Bnr1ΔL1 localization was entirely dependent upon Gin4 (Figure 2D). The steady-state protein expression of these constructs was comparable in wild-type cells. Although there was decreased expression of all three Bnr1 constructs in the gin4Δ strains, the expression difference was more pronounced for full-length 3GFP-Bnr1 than for 3GFP-Bnr1-ΔL1 or 3GFP-Bnr1-ΔL2 (Supplemental Figure S2E). Therefore the analysis of these constructs is consistent with the data from the overexpression experiments: the L2 domain of Bnr1 specifically requires Gin4 for its ability to localize to the bud neck.
Gin4, Elm1, and Shs1 are required for the ability of Bnr1 to assemble actin cables
Although neither BNR1 nor BNI1 is essential for the viability of yeast cells, cells lacking both formins are inviable because of the inability to undergo polarized morphogenesis (Imamura et al., 1997; Kamei et al., 1998; Evangelista et al., 2002; Sagot et al., 2002a). Thus synthetic lethality with deletions of BNI1 or mutations that conditionally inactivate Bni1 protein identifies candidate genes that might collaborate with BNR1. Large-scale synthetic genetic array data identified elm1Δ, gin4Δ, nap1Δ, and shs1Δ as having synthetic growth defects with bni1Δ (Tong et al., 2001, 2004; Costanzo et al., 2010). By directed genetic crosses, we confirmed these findings and found that a gin4Δ bni1Δ double deletion is nearly lethal, similar to the growth phenotype of bni1Δ bnr1Δ (Supplemental Figure S3A). The double-deletion strains shs1Δ bni1Δ and elm1Δ bni1Δ had temperature-sensitive synthetic growth defects at 30 and 35°C (Supplemental Figure S3A). In contrast, relative to the single-mutant strains, nap1Δ bni1Δ double-mutant strains did not display a synthetic growth defect (Supplemental Figure S3A). Because the aforementioned double-deletion strains rapidly developed suppressor mutations, elm1Δ, gin4Δ, nap1Δ, and shs1Δ were combined with the bni1-1 temperature-sensitive allele. The bni1-1 allele contains a mutation in the coding sequence for the FH2 domain; bnr1Δ bni1-1 strains are inviable at 34°C and lose all actin cables within a 4-min shift to the nonpermissive temperature (Sagot et al., 2002a). The gin4Δ bni1-1 and elm1Δ bni1-1 strains were, to differing degrees, temperature sensitive for growth: gin4Δ bni1-1 failed to grow at 35°C, whereas elm1Δ bni1-1 had a slow-growth phenotype at 35°C and failed to grow at 37°C (Figure 3A and Supplemental Figure S3B). By contrast, nap1Δ bni1-1 strains grew normally at all temperatures (Figure 3A). These data may be explained by the requirement for these genes to localize Bnr1 because of the close correlation of the growth phenotype with the requirement for Bnr1 bud neck localization. Of interest, the shs1Δ bni1-1 strains also had a growth defect at 35°C and were inviable at 37°C (Figure 3A), despite the fact that Bnr1 localizes normally at all temperatures in strains lacking Shs1. This raises the possibility that Gin4 and Elm1 are required for both Bnr1 function and localization, whereas Shs1 may be specifically required for Bnr1 function.
FIGURE 3:

Bnr1-mediated actin cable assembly requires Gin4, Elm1, and Shs1. (A) Temperature-sensitive growth of gin4Δ bni1-1, elm1Δ bni1-1, and shs1Δ bni1-1 strains. Fivefold dilutions of cells were spotted on yeast extract/peptone/dextrose plates and grown for 1 d at 35°C (middle) or 37°C (bottom) or for 2 d at 24°C (top). (B) Myo2-GFP localization is depolarized in gin4Δ bni1-1, elm1Δ bni1-1, and shs1Δ bni1-1 after a 15-min shift to 35°C. Cells were imaged in three dimensions with five 0.5-μm stacks using the same exposure conditions for all experiments. Images are maximum-projection images; scale bar, 5 μm. (C) Quantification of Myo2-GFP localization from experiments in B. Cells of the indicated genotype were scored for Myo2-GFP localization to the bud tip (white with dots) or the bud neck (black) or unpolarized localization (gray). Experiments were performed three times (n > 200 cells for each repetition). For each double mutant, at least two independent clones from different spores were analyzed (data not shown).
The morphogenesis checkpoint delays the cell cycle of budding yeast when bud formation is impaired. The Swe1 kinase is the central mediator of this checkpoint (Lew, 2003). Septin perturbations lead to Swe1-dependent cell cycle delays. Deletion of Swe1 in strains lacking septins or septin-associated components can suppress the elongated morphology and growth defects that are characteristic of these strains. However, swe1Δ does not rescue the septin defects in these strains (Barral et al., 1999; Sreenivasan and Kellogg, 1999; Longtine et al., 2000). Genetic crosses were performed to determine whether the synthetic growth defect of gin4Δ bni1-1, elm1Δ bni1-1, and shs1Δ bni1-1 strains could be explained by the activation of Swe1. However, the additional deletion of SWE1 did not rescue the temperature sensitivity of these double mutant strains (Supplemental Figure S3C) and, in fact, resulted in a more pronounced growth defect. This additional growth defect may be explained by the previously described growth defect of swe1∆ bni1∆ strains (Tong et al., 2004). Therefore the synthetic growth defects of gin4Δ bni1-1, elm1Δ bni1-1, and shs1Δ bni1-1 strains are not due to activation of the morphogenesis checkpoint.
Next we determined whether Gin4, Elm1, and Shs1 promote Bnr1-dependent actin cable assembly by labeling gin4Δ bni1-1, elm1Δ bni1-1, and shs1Δ bni1-1 strains with Alexa 568–phalloidin. In common with bnr1Δ bni1-1 strains (Sagot et al., 2002a), gin4Δ bni1-1 and elm1Δ bni1-1 strains lost most actin cables after a 15-min shift to 35°C. Of interest, shs1Δ bni1-1 cells also displayed a loss of actin cables after the temperature shift (Supplemental Figure S3C). In parallel, we used an endogenous-level GFP-tagged copy of the type V myosin, Myo2, to evaluate the presence of intact actin cables. Most polarized transport in budding yeast is carried by Myo2 along actin cables (Pruyne and Bretscher, 2000; Pruyne et al., 2004b). As expected, after a 15-min shift to 35°C of a bnr1Δ bni1-1 strain, the bud tip polarization of Myo2 was lost. Myo2 polarization was similarly disrupted in gin4Δ bni1-1, elm1Δ bni1-1, and shs1Δ bni1-1 strains at the restrictive temperature (Figure 3, B and C). Thus Elm1, Gin4, and Shs1 are all required for normal actin cable assembly by Bnr1.
Gin4 kinase activity is not required for actin cable assembly by Bnr1 in vivo
We next examined whether the kinase activity of Gin4 or its role in septin organization promotes Bnr1 function. The possibility that Gin4 kinase activity could be required was suggested by the fact that Elm1 is an upstream kinase for Gin4 that promotes its kinase activity (Sreenivasan and Kellogg, 1999; Asano et al., 2006). However, the kinase activity of Gin4 is not absolutely required for the role of Gin4 in regulating septin architecture (Longtine et al., 1998a; Mortensen et al., 2002). Constructs encoding full-length Gin4 or a Gin4 deletion lacking the kinase domain (amino acids 17–280; Iwase and Toh-e, 2004) were introduced into a gin4Δ bni1-1 strain. Full-length Gin4-2HA and Gin4Δkinase-2HA expression complemented the growth defect of gin4Δ bni1-1 strains comparably at the restrictive temperature (35°C; Figure 4B). Moreover, Gin4Δkinase-2HA expression also corrected the defect in the bud neck localization of Bnr1 (Supplemental Figure S4A), as well as the defect in polarized Myo2 localization (Figure 4, C and D). The epitope-tagged, full-length Gin4 does not complement as well as the untagged control, but the tagged Gin4Δkinase complements as well as the tagged full-length Gin4. Note that the tagged proteins are expressed at lower steady-state levels than the untagged control and that the Gin4Δkinase-2HA protein is expressed at lower levels than the full-length Gin4-2HA (Figure 4C). However, despite its lower expression, Gin4Δkinase-2HA complements in the functional assays comparably to the better-expressed full-length Gin4-2HA. Thus the regulation of Bnr1 by Gin4 does not require the Gin4 kinase activity.
FIGURE 4:
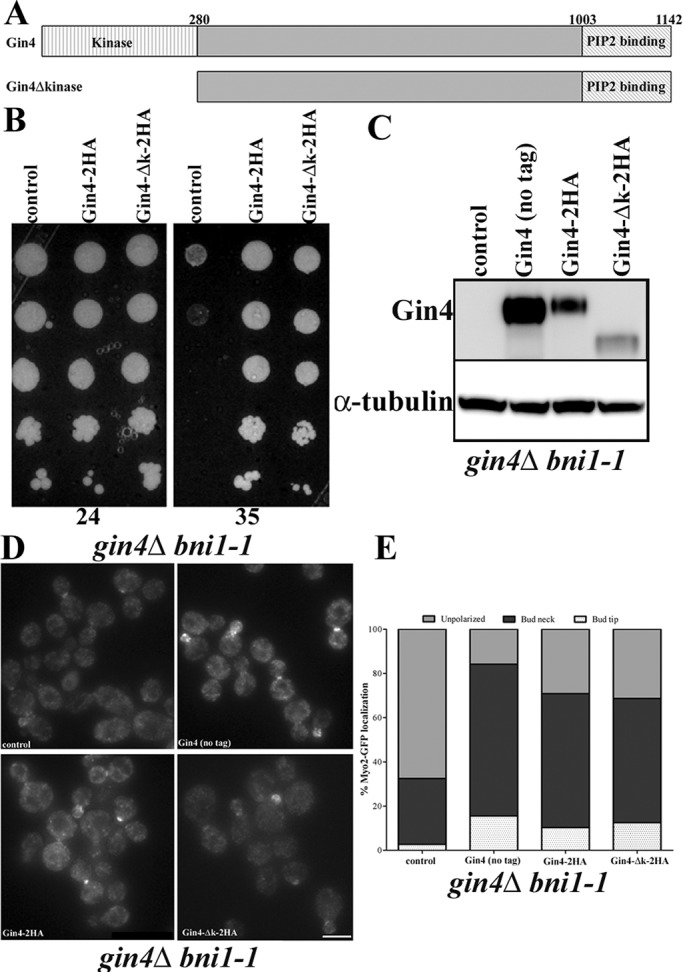
The role of Gin4 in localization and activation of Bnr1 is independent of its kinase domain. (A) Domain architecture of Gin4. The kinase domain of Gin4 (18–280) and phospholipid-binding domain are indicated. (B) The temperature-sensitive growth defect of gin4Δ bni1-1 strains is rescued by both [pGIN4-GIN4-2HA] and [pGIN4-Δkinase-2HA] but not by the control vector. Fivefold dilutions of cells were spotted on –Ura plates and grown for 1 d at 35°C (right) or 2 d at 24°C (left). (C) Comparative Western blotting with anti-Gin4 in gin4Δ bni1-1 MYO2-GFP strains shows that the level of Gin4 correlates with the rescue of polarized assembly observed in D and E. (D) Myo2-GFP localization is polarized when the gin4Δ bni1-1 strain is rescued by either [pGIN4-GIN4-2HA] or [pGIN4-Δkinase-2HA]. We also compared complementation with an untagged [pGIN4-GIN4], which showed similar Myo2-GFP localization to the tagged Gin4 plasmids. Images were collected and analyzed as in Figure 3B. Scale bar, 5 μm. (E) Quantification of Myo2-GFP localization from experiments in D. Cells of the indicated genotype were scored for Myo2-GFP localization to the bud tip (white with dots) or the bud neck (black) or unpolarized localization (gray). Experiments were performed three times (n > 200 cells for each repetition).
Gin4 binds directly to the Bnr1-758 minimal localization domain
Next we examined whether Elm1 and Gin4 interact with Bnr1 in vivo. We generated a BNR1-758-13myc strain with either Elm-TAP– or Gin4-TAP–tagged protein. The TAP tag consists of a calmodulin-binding peptide, a TEV cleavage site, and two immunoglobulin G–binding domains of Staphylococcus aureus protein A. We performed immunoprecipitations with an anti–calmodulin binding protein (CBP) antibody and probed Western blots with an anti-CBP antibody (to detect Elm1 or Gin4) and an anti-myc antibody (to detect Bnr1). Bnr1-758 bound Gin4-TAP in buffer containing 150 mM KCl (Figure 5A). This binding is specific because Elm1 does not bind to Bnr1-758 in these conditions.
FIGURE 5:

Bnr1-N interacts with Gin4-C in vivo and in vitro. (A) Immunoprecipitations from yeast extracts using an anti-CBP antibody in BNR1-758-13myc strains that also expressed either Elm1-TAP or Gin4-TAP. In 150 mM KCl, Bnr1-758 bound to Gin4 but not Elm1. (B) Bnr1-758 interacts with Gin4 in a kinase domain–independent manner. Immunoprecipitations from yeast extracts using an anti-HA antibody in gin4Δ BNR1-758-13myc strains that were complemented with CEN URA plasmids bearing no insert (control), [pGIN4-GIN4-2HA], or [pGIN4-Δkinase-2HA]. At 75 mM KCl, we detected binding of Bnr1-758-13myc to Gin4Δkinase-2HA (Gin4Δk) but little if any binding to full-length Gin4-2HA. At 150 mM KCl, we detected robust binding of Bnr1-758-13myc to Gin4-2HA but less to Gin4∆k-2HA. The immunoprecipitations in A and B were performed at least three times; representative Western blots are shown. (C, D) Full-length Bnr1 and Bnr1-N bind directly to Gin4 in a kinase-independent manner. Bnr1 was present at 56 nM, and all Gin4 constructs were present at 120 nM. (C) We used glutathione agarose to pull-down full-length GST-Gin4 or GST-Gin4Δkinase in the presence of 6His-Bnr1 and probed Western blots with anti-His antibodies. Bnr1* denotes the N-terminal proteolysis product that occurs during the purification of full-length Bnr1. Both Bnr1 full-length and Bnr1* bound more robustly to GST-Gin4 full length at 150 mM KCl than at 75 mM KCl, whereas they bound more robustly to GST-Gin4Δkinase at 75 mM KCl. (D) Pull-down of 6His-Bnr1 full-length and 6His-Bnr1-758 (Bnr1-N) with pCAL-n-FLAG-Gin4-C (amino acids 540–1142); controls (–) were performed without Gin4-C added. At 75 mM KCl, we found that Bnr1 full-length, Bnr1*, and Bnr1-N bound to Gin4-C; Bnr1-N and Bnr1* bound more robustly than full-length Bnr1. Note that we consistently observed an additional band in the anti-His Western blots of the pull-downs (marked with an asterisk) when both Bnr1 and Gin4-C were combined; this band was not present in the inputs of any of the individual proteins. Note that in D the inputs are diluted to 56 nM and 120 nM, whereas in C the Bnr1 input is present at 120 nM. The pull-downs were resuspended in minimal volumes of sample buffer before resolving by SDS–PAGE, which results in the apparent increase in the concentration of protein in the pull-down lanes. All pull-down experiments were performed at least two times; representative Western blots are shown. (E) Coomassie-stained gel of the purified proteins used in the pull-down experiments. Note that these represent three different SDS–PAGE gels for the three different sets of proteins. For Western blots and gels, lanes that were not relevant to the experiments here may have been removed.
To further investigate the interaction of Gin4 with Bnr1 in vivo, we generated a gin4Δ strain that expressed Bnr1-758-13myc from the BNR1 locus and introduced centromeric plasmids to express Gin4-2HA and Gin4Δkinase-2HA from the GIN4 promoter. We performed immunoprecipitations with an anti-hemagglutinin (HA) antibody and probed Western blots with anti-HA antibody (Gin4) and anti-myc antibody (Bnr1). At 75 mM KCl, we detected binding of Bnr1-758-13myc to Gin4Δkinase-2HA but not to full-length Gin4-2HA (Figure 5B). At 150 mM KCl, we also detected binding of Bnr1-758-13myc to Gin4-2HA (Figure 5B).
Pull-down experiments with purified proteins were performed to determine whether the interaction between Bnr1 and Gin4 was direct. Hexahistidine (6His)-Bnr1 was purified from yeast; glutathione S-transferase (GST)–Gin4 and GST-Gin4Δkinase were purified from Escherichia coli (Figure 5E). We used glutathione agarose to pull-down GST-Gin4 full length or GST-Gin4Δkinase in the presence of 6His-Bnr1 and probed Western blots with anti-His antibodies. Bnr1 was at 56 nM; all GST proteins were at 120 nM. Note that the purification of full-length Bnr1 yields a proteolysis product (Bnr1*), which is an N-terminal fragment similar in size to the Bnr1-758 used in the immunoprecipitation experiments. Both Bnr1 full length and Bnr1* bound more robustly to full-length Gin4 at 150 mM KCl than at 75 mM KCl. In contrast, they bound more robustly to Gin4Δkinase at 75 mM KCl (Figure 5C). The basis for these salt-dependent effects is not clear, but the following points may be relevant. First, the results are highly reproducible, and we note that the salt-dependent effects are seen with the immunoprecipitations from extracts (Figure 5B) as well as with purified proteins. Second, some effects may be explained by the known regulatory autoinhibition of Gin4, which may be in a more open conformation at higher salt concentration (Barral et al., 1999; Mortensen et al., 2002). If this were the case, we would predict that the Gin4 construct lacking the kinase domain would be able to interact at the lower salt concentration; indeed, our experiments are consistent with this prediction (Figure 5, B and C). What remains unclear is why the interaction of Gin4Δkinase with Bnr1-758 is inhibited by higher salt concentration. This mutant may not properly fold in this condition or might bind another protein in the extract. Nevertheless, our data demonstrate that the N-terminal minimal localization domain of Bnr1 binds to Gin4 in a kinase-independent manner. Moreover, our experiments suggest that the interaction is direct.
To independently confirm these results, we performed a pull-down of full-length 6His-Bnr1 and 6His-Bnr1-758 (Bnr1-N) with a different Gin4 construct (Gin4-C), which contained amino acids 540–1142. At 75 mM KCl, we found that both Bnr1 full length and Bnr1-N bound to Gin4-C; Bnr1-N and Bnr1* bound more robustly than full-length Bnr1 (Figure 5D). We speculate that this is due to autoinhibition of full-length Bnr1. Taken together, our data suggest that the N-terminal minimal localization domain of Bnr1 binds directly to the C-terminal of Gin4.
A defect in the function of Bnr1 explains the growth defect of the gin4Δ bni1-1 and shs1Δ bni1-1 strains
Our genetic data raised the possibility that Shs1 and Gin4 are required for the activation of Bnr1. To test this hypothesis directly, we determined whether constitutively active Bnr1 would suppress the temperature-sensitive growth of strains lacking either Gin4 or Shs1 in combination with Bni1. Prior work suggests that, like other DRFs, Bnr1 is negatively regulated by autoinhibition due to the interaction of the N-terminal RBD/DID with the C-terminal DAD domain. For DRFs, including Bnr1, disruption of either the DID or the DAD domain results in the constitutive activation of actin filament assembly (Dong et al., 2003; Chesarone et al., 2009). Bnr1∆L1 lacks amino acids 1–466, which include the RBD and DID domains. In common with other constitutively active formins in budding yeast, overexpression of Bnr1∆L1 is lethal (Gao and Bretscher, 2008; our unpublished data). Strikingly, expression of 3GFP-Bnr1ΔL1, but not 3GFP-Bnr1, strongly suppressed the growth defect of the gin4Δ bni1-1 bnr1Δ or the shs1Δ bnr1Δ bni1-1 strain (Figure 6A). However, expression of 3GFP-Bnr1ΔL2 did not suppress the growth defect of the gin4Δ bni1-1 bnr1Δ strains (Supplemental Figure S5). Note that for this experiment all constructs are slightly (twofold to threefold) overexpressed because they are carried on centromeric plasmids. Presumably, this is why there was a small degree of complementation by the full-length 3GFP-Bnr1 in the gin4Δ bni1-1 bnr1Δ strain and may explain why the shs1Δ bni1-1 bnr1Δ strains show a growth defect at the permissive temperature. However, Western blotting demonstrated that the phenotypes of these strains could not be explained by the expression levels of the constructs (Supplemental Figure S2E). Most significantly, Bnr1∆L1 and Bnr1∆L2 are expressed comparably in all strains, but only Bnr1∆L1 suppresses the growth defect. Finally, Alexa 568–phalloidin labeling demonstrated that the Bnr1∆L1 but not the full-length protein restored actin cable assembly in the gin4Δ bni1-1 bnr1Δ or shs1Δ bnr1Δ bni1-1 strain (Figure 6B).
FIGURE 6:

Expression of activated Bnr1 rescues gin4Δ bnr1Δ bni1-1 and shs1Δ bnr1Δ bni1-1 growth defect and actin polarity defect. (A) Expression of [pBNR1-3GFP-BNR1ΔL1] rescues the temperature-sensitive phenotype of gin4Δ bnr1Δ bni1-1 and shs1Δ bnr1Δ bni1-1 strains, but [pBNR1-3GFP-BNR1] expression has little effect. Tenfold dilutions of cells were spotted on −Ura plates and grown for 2 d at 33°C (bottom) or 3 d at 24°C (top). (B) Phalloidin actin staining in cells fixed after 5 min at 33°C. Images are deconvolved and maximum-projection images generated from 0.3-μm stacks. The percentage of cells with actin cable staining is given in the lower left corner; cells were scored as positive for actin cables when any linear actin structures were detected. Arrowheads indicate cells with actin cables. Experiments were performed two times (n > 100 cells for each experiment). For each strain, we analyzed two independent isolates, which both showed similar effects on growth and actin cable staining. Scale bar, 5 μm.
3GFP-Bnr1ΔL1 is not localized to the bud neck in a gin4Δ strain (Figure 2, C and D); however, 3GFP-Bnr1ΔL1 localization in a shs1Δ strain is normal (unpublished data). Therefore, to confirm the rescue of the gin4Δ bni1-1 with the activated Bnr1, we generated a Bnr1 deletion lacking the DAD domain. Bnr1∆DAD-GFP displays partial localization in a gin4Δ strain (unpublished data). Cells that express Bnr1ΔDAD-3HA contain disorganized arrays of short cables and display impaired movement of GFP-Sec4 particles (Chesarone et al., 2009). We generated a strain expressing Bnr1ΔDAD-3HA at the endogenous level as in Chesarone et al. (2009). The strain bnr1ΔDAD-3HA gin4Δ bni1-1 exhibited improved growth at 35°C relative to the gin4Δ bni1-1 control strain (Figure 7A and Supplemental Figure S6A). Thus truncating the DAD domain of Bnr1 partially rescued the defect in Myo2 polarization of the gin4Δ bni1-1 strain at the restrictive temperature (Figure 7, B and C). Finally, Alexa 568–phalloidin labeling demonstrated that the Bnr1∆DAD truncation restored actin cable assembly in the gin4Δ bni1-1 strain (Supplemental Figure S6B). Taken together, our data suggest a novel role for Gin4 and Shs1 in the activation of Bnr1.
FIGURE 7:

Expression of activated Bnr1 rescues gin4Δ bni1-1 growth defect and actin polarity defect. (A) Expression of bnr1ΔDAD-3HA partially rescues the temperature-sensitive phenotype of a gin4Δ bni1-1 strain. Fivefold dilutions of cells were spotted on yeast extract/peptone/dextrose plates and grown for 1 d at 35°C (bottom) or 2 d at 24°C (top). (B) Myo2-GFP localization is unpolarized in gin4Δ bni1-1 but shows an increased polarization in gin4Δ bni1-1 bnr1ΔDAD-3HA. Images were acquired and analyzed as in Figure 3B. Scale bar, 5 μm. (C) Quantification of Myo2-GFP localization from experiments in B. Cells of the indicated genotype were scored for Myo2-GFP localization to the bud tip (white with dots) or the bud neck (black) or unpolarized localization (gray). Experiments were performed two times (n > 100 cells for each repetition). For gin4Δ bni1-1 bnr1ΔDAD-3HA strains, we analyzed two independent isolates, which both showed similar effects on growth and Myo2-GFP localization.
DISCUSSION
In this study, we demonstrated that the MARK/Par1-family, septin-associated kinase Gin4 has an important role in localizing and activating Bnr1 at the bud neck. In addition, our experiments reveal an unexpected requirement for the septin Shs1 in the activation of Bnr1.
Control of Bnr1 localization by septins and Gin4
Bnr1 has two independent localization signals; each one controls a distinct element of the localization of Bnr1. L1 specifies the spatial (mother-side bias) localization; L2 is required for the accurate temporal localization of Bnr1 through the cell cycle (Gao et al., 2010). Both L1 and L2 require an intact septin cytoskeleton for localization to the bud neck (Gao et al., 2010). However, two-hybrid experiments failed to detect an interaction of the Bnr1 N-terminus, L1, or L2 with individual septin subunits (Kikyo et al., 1999; Gao et al., 2010). Here we show that Bnr1 L2 requires Gin4 for its localization to the bud neck. Gin4 localization to the bud neck is septin dependent (Longtine et al., 1998a), which is consistent with the observation that localization of Bnr1 L2 requires an intact septin cytoskeleton for its localization (Gao et al., 2010). The activity and localization of Gin4 are highly regulated through the cell cycle (Altman and Kellogg, 1997; Longtine et al., 1998a; Mortensen et al., 2002; Asano et al., 2006). Like Bnr1, Gin4 localizes to the bud neck at the onset of bud emergence and leaves the bud neck at the start of cytokinesis (Longtine et al., 1998a; our unpublished data). Gin4 is not localized in cells treated with α-factor (Longtine et al., 1998a); similarly, Bnr1 is not localized and is not required for the assembly of actin cables in mating projections (Evangelista et al., 1997; our unpublished data). Therefore Gin4 is an excellent candidate to mediate the cell cycle–specific and developmental regulation of Bnr1 localization.
L2 is located in a previously uncharacterized region of Bnr1. We performed homology scanning of Bnr1 sequences from sensu lato yeast and fungi, as well as with the formins Bni1 from budding yeast and cdc12 from fission yeast (see Materials and Methods for details). This method examines the alignments of protein sequences and scores the relative similarity along the length of the protein sequences (Crutchley et al., 2009). We chose to compare sensu lato yeast and fungal Bnr1 sequences, as they all have Gin4 homologues. In contrast, there is no evidence that either Bni1 or Cdc12 requires the septin cytoskeleton for localization. Supplemental Figure S7 shows the sequence similarity of Bnr1 1–758 from sensu lato yeast (blue), fungi (pink), and Bni1 and Cdc12 (yellow). Of interest, in sensu lato yeast, there is a well-conserved peak at 639−660 (marked with two asterisks), which is not present in Bni1 or Cdc12 but is slightly similar in fungal Bnr1 sequences. We speculate that this region may be important for binding of Bnr1 to Gin4. Indeed, our structure–function analysis (Figure 2) is consistent with this hypothesis.
The binding partner of Bnr1 L1 remains to be identified. Deletion of Rho4, a known binding partner of Bnr1 (Kikyo et al., 1999), did not affect the localization of full-length Bnr1, Bnr1 L1, or Bnr1 L2 (unpublished data). Our homology scanning data showed that L1 (amino acids 35–501) contains one peak that is well conserved among sensu lato Bnr1 sequences and fungal Bnr1 sequences but is not present in Bni1 or Cdc12 (Supplemental Figure S7, marked with an asterisk). This sequence (amino acids 100–135) contains a high number of basic residues and has a pI of 9.5. The presence of a basic-rich region (BRR) is consistent with published reports that demonstrate that a BRR on the N-terminus of mDia1 and mDia2 binds to phospholipids (Ramalingam et al., 2010; Gorelik et al., 2011). Alternatively, the peak in L1 may be a novel septin-binding domain.
Regulation of Bnr1 by Gin4 and Shs1
Our data demonstrate that Gin4 is at least required for localization of Bnr1 to the bud neck. Moreover, a gin4Δ bni1-1 strain showed temperature-sensitive growth caused by a loss of polarized actin cables. Of interest, Shs1 did not affect the localization of Bnr1 to the bud neck at any temperature tested, but a shs1Δ bni1-1 strain showed a temperature-sensitive growth defect and a loss of polarized actin cables. Bnr1 can be activated by truncation of the N-terminal RBD or the C-terminal DAD (Gao and Bretscher, 2008, 2009; Chesarone et al., 2009). Deletion of the RBD/DID domains of Bnr1 can suppress the temperature-sensitive growth and the polarized actin defect of both the gin4Δ bni1-1 and shs1Δ bni1-1 strains. Moreover, activation of Bnr1 by truncation of the DAD domain can partially rescue the growth and actin assembly defects of the gin4Δ bni1-1 strain. There are several possible explanations for the partial rescue with the truncation of the DAD domain. The cables generated by Bnr1ΔDAD are short but numerous and have defects in Sec4 particle movement (Chesarone et al., 2009). In addition, the DAD domain can bind to G-actin and has a role in the nucleation of actin filaments (Gould et al., 2011). Finally, our localization data suggest that bnr1ΔDAD-3HA gin4Δ bni1-1 strains would only have partial localization of Bnr1 to the bud neck (∼67% at room temperature). Thus it is possible that only some cells would have cables present at the bud neck, and those cables would be less organized than full-length Bnr1.
One unresolved issue from our work is why Elm1 shows synthetic lethality and actin polarization defects in combination with bni1-1. One might expect that the requirement for Elm1 is due to its ability to phosphorylate and activate Gin4 (Sreenivasan and Kellogg, 1999). However, the kinase domain of Gin4 is not required for the localization or activity of Bnr1. Thus there may be an additional role for Elm1 in the regulation of Bnr1 that is independent of its established activity as a regulator of Gin4.
In general, the relationship between the localization and activation of formins is poorly understood. In budding yeast, nonlocalized formins can support growth, albeit with compromised organization of actin cables and defects in polarized growth (Sagot et al., 2002a; Gao and Bretscher, 2009). Increasing evidence suggests that localization is a prerequisite for the release of autoinhibition. For mDia1, binding to active Rho regulates the activation and localization to the site of actin assembly (Seth et al., 2006). The fission yeast formin for3 must be localized for the release of autoinhibition to occur (Martin et al., 2007). Furthermore, reports suggest that the N-terminal BRR of mDia1 and mDia2 is involved in recruiting mDia to the plasma membrane, permitting activation by the binding of Rho-GTPase to the DID/RBD (Ramalingam et al., 2010; Gorelik et al., 2011). These data suggest that activation of formins may be controlled by a coincidence detection mechanism by which multiple inputs contribute to the complete activation of the formin; such a mechanism would ensure that actin nucleation occurs only at appropriate sites of actin assembly (Gorelik et al., 2011).
Our results demonstrate that the budding yeast formin Bnr1 is regulated by an autoinhibition mechanism, similar to other DRFs. Our genetic data suggest that the loss of Gin4 can be overcome by activation of Bnr1. Because Gin4 is required for the localization of Bnr1, this result might simply be explained by a requirement for the localization of Bnr1 for the relief of autoinhibition. Gin4 likely interacts with the L2 domain of Bnr1, which is outside the typical domains involved in autoinhibition; therefore Gin4 may be required for the localization of Bnr1 to the bud neck, which then allows the relief of autoinhibition by another factor. The localization of Gin4 closely mirrors that of Bnr1; moreover, Gin4 is highly regulated through the cell cycle. Thus one major function of Gin4 is to control the localization of Bnr1.
Our results also demonstrate that Shs1 is not required for the localization of Bnr1 but is required for its actin assembly activity. Moreover, activated Bnr1 overcomes the requirement for Shs1. We speculate that Shs1, perhaps in combination with other septins, may bind to the L1 domain of Bnr1 and relieve autoinhibition. One simple interpretation of our data would be a stepwise model: Gin4 (in a cell cycle–dependent manner) localizes Bnr1 (via the L2 domain) to the bud neck and then Shs1, together, with Rho-GTPases, contributes to the release of Bnr1 autoinhibition. We also do not exclude the possibility that Gin4 has a direct role in Bnr1 activation in addition to the role we defined in Bnr1 localization. The hypothesis that septins and septin-associated proteins are involved in the regulation of a formin is particularly interesting in light of the established connections between the actin and septin cytoskeletons (Mostowy and Cossart, 2012). Therefore our results may have implications for the regulation of formins in other systems.
MATERIALS AND METHODS
Strains and plasmids
Media and genetic techniques were as previously described (Rose et al., 1990). BNR1-GFP and MYO1-CFP strains were generated as previously described (Buttery et al., 2007). Deletion strains were obtained from a haploid deletion library (Open Biosystems, Lafayette, CO) and were crossed with strains of interest. Myo2-GFP was obtained from a GFP library (Open Biosystems) and was crossed into the strains of interest. The bnr1-ΔDAD-3HA construct was generated as described (Chesarone et al., 2009) and transformed into a wild-type diploid of our strain background (Research Genetics S288C BY4730). GFP-tagged Bnr1 truncations were generated using established methods (Longtine et al., 1998b). See Supplemental Table S1 for complete details about the yeast strains used; additional details are available upon request.
Gin4 plasmids [pGIN4-GIN4-2HA] and [pGIN4-Δkinase-2HA] were obtained from the Iwase lab (Iwase and Toh-e, 2004). The Gin4 untagged plasmid was a gift from Mark Longtine (Longtine et al., 1998a). [pBNR1-3GFP-Bnr1] was previously described (Buttery et al., 2007). [pBNR1-3GFP-Bnr1-ΔL1] and [pBNR1-3GFP-Bnr1-ΔL2] plasmids were obtained from Anthony Bretscher (Gao et al., 2010). See Supplemental Table S2 for the complete details on the plasmids used; additional details are available upon request.
Microscopy
All live-cell imaging was performed as previously described (Buttery et al., 2007). Cells were fixed with 3.7% formaldehyde for 7 min at room temperature, washed three times in phosphate-buffered saline (PBS), and resuspended in a final volume of 50 μl. For galactose-inducible strains, cells were grown overnight in yeast extract/peptone (YEP) with 2% raffinose, refreshed to OD600 0.2 in YEP raffinose, grown to an OD600 of 0.4, and induced with 2% galactose in YEP media for 1 h or 4 h, as indicated.
Staining of the actin cytoskeleton using Alexa 568–phalloidin actin was performed as described (Sagot et al., 2002a). For the experiments in Supplemental Figure S3, Figure 6, and Supplemental Figure S6, we imaged cells with at least fifteen 0.3-μm stacks. We scored cells for actin cables by examining every stack in the series; cells were scored as positive for actin cables when any discernible linear actin structures were present.
Analysis of microscopy data
All image manipulations and fluorescence intensity measurements were carried out using SlideBook (Intelligent Imaging Innovations, Denver, CO) software; data were exported to and analyzed in Excel (Microsoft, Redmond, WA) or Prism software (GraphPad Software, La Jolla, CA). For each experiment, images to be compared were obtained using the same exposure time and the same number of z-stacks; projection images were generated, and the fluorescence intensities were set to the same minimum and maximum values, thereby normalizing the images for accurate comparison. Fluorescence intensity measurements were made using the mask feature in SlideBook. In cells that were positive for Myo1-CFP signal at the bud neck, we measured the mean fluorescence intensity of both Bnr1 and Myo1 with a mask of 12–18 pixels at the bud neck. We subtracted the background fluorescence, which was calculated by taking the average of the mean fluorescence intensity for three 6-pixel spots in the cytoplasm of each cell. The ratio of yellow fluorescent protein (YFP; Bnr1) to CFP (Myo1) was then determined, and the mean of the samples was calculated. In Figure 1C, each data point represents the ratio of the background-corrected Bnr1-GFP at the bud neck to the background-corrected Myo1-CFP at the bud neck. We judged the statistical significance of the mean of the Bnr1:Myo1 ratio. In the experiments in Figure 1, Bnr1-GFP was visualized with a YFP filter to eliminate the signal from Myo1-CFP. For Figure 2E, we used a similar method to measure the fluorescence intensity of the various Bnr1 constructs at the bud neck and subtracted the background fluorescence intensity, calculated as described.
Western blotting and SDS–PAGE
Standard procedures were used for SDS–PAGE and Western blotting. Mouse anti-myc antibody (9E10; Roche, Basel, Switzerland), rat anti–tubulin α antibody (Accurate Chemical, Westbury, NY), mouse anti-GFP antibody (7.1 and 13.1; Roche, Basel, Switzerland), monoclonal anti-His antibody (GE Healthcare, Little Chalfont, United Kingdom), anti-HA monoclonal (12CA5, Roche), and anti-CBP monoclonal (Millipore, Darmstadt, Germany) were obtained from commercial sources. The anti-Gin4 polyclonal antibody was a gift from Doug Kellogg, University of California, Santa Cruz.
Protein purification
Gin4.
BL21* cells carrying the GST-GIN4 plasmid (obtained from Jeremy Thorner, University of California, Berkeley) were grown and purified according to the methods previously described (Carroll et al., 1998), with the following modifications. Gin4-C (amino acids 540–1142) was expressed with a pCAL-n-FLAG tag (Agilent, Santa Clara, CA) on the N-terminal. Cells carrying this plasmid were grown at 24ºC to an optical density of 0.6–0.8; expression was induced for 3 h with 0.5 mM isopropyl-β-d-thiogalactoside. After expression of each of the Gin4 constructs, cells were flash frozen in liquid nitrogen and then lysed using the coffee grinder method and stored at −80°C. Purification of GST-Gin4Δkinase was performed as for full-length Gin4, except that 20 mM glutathione was used for the elution. For Gin4-C, the manufacturer's protocol was used for purification with calmodulin affinity resin, except that the binding buffer contained 300 mM NaCl and the elution buffer contained 1 M NaCl.
Bnr1.
The [GAL1,10::6His-BNR1::URA] 2 μ plasmid was transformed into a protease-deficient strain (Jones, 1990). Constructs encoding truncations of [GAL1,10::6His-BNR1::URA] 2 μ were generated by direct disruption of the plasmid in a bnr1Δ strain of the Research Genetics background. For expression and purification of both full-length and N-terminal Bnr1, we modified previously described protocols (Moseley et al., 2006). Cultures were grown in SC-URA with 2% raffinose; cells were expanded from single colonies to 1 l over the course of 3 d. At OD600 0.8, expression was induced with 2% galactose for 8 h. Cells were collected by centrifugation and flash frozen on liquid nitrogen. Cells were lysed in liquid nitrogen with a coffee grinder; powders were stored as 5-g aliquots at −80ºC. Five grams of frozen powder was slowly warmed to room temperature and resuspended in 50 ml of lysis buffer (1× PBS, 100 mM NaCl, 25 mM Tris, pH 8, 20 mM imidazole, 0.8 mM dithiothreitol [DTT], and 5% glycerol with 2× protease inhibitor cocktail [Complete EDTA-free; Roche]). The lysates were centrifuged at 60,000 rpm for 1 h at 4ºC in a Ti70 rotor. We added 0.5 ml 50% nickel-nitriloacetic acid (Ni-NTA) agarose (Qiagen, Valencia, CA) to each 5-g lysate and incubated with end-over-end rotation for 1.5–2 h at 4ºC. Ni-NTA agarose was separated by centrifugation and washed with 5× volumes with wash buffer (1× PBS, 350 mM NaCl, 25 mM Tris, pH 8, 20 mM imidazole, 0.8 mM DTT, 5% glycerol, 1× protease inhibitors). The resin was then loaded onto a disposable column (Bio-Rad, Hercules, CA) to remove any remaining wash buffer; elution buffer (1× PBS, 100 mM NaCl, 25 mM Tris, pH 8, 350 mM imidazole, 1.0 mM DTT, 5% glycerol, 1× protease inhibitors) was then added at one-half the volume of the original agarose. The buffer in the resulting fraction was exchanged for HEKG5 (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]-K, pH 7.4, 1 mM EDTA, 50 mM KCl, 5% glycerol) via Zeba desalt columns (Pierce, Rockford, IL).
Protein concentrations were estimated via SDS–PAGE gel densitometry using dilutions of bovine serum albumin as a standard. We confirmed that purified proteins were monodispersed using gel filtration chromatography with Sepharose 200 (GE Healthcare), which was run at 0.4 ml/min with HEKG5 buffer with 1 mM DTT.
Immunoprecipitation and pull-downs
Immunoprecipitations of Bnr1-758-13myc with either anti-CBP (Millipore) or anti-HA were performed as previously described (Carroll et al., 1998; Kellogg and Moazed, 2002), except that the IP buffer contained 50 mM HEPES-K, pH 7.6, 1 mM ethylene glycol tetraacetic acid, 1 mM MgCl2, and 0.1% Tween-20 with either 75 or 150 mM KCl, as indicated.
The pull-down assay using GST-Gin4 or GST-Gin4Δkinase was performed as follows. Full-length 6His-Bnr1 was combined with GST alone, GST-Gin4, or GST-Gin4Δkinase prebound to beads and normalized for protein concentration. HEKG5, with a final concentration of either 75 or 150 mM KCl, was added to a final volume of 100 μl. Reactions were incubated for 1 h at 4ºC with end-over-end rotation. Beads were washed five times with 250 μl of HEKG5 with either 75 or 150 mM KCl. Beads were resuspended in 10 μl of 2× SDS–PAGE sample buffer (Boston Bioproducts, Ashland, MA). The samples were boiled for 5 min and centrifuged at maximum speed before loading. Similarly, full-length or Bnr1-N was combined with FLAG affinity beads with or without pCAL-n-FLAG-Gin4-C.
Homology scanning
We used methods described previously (Crutchley et al., 2009) to analyze the percentage identity of sequences with a sliding window. We used the budding yeast sequence of Bnr1 as the base sequence. For the data set of sensu lato yeast, we aligned S. cerevisiae, Saccharomyces bayanus, and Saccharomyces castelli. For the fungal data set, we aligned Kluyveromyces lactis, Candida albicans, Candida glabrata, and Ashbya gossypii. The output of the alignment program was then copied into Excel and used to generate graphical data.
Supplementary Material
Acknowledgments
We thank A. Bretscher, B. Goode, J. Thorner, and M. Iwase for strains and/or plasmids. We thank D. Kellogg for plasmids and the Gin4 antibody. We are grateful to Chiwa Kuroda for technical support. We acknowledge members of the Pellman lab for helpful discussions. S.M.B. was supported by National Institutes of Health National Research Service Award F32 GM076895. D.P. was supported by National Institutes of Health Grant GM61345.
Abbreviations used:
- DAD
Diaphanous autoregulatory domain
- DID
Diaphanous inhibitory domain
- DRF
Diaphanous-related formin
- RBD
Rho-binding domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0395) on August 23, 2012.
REFERENCES
- Altman R, Kellogg D. Control of mitotic events by Nap1 and the Gin4 kinase. J Cell Biol. 1997;138:119–130. doi: 10.1083/jcb.138.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang SF, Zhao ZS, Lim L, Manser E. DAAM1 is a formin required for centrosome re-orientation during cell migration. PLoS One. 2010;5(e):13064. doi: 10.1371/journal.pone.0013064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Park JE, Yu LR, Zhou M, Sakchaisri K, Park CJ, Kang YH, Thorner J, Veenstra TD, Lee KS. Direct phosphorylation and activation of a Nim1-related kinase Gin4 by Elm1 in budding yeast. J Biol Chem. 2006;281:27090–27098. doi: 10.1074/jbc.M601483200. [DOI] [PubMed] [Google Scholar]
- Barral Y, Parra M, Bidlingmaier S, Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187. doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini F, Gundersen GG. Formins and microtubules. Biochim Biophys Acta. 2011;1803:164–173. doi: 10.1016/j.bbamcr.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini F, Moseley JB, Schmoranzer J, Cassimeris L, Goode BL, Gundersen GG. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J Cell Biol. 2008;181:523–536. doi: 10.1083/jcb.200709029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquin N, Barral Y, Courbeyrette R, Blondel M, Snyder M, Mann C. Regulation of cytokinesis by the Elm1 protein kinase in Saccharomyces cerevisiae. J Cell Sci. 2000;113:1435–1445. doi: 10.1242/jcs.113.8.1435. [DOI] [PubMed] [Google Scholar]
- Buttery SM, Yoshida S, Pellman D. Yeast formins Bni1 and Bnr1 utilize different modes of cortical interaction during the assembly of actin cables. Mol Biol Cell. 2007;18:1826–1838. doi: 10.1091/mbc.E06-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11:237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Altman R, Schieltz D, Yates JR, Kellogg D. The septins are required for the mitosis-specific activation of the Gin4 kinase. J Cell Biol. 1998;143:709–717. doi: 10.1083/jcb.143.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarone M, Gould CJ, Moseley JB, Goode BL. Displacement of formins from growing barbed ends by bud14 is critical for actin cable architecture and function. Dev Cell. 2009;16:292–302. doi: 10.1016/j.devcel.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Myers CL, Andrews B, Boone C. Charting the genetic interaction map of a cell. Curr Opin Biotechnol. 2010;22:66–74. doi: 10.1016/j.copbio.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Crutchley J, King KM, Keaton MA, Szkotnicki L, Orlando DA, Zyla TR, Bardes ES, Lew DJ. Molecular dissection of the checkpoint kinase Hsl1p. Mol Biol Cell. 2009;20:1926–1936. doi: 10.1091/mbc.E08-08-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Pruyne D, Bretscher A. Formin-dependent actin assembly is regulated by distinct modes of Rho signaling in yeast. J Cell Biol. 2003;161:1081–1092. doi: 10.1083/jcb.200212040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista M, Blundell K, Longtine MS, Chow CJ, Adames N, Pringle JR, Peter M, Boone C. Bni1p, a yeast formin linking cdc42p and the actin cytoskeleton during polarized morphogenesis. Science. 1997;276:118–122. doi: 10.1126/science.276.5309.118. [DOI] [PubMed] [Google Scholar]
- Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol. 2002;4:32–41. doi: 10.1038/ncb718. [DOI] [PubMed] [Google Scholar]
- Faix J, Grosse R. Staying in shape with formins. Dev Cell. 2006;10:693–706. doi: 10.1016/j.devcel.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Fang X, Luo J, Nishihama R, Wloka C, Dravis C, Travaglia M, Iwase M, Vallen EA, Bi E. Biphasic targeting and cleavage furrow ingression directed by the tail of a myosin II. J Cell Biol. 2010;191:1333–1350. doi: 10.1083/jcb.201005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faty M, Fink M, Barral Y. Septins: a ring to part mother and daughter. Curr Genet. 2002;41:123–131. doi: 10.1007/s00294-002-0304-0. [DOI] [PubMed] [Google Scholar]
- Gao L, Bretscher A. Analysis of unregulated formin activity reveals how yeast can balance F-actin assembly between different microfilament-based organizations. Mol Biol Cell. 2008;19:1474–1484. doi: 10.1091/mbc.E07-05-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Bretscher A. Polarized growth in budding yeast in the absence of a localized formin. Mol Biol Cell. 2009;20:2540–2548. doi: 10.1091/mbc.E09-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Liu W, Bretscher A. The yeast formin Bnr1p has two localization regions that show spatially and temporally distinct association with septin structures. Mol Biol Cell. 2010;21:1253–1262. doi: 10.1091/mbc.E09-10-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G 3rd, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, Nogales E. Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol. 2011;195:993–1004. doi: 10.1083/jcb.201107123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladfelter AS, Kozubowski L, Zyla TR, Lew DJ. Interplay between septin organization, cell cycle and cell shape in yeast. J Cell Sci. 2005;118:1617–1628. doi: 10.1242/jcs.02286. [DOI] [PubMed] [Google Scholar]
- Gladfelter AS, Zyla TR, Lew DJ. Genetic interactions among regulators of septin organization. Eukaryot Cell. 2004;3:847–854. doi: 10.1128/EC.3.4.847-854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- Gorelik R, Yang C, Kameswaran V, Dominguez R, Svitkina T. Mechanisms of plasma membrane targeting of formin mDia2 through its amino terminal domains. Mol Biol Cell. 2011;22:189–201. doi: 10.1091/mbc.E10-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould CJ, Maiti S, Michelot A, Graziano BR, Blanchoin L, Goode BL. The formin DAD domain plays dual roles in autoinhibition and actin nucleation. Curr Biol. 2011;21:384–390. doi: 10.1016/j.cub.2011.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs HN. Formin proteins: a domain-based approach. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Imamura H, Tanaka K, Hihara T, Umikawa M, Kamei T, Takahashi K, Sasaki T, Takai Y. Bni1p and Bnr1p: downstream targets of the Rho family small G-proteins which interact with profilin and regulate actin cytoskeleton in Saccharomyces cerevisiae. EMBO J. 1997;16:2745–2755. doi: 10.1093/emboj/16.10.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase M, Toh-e A. Ybr267w is a new cytoplasmic protein belonging to the mitotic signaling network of Saccharomyces cerevisiae. Cell Struct Funct. 2004;29:1–15. doi: 10.1247/csf.29.1. [DOI] [PubMed] [Google Scholar]
- Jones EW. Vacuolar proteases in yeast Saccharomyces cerevisiae. Methods Enzymol. 1990;185:372–386. doi: 10.1016/0076-6879(90)85033-k. [DOI] [PubMed] [Google Scholar]
- Kadota J, Yamamoto T, Yoshiuchi S, Bi E, Tanaka K. Septin ring assembly requires concerted action of polarisome components, a PAK kinase Cla4p, and the actin cytoskeleton in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:5329–5345. doi: 10.1091/mbc.E04-03-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei T, Tanaka K, Hihara T, Umikawa M, Imamura H, Kikyo M, Ozaki K, Takai Y. Interaction of Bnr1p with a novel Src homology 3 domain-containing Hof1p. Implication in cytokinesis in Saccharomyces cerevisiae. J Biol Chem. 1998;273:28341–28345. doi: 10.1074/jbc.273.43.28341. [DOI] [PubMed] [Google Scholar]
- Kellogg DR, Moazed D. Protein- and immunoaffinity purification of multiprotein complexes. Methods Enzymol. 2002;351:172–183. doi: 10.1016/s0076-6879(02)51847-4. [DOI] [PubMed] [Google Scholar]
- Kikyo M, Tanaka K, Kamei T, Ozaki K, Fujiwara T, Inoue E, Takita Y, Ohya Y, Takai Y. An FH domain-containing Bnr1p is a multifunctional protein interacting with a variety of cytoskeletal proteins in Saccharomyces cerevisiae. Oncogene. 1999;18:7046–7054. doi: 10.1038/sj.onc.1203184. [DOI] [PubMed] [Google Scholar]
- Kohno H, et al. Bni1p implicated in cytoskeletal control is a putative target of Rho1p small GTP binding protein in Saccharomyces cerevisiae. EMBO J. 1996;15:6060–6068. [PMC free article] [PubMed] [Google Scholar]
- Lee L, Klee SK, Evangelista M, Boone C, Pellman D. Control of mitotic spindle position by the Saccharomyces cerevisiae formin Bni1p. J Cell Biol. 1999;144:947–961. doi: 10.1083/jcb.144.5.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ. The morphogenesis checkpoint: how yeast cells watch their figures. Curr Opin Cell Biol. 2003;15:648–653. doi: 10.1016/j.ceb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Li F, Higgs HN. The mouse Formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003;13:1335–1340. doi: 10.1016/s0960-9822(03)00540-2. [DOI] [PubMed] [Google Scholar]
- Li F, Higgs HN. Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. J Biol Chem. 2005;280:6986–6992. doi: 10.1074/jbc.M411605200. [DOI] [PubMed] [Google Scholar]
- Liu W, Sato A, Khadka D, Bharti R, Diaz H, Runnels LW, Habas R. Mechanism of activation of the formin protein Daam1. Proc Natl Acad Sci USA. 2008;105:210–215. doi: 10.1073/pnas.0707277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, Fares H, Pringle JR. Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J Cell Biol. 1998a;143:719–736. doi: 10.1083/jcb.143.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998b;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, Lew DJ. Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y. FORMIN a link between kinetochores and microtubule ends. Trends Cell Biol. 2011;21:625–629. doi: 10.1016/j.tcb.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SG, Rincon SA, Basu R, Perez P, Chang F. Regulation of the Formin for3p by cdc42p and bud6p. Mol Biol Cell. 2007;18:4155–4167. doi: 10.1091/mbc.E07-02-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Thorner J. Septins: molecular partitioning and the generation of cellular asymmetry. Cell Div. 2009;4:18. doi: 10.1186/1747-1028-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen EM, McDonald H, Yates J 3rd, Kellogg DR. Cell cycle-dependent assembly of a Gin4-septin complex. Mol Biol Cell. 2002;13:2091–2105. doi: 10.1091/mbc.01-10-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley JB, Maiti S, Goode BL. Formin proteins: purification and measurement of effects on actin assembly. Methods Enzymol. 2006;406:215–234. doi: 10.1016/S0076-6879(06)06016-2. [DOI] [PubMed] [Google Scholar]
- Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol. 2012;13:183–194. doi: 10.1038/nrm3284. [DOI] [PubMed] [Google Scholar]
- Ozaki-Kuroda K, Yamamoto Y, Nohara H, Kinoshita M, Fujiwara T, Irie K, Takai Y. Dynamic localization and function of Bni1p at the sites of directed growth in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:827–839. doi: 10.1128/MCB.21.3.827-839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Bretscher A. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J Cell Sci. 2000;113(Pt 3):365–375. doi: 10.1242/jcs.113.3.365. [DOI] [PubMed] [Google Scholar]
- Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C. Role of formins in actin assembly: nucleation and barbed-end association. Science. 2002;297:612–615. doi: 10.1126/science.1072309. [DOI] [PubMed] [Google Scholar]
- Pruyne D, Gao L, Bi E, Bretscher A. Stable and dynamic axes of polarity use distinct formin isoforms in budding yeast. Mol Biol Cell. 2004a;15:4971–4989. doi: 10.1091/mbc.E04-04-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Legesse-Miller A, Gao L, Dong Y, Bretscher A. Mechanisms of polarized growth and organelle segregation in yeast. Annu Rev Cell Dev Biol. 2004b;20:559–591. doi: 10.1146/annurev.cellbio.20.010403.103108. [DOI] [PubMed] [Google Scholar]
- Ramalingam N, Zhao H, Breitsprecher D, Lappalainen P, Faix J, Schleicher M. Phospholipids regulate localization and activity of mDia1 formin. Eur J Cell Biol. 2010;89:723–732. doi: 10.1016/j.ejcb.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Sagot I, Klee SK, Pellman D. Yeast formins regulate cell polarity by controlling the assembly of actin cables. Nat Cell Biol. 2002a;4:42–50. doi: 10.1038/ncb719. [DOI] [PubMed] [Google Scholar]
- Sagot I, Rodal AA, Moseley J, Goode BL, Pellman D. An actin nucleation mechanism mediated by Bni1 and profilin. Nat Cell Biol. 2002b;4:626–631. doi: 10.1038/ncb834. [DOI] [PubMed] [Google Scholar]
- Seth A, Otomo C, Rosen MK. Autoinhibition regulates cellular localization and actin assembly activity of the diaphanous-related formins FRLalpha and mDia1. J Cell Biol. 2006;174:701–713. doi: 10.1083/jcb.200605006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasan A, Kellogg D. The elm1 kinase functions in a mitotic signaling network in budding yeast. Mol Cell Biol. 1999;19:7983–7994. doi: 10.1128/mcb.19.12.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Taylor JM, Mack CP. Enhancement of mDia2 activity by Rho-kinase-dependent phosphorylation of the diaphanous autoregulatory domain. Biochem J. 2011;439:57–65. doi: 10.1042/BJ20101700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolliday N, VerPlank L, Li R. Rho1 directs formin-mediated actin ring assembly during budding yeast cytokinesis. Curr Biol. 2002;12:1864–1870. doi: 10.1016/s0960-9822(02)01238-1. [DOI] [PubMed] [Google Scholar]
- Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, Barral Y. The septin family of GTPases: architecture and dynamics. Nat Rev Mol Cell Biol. 2008;9:478–489. doi: 10.1038/nrm2407. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Kono K, Lowery DM, Bartolini S, Yaffe MB, Ohya Y, Pellman D. Polo-like kinase Cdc5 controls the local activation of Rho1 to promote cytokinesis. Science. 2006;313:108–111. doi: 10.1126/science.1126747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.