

Substratum stiffness controls the subcellular localization of Rac1b, a highly activated splice variant of the small GTPase Rac1. On stiff substrata, Rac1b localizes to the plasma membrane, forming a complex with NADPH oxidase and generating ROS, thus inducing the expression of the transcription factor Snail and downstream signaling to EMT.
Abstract
Epithelial–mesenchymal transition (EMT) is a form of epithelial plasticity implicated in fibrosis and tumor metastasis. Here we show that the mechanical rigidity of the microenvironment plays a pivotal role in the promotion of EMT by controlling the subcellular localization and downstream signaling of Rac GTPases. Soft substrata, with compliances comparable to that of normal mammary tissue, are protective against EMT, whereas stiffer substrata, with compliances characteristic of breast tumors, promote EMT. Rac1b, a highly activated splice variant of Rac1 found in tumors, localizes to the plasma membrane in cells cultured on stiff substrata or in collagen-rich regions of human breast tumors. At the membrane, Rac1b forms a complex with NADPH oxidase and promotes the production of reactive oxygen species, expression of Snail, and activation of the EMT program. In contrast, soft microenvironments inhibit the membrane localization of Rac1b and subsequent redox changes. These results reveal a novel mechanotransduction pathway in the regulation of epithelial plasticity via EMT.
INTRODUCTION
Epithelial plasticity permits simple or stratified epithelial tissues to actively modulate their cohesiveness and differentiated phenotypes in response to extracellular stimuli. An exemplary form of epithelial plasticity is the epithelial–mesenchymal transition (EMT), a phenotypic switch in which epithelial cells detach from their neighbors, acquire mesenchymal attributes, and become motile and invasive. This switch in cell fate is driven by characteristic changes in gene expression, including decreases in E-cadherin and increases in Snail-family transcription factors. During embryonic development, EMT endows the epithelium with sufficient plasticity to form the mesoderm, the neural crest, and the heart valves (Nieto, 2011). EMT likewise promotes epithelial plasticity during postnatal morphogenesis, particularly in mammary branching (Lee et al., 2011). Recent evidence suggests that EMT may also be activated in the adult under pathological conditions, including fibrosis and cancer (Acloque et al., 2009; Kalluri and Weinberg, 2009). Tumors that express mesenchymal markers have a greater tendency to be invasive and metastasize than those that do not and are associated with a poor prognosis (Fang et al., 2011; Kim et al., 2011).
These normal and pathological transitions in epithelial cell fate are highly regulated by soluble signals in the microenvironment. Matrix metalloproteinases (MMPs) are extracellular enzymes important for a wide variety of developmental and pathological processes associated with extensive tissue remodeling (Khokha and Werb, 2011). MMPs play a critical role in the development of many organs, including the mammary gland, where these enzymes control morphogenesis during puberty and epithelial remodeling during postlactational involution (Talhouk et al., 1991; Witty et al., 1995; Wiseman et al., 2003). MMP3 (stromelysin-1) and MMP7 (matrilysin-1) have been shown to induce EMT-associated fibrosis and carcinogenesis in adult transgenic mice (Lochter et al., 1997a, b; Rudolph-Owen et al., 1998; Sternlicht et al., 1999; Noe et al., 2001), and are associated with a number of human malignancies, including breast cancer (Heppner et al., 1996; Nakopoulou et al., 1999; Ghilardi et al., 2002). In mammary epithelial cells cultured on noncompliant substrata such as glass or polystyrene, treatment with MMP3 induces the expression of Rac1b (Radisky et al., 2005), which is a splice variant of the small GTPase Rac1 that contains an additional 19 amino acids adjacent to the switch II domain (a region important for interaction with effectors), resulting in impaired intrinsic GTPase activity (Singh et al., 2004). Rac1b is expressed in a variety of human cancers, including carcinomas of the breast and colon, where its expression sustains tumor cell survival (Matos and Jordan, 2005, 2008; Matos et al., 2008) and is highly correlated with malignant progression (Jordan et al., 1999; Schnelzer et al., 2000; Singh et al., 2004). Sustained expression of Rac1b elevates levels of cellular reactive oxygen species (ROS) and promotes expression of the EMT-associated transcription factor Snail (Radisky et al., 2005). In vivo, these alterations cause excessive deposition of extracellular matrix (ECM), disruptions in tissue structure, and fibrotic stiffening (Sternlicht et al., 1999). Although misexpression in the adult can have deleterious consequences, MMPs are expressed at high levels during development without causing pathological EMT or fibrosis, suggesting the existence of protective mechanisms in the microenvironment of normal tissues.
Mechanical signals from the microenvironment are important contributors to both normal and pathological developmental processes. The mechanical compliance, or rigidity, of the ECM substratum profoundly affects cell morphology, with stiffer substrata promoting integrin clustering, focal adhesion formation, cell spreading, and intracellular tension (Wang et al., 2002; Engler et al., 2004; Yeung et al., 2005; Guo et al., 2006). Substratum rigidity also regulates cell behaviors, including motility, proliferation, and differentiation (Pelham and Wang, 1997; Lo et al., 2000; Peyton and Putnam, 2005; Engler et al., 2006; Klein et al., 2009; Lui et al., 2011). In the mammary gland, stiff matrices with compliances characteristic of the average mammary tumor have been shown to disrupt mammary ductal architecture (Provenzano et al., 2008) and to promote neoplastic progression and a malignant phenotype (Paszek et al., 2005; Provenzano et al., 2008; Levental et al., 2009). Conversely, soft matrices with compliances comparable to that of the normal mammary gland promote normal mammary functional differentiation (Alcaraz et al., 2008). The molecular basis for these stiffness-mediated effects remains ill defined but is usually attributed to possible alterations in the transmission of force from focal adhesions through the actin cytoskeleton to the nucleus (Schwartz, 2010; Eyckmans et al., 2011).
Here we examine the role of the mechanical properties of the microenvironment in the regulation of epithelial plasticity via EMT. We use synthetic polyacrylamide substrata to mimic the mechanical properties of the normal murine mammary gland, which is refractory to EMT induction via MMP3, and those of the average mammary tumor, in which these signals promote EMT. Using this system, we show that stiffness-dependent control of epithelial plasticity is mediated by membrane targeting of Rac1b and consequent assembly and activation of NADPH oxidase to catalyze the production of ROS. Consistent with these observations, immunohistochemical analysis of Rac1b in human breast tissue biopsies reveals increased expression and altered cellular localization in desmoplastic regions of human breast tumors as compared with normal breast tissue. We further find that exogenously controlling the membrane localization of Rac1b bypasses signals from the mechanical microenvironment for induction of EMT. This study demonstrates that matrix compliance can drive EMT by controlling the localization and activity of molecular switches and unveils a novel mechanotransduction pathway independent of the direct transmission of force through the cytoskeleton.
RESULTS
The mechanical microenvironment controls induction of EMT by MMP3
Treatment of mammary epithelial cells with MMP3 results in cell scattering (Figure 1A) and gene expression changes consistent with an EMT program, including down-regulation of epithelial markers such as keratins and E-cadherin and up-regulation of the mesenchymal markers vimentin and α-smooth muscle actin (αSMA) (Figure 1, B and C). To investigate the role of substratum compliance in MMP3-induced EMT, we cultured cells on ECM-coated polyacrylamide substrata with a range of compliances (reported as Young's modulus, E) spanning those of the normal mammary gland and the average mammary tumor (Figure 1D and Supplemental Figure S1). On stiff substrata, exposure to MMP3 resulted in dissolution of intercellular adhesions and cell scattering (Figure 1E), as well as down-regulation of epithelial keratins and E-cadherin and up-regulation of Snail, vimentin, and αSMA at both the transcript and protein levels (Figure 1, F–H, and Supplemental Figure S2). Conversely, cells cultured on soft substrata were unresponsive to treatment with MMP3 and failed to scatter or express the EMT proteome (Figure 1, E–H, and Supplemental Figure S2). These data show that a microenvironment with physiologically normal compliance is protective of the epithelial cell phenotype and prevents EMT in response to MMP3.
FIGURE 1:
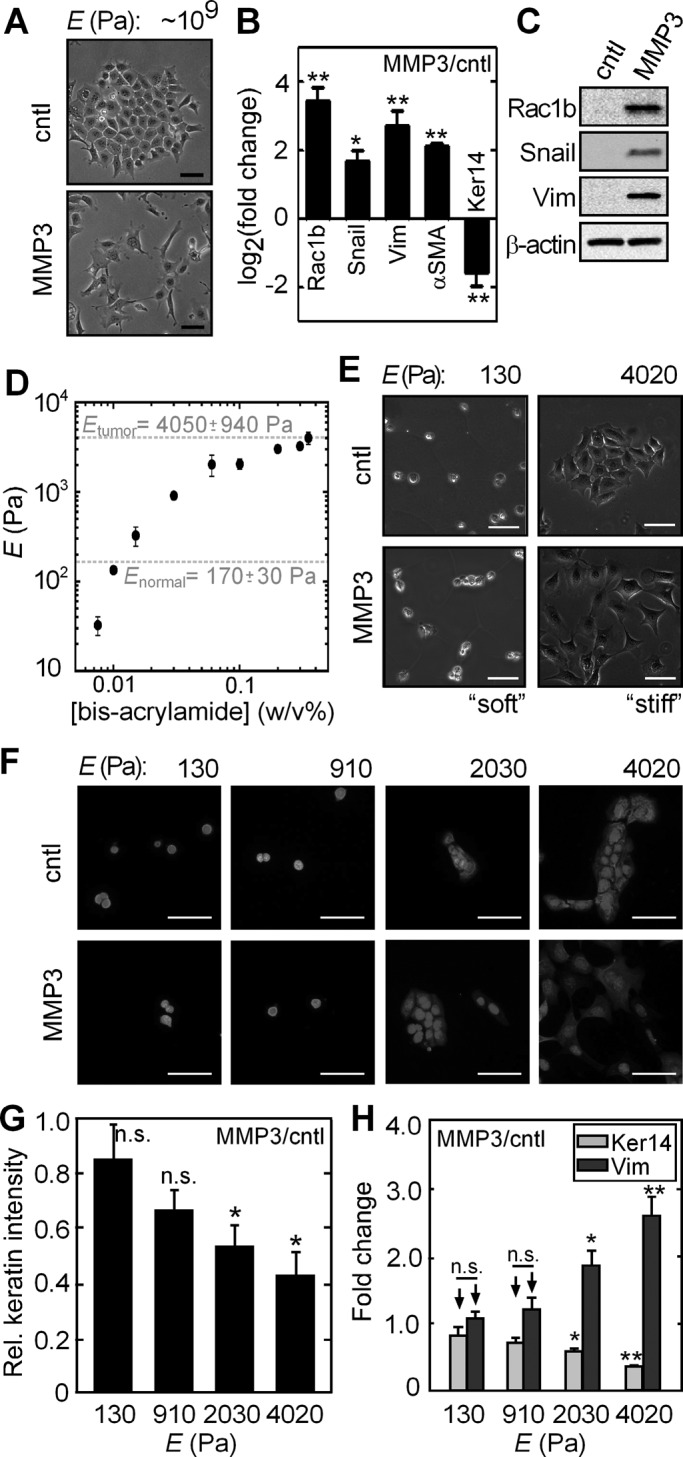
Soft, compliant substratum protects against MMP3-induced EMT. (A) Phase contrast images of control and MMP3-treated mammary epithelial cells on tissue culture–grade polystyrene. (B) Fold change in the transcript levels of EMT markers upon treatment with MMP3 in cells on polystyrene (n = 4). (C) Immunoblot analysis for EMT markers in control and MMP3-treated mammary epithelial cells on polystyrene. (D) Young's modulus (E) of polyacrylamide substrata as a function of bis-acrylamide concentration. Values reported for Enormal and Etumor are from Paszek et al. (2005). Error bars represent SD. (E) Phase contrast images of control and MMP3-treated mammary epithelial cells cultured on polyacrylamide substrata of soft (E ∼ 130 Pa) and stiff (E ∼ 4020 Pa) compliances. (F) Fluorescence images of control and MMP3-treated mammary epithelial cells cultured on polyacrylamide substrata of various compliances stained for keratins (red) and DNA (blue). (G) Relative keratin expression in MMP3-treated mammary epithelial cells on substrata of various compliances. (H) Fold change in the transcript levels of keratin-14 and vimentin upon treatment with MMP3 on substrata of various compliances (n = 4). Error bars represent SEM. *p < 0.05, **p < 0.01. Scale bars, 50 μm.
Soft substrata block production of ROS downstream of MMP3 and Rac1b
To determine how matrix compliance regulates MMP3-induced EMT, we evaluated the effect of substratum stiffness on downstream signaling. We found that treatment with MMP3 induced an increase in the levels of Rac1b that was independent of substratum rigidity (Figure 2A); thus cells cultured on soft substrata still respond to MMP3 by increasing expression of Rac1b. Moreover, ectopic expression of Rac1b in the absence of MMP3 using a recombinant adenovirus (Ad-Rac1b; Figure 2B) efficiently induced EMT in mammary epithelial cells cultured on polystyrene (Supplemental Figure S3) and on stiff polyacrylamide substrata (Figure 2C and Supplemental Figure S4) but did not induce EMT in cells cultured on soft substrata (Figure 2C and Supplemental Figure S4). We found that other downstream effects of MMP3-induced Rac1b expression, namely the increased cell spreading and production of ROS, were also only induced on stiff substrata, as cells cultured on soft substrata and treated with MMP3 showed significantly decreased cell spreading (Figure 2D) and ROS induction (Figure 2E) as compared with cells cultured on stiff substrata and treated with MMP3. Nonetheless, cells on soft substrata were still capable of undergoing EMT. Treatment with H2O2 to elevate the levels of ROS in the absence of MMP3 promoted EMT on all substrata, regardless of compliance (Figure 2F). Similarly, ectopic expression of Snail using a recombinant adenovirus (Ad-Snail; Kajita et al., 2004; Figure 2G) also promoted EMT on all substrata, regardless of compliance (Figure 2H). Soft substrata therefore block MMP3-induced EMT immediately downstream of Rac1b, suggesting that Rac1b is prevented from inducing ROS production in cells in soft, compliant microenvironments (Figure 2I).
FIGURE 2:

Soft, compliant substratum blocks MMP3-mediated induction of cell spreading and generation of ROS. (A) Fold change in the transcript levels of Rac1b for MMP3-treated cells cultured on polyacrylamide substrata of varying compliances (n = 4). (B) Immunoblot analysis of Rac1b, Rac1, and β-actin in cells transduced with Ad-Rac1b or Ad-GFP control. (C) Fold change in the transcript levels of keratin-14 and vimentin for cells ectopically expressing Rac1b on substrata of various compliances (n = 4). (D) Projected cell area as a function of substratum compliance (n = 4; 50 cells/n). (E) Relative change in cellular ROS levels (assessed by DCFDA) in cells cultured on polyacrylamide substrata of various compliances and treated with MMP3 (n = 4; 50 cells/n). (F) Fold change in the transcript levels of keratin-14 and vimentin for cells treated with H2O2 (n = 4). (G) Immunoblot analysis of Snail, E-cadherin, and β-actin in cells transduced with Ad-Snail or Ad-GFP control. (H) Fold change in the transcript levels of keratin-14, vimentin, and E-cadherin for cells ectopically expressing Snail on substrata of various compliances (n = 3). (I) Scheme outlining EMT induction pathway for MMP3. Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Transforming growth factor-β (TGFβ) is well recognized as a mediator of EMT in a variety of physiological contexts (Bhowmick et al., 2001; Iwano et al., 2002; Kim et al., 2006) and can be activated by treatment with H2O2 (Iglesias-De La Cruz et al., 2001). To assess whether TGFβ activation plays a role in promoting MMP3-induced EMT in cells on stiff microenvironments, we monitored the amount of active and total TGFβ through the use of a luciferase reporter system (Abe et al., 1994). We found no difference in TGFβ activation or expression in the conditioned medium obtained from cells cultured on the different substrata (Supplemental Figure S5). Although this assay could not rule out differences in activated TGFβ that may have remained associated with the cell membrane or ECM, blocking TGFβ signaling by treating with a pharmacological inhibitor of the type I TGFβ receptor kinase did not affect the MMP3- or H2O2-induced EMT, as defined by down-regulation of keratin-14 and E-cadherin, or up-regulation of vimentin, αSMA, and Snail (Supplemental Figure S5).
Soft substrata inhibit ROS production by blocking assembly of Rac1b with NADPH oxidase
Treatment with ROS-quenching agents completely abrogates MMP3- and Rac1b-mediated induction of EMT in mammary epithelial cells (Radisky et al., 2005; Nelson et al., 2008). Rac-family members can regulate the production of ROS by associating with the multimolecular membrane-associated enzyme, NADPH oxidase (Hordijk, 2006). Upon translocation to the membrane, activated Rac binds to the Nox activator p67phox to form an activated complex (Diekmann et al., 1994). Rac1b had not previously been shown to interact with NADPH oxidase, but we hypothesized that, as an activated GTPase, it may facilitate assembly of this enzyme. To characterize the role of matrix compliance in Rac1b-induced ROS production, we determined the expression levels of NADPH oxidase subunits in cells cultured on substrata of varying compliance. We found that components of NADPH oxidase, including the catalytic subunit gp91phox/Nox2, the Nox organizer p47phox, and the Nox activator p67phox, were expressed in mammary epithelial cells independent of substratum rigidity or Rac1b expression (Figure 3A). Furthermore, Rac1b coimmunoprecipitated with components of the NADPH oxidase complex, including p67phox and p47phox, when cells were cultured on stiff substrata (Figure 3B). However, we found that formation of this complex was significantly diminished when cells were cultured on soft substrata (Figure 3B). Similar stiffness-dependent assembly was observed in MCF10A human mammary epithelial cells (Supplemental Figure S6), suggesting that soft matrices inhibit the assembly of NADPH oxidase by blocking the interaction between Rac1b and p67phox. Consistent with these results, Rac1b-expressing cells cultured on soft substrata were prevented from producing ROS (Figure 3C). Moreover, inhibiting NADPH oxidase using diphenylene iodonium (DPI) blocked induction of EMT by MMP3 (Figure 3D) and Rac1b (Figure 3E). These data show that soft, compliant substrata protect cells from MMP3-induced EMT by disrupting the interaction between Rac1b and NADPH oxidase and thereby inhibiting production of ROS.
FIGURE 3:

NADPH oxidase associates with Rac1b to mediate MMP3-induced EMT. (A) Immunoblot analysis of gp91phox, p67phox, p47phox, Rac1b, GFP, and β-actin for cells ectopically expressing Rac1b or control and cultured on substrata of various compliances. (B) SCp2 cells were transduced with GFP or YFP-Rac1b and grown on soft (130 Pa) or stiff (4020 Pa) substrata. Immunoprecipitation was performed using anti-GFP antibody. Whole-cell lysates and IP products were assayed by immunoblot analysis to detect p67phox, p47phox, Rac1b, and GFP. (C) Fold change in cellular ROS levels in cells transduced with Ad-Rac1b or Ad-GFP control and grown on soft or stiff substrata (n = 4; 60 cells/n). (D) Changes in EMT marker expression in cells treated with and without MMP3 and dimethyl sulfoxide (vehicle control) or the NADPH oxidase inhibitor DPI (n = 5). (E) Changes in EMT marker expression in cells transduced with Ad-Rac1b or Ad-GFP control and treated with vehicle or DPI (n = 4). Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Soft substrata block localization of Rac1b to the plasma membrane
Rac1b is a self-activating splice variant of Rac1 (Fiegen et al., 2004). That its association with NADPH oxidase and promotion of cellular ROS were inhibited by culture on soft substrata suggested that Rac1b signaling is regulated by mechanisms other than activation. Membrane localization of Rac1 is mediated by posttranslational prenylation (Didsbury et al., 1990) and is required for activation of its downstream effectors (del Pozo et al., 2000, 2002), including NADPH oxidase–mediated generation of ROS (Ando et al., 1992; Heyworth et al., 1993). We therefore monitored the membrane localization of Rac1b in mammary epithelial cells by expressing ectopically a yellow fluorescent protein (YFP) fusion protein (YFP-Rac1b). In cells cultured on glass or stiff polyacrylamide substrata, YFP-Rac1b localized to the cell periphery and in membrane ruffles (Figure 4A). However, in cells cultured on soft substrata, YFP-Rac1b was primarily internalized within a central region of the cytoplasm (Figure 4A), similar to previously reported descriptions of the localization of Rac1 within cholesterol-enriched membrane microdomains in suspended cells (del Pozo et al., 2004, 2005). These effects on the subcellular localization of Rac1b were confirmed via cellular fractionation followed by immunoblotting analysis (Supplemental Figure S6). These results indicate that soft, compliant substratum blocks the localization of Rac1b to the plasma membrane.
FIGURE 4:

Membrane localization of Rac1b is required for MMP3-induced EMT. (A) Fluorescence images of GFP, YFP-Rac1b, YFP-Rac1b-SAAX, and YFP-Rac1b-myr in SCp2 cells grown on substrata of various compliances. (B) SCp2 cells were transduced with GFP, YFP-Rac1b, YFP-Rac1b-SAAX, and YFP-Rac1b-myr and grown on soft (130 Pa) or stiff (4020 Pa) substrata. Immunoprecipitation was performed using anti-GFP antibody. Whole-cell lysates and immunoprecipitated products were assayed by immunoblot analysis to detect p67phox, Rac1b, and GFP. (C) Fold change in cellular ROS levels in cells transduced with GFP, Rac1b, Rac1b-myr, or Rac1b-SAAX (n = 4; 60 cells/n). (D, E) Fold change in the transcript levels of keratin-14 and vimentin for cells ectopically expressing YFP-Rac1b-SAAX (D) or YFP-Rac1b-myr (E; n = 4). (F) Disrupting isoprenoid biosynthesis using mevinolin reduces cholesterol levels (stained with filipin) and YFP-Rac1b localization to the membrane. (G) Marker transcript levels for cells treated with and without MMP3 and mevinolin; statistics indicate vehicle vs. mevinolin (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars, 10 μm.
Isoprenylation of Rac1 occurs on a carboxy-terminal CAAX motif (Kinsella et al., 1991), which is located within the sixth coding exon and is conserved in the Rac1b splice variant; isoprenylation facilitates localization of Rac1 to the plasma membrane (Jordan et al., 1999; Schnelzer et al., 2000). To determine whether the membrane localization of Rac1b was required for its induction of EMT, we generated mutants that either blocked posttranslational isoprenylation by replacing the prenyl-attachment cysteine with a serine residue (Rac1b-SAAX) or had an additional membrane-targeting myristoylation signal (Rac1b-myr), thus either blocking or promoting association with the plasma membrane (del Pozo et al., 2000; Michaelson et al., 2008). Rac1b-SAAX was sequestered from the membrane on all culture surfaces and predominantly localized to the nucleus (Figure 4A), consistent with previous reports showing that the carboxy-terminal polybasic region of Rac1 can act as a nuclear localization sequence (Lanning et al., 2004). In contrast, Rac1b-myr was targeted to the plasma membrane on both stiff and soft substrata (Figure 4A). These imaging results were confirmed by immunoblotting analysis of Rac1b in membrane fractions of the cell lysates (Supplemental Figure S6). Blocking membrane localization using Rac1b-SAAX inhibited the association between Rac1b and NADPH oxidase (Figure 4B) and production of ROS (Figure 4C). In contrast, promoting membrane localization using Rac1b-myr promoted the association between Rac1b and the NADPH oxidase complex even on soft substrata (Figure 4B). To determine whether the membrane localization of Rac1b and subsequent production of ROS are required for Rac1b-induced EMT, we determined the expression levels of genes associated with EMT. Sequestering Rac1b away from the plasma membrane using Rac1b-SAAX blocked the induction of EMT in cells on all culture surfaces, including stiff substrata (Figure 4D and Supplemental Figures S4 and S7). Conversely, targeting Rac1b to the membrane using Rac1b-myr induced EMT on both soft and stiff substrata (Figure 4E and Supplemental Figures S4 and S7). Consistent with these results, disrupting isoprenoid biosynthesis using mevinolin (lovastatin) blocked YFP-Rac1b localization to the membrane (Figure 4F) and abrogated the induction of EMT by MMP3 (Figure 4G). These data suggest that localization of Rac1b to the plasma membrane is required for its downstream signaling and induction of EMT.
Rac1b localizes to the membrane in human breast tumors but not normal mammary gland
To determine how substratum stiffness–dependent regulation of Rac1b localization functions in vivo, we examined the distribution of the protein in situ in biopsies taken from nonmalignant human breast tissue and collagen-rich regions of human breast tumors, which are regions of enhanced stiffness (Provenzano et al., 2008; Levental et al., 2009; Lopez et al., 2011). Immunohistochemical analysis revealed that the endogenously low levels of Rac1b were primarily cytoplasmic in normal mammary tissue (Figure 5A). In marked contrast, Rac1b showed an apparent relocalization in the epithelial cells in collagen-rich regions of human breast tumors (Figure 5B), consistent with our findings in cell culture models that stiff microenvironments promote Rac1b membrane localization and downstream signaling.
FIGURE 5:

Rac1b is localized to the membrane in human breast tumor epithelium but not nontumorigenic epithelium. Immunohistochemical analysis of collagen and Rac1b in (A) normal human mammary epithelium and (B) tumorigenic epithelium. Scale bars, 100 μm.
Soft substrata control Rac1b localization through integrin clustering
Rac1 is recruited to the plasma membrane and maintained in lipid rafts through signaling involving integrins and phosphorylated caveolin (del Pozo et al., 2004, 2005). Integrins block the endocytosis of lipid rafts and maintain Rac1 at the surface by retaining phosphorylated caveolin within focal adhesions. That Rac1b was sequestered from the membrane when cells were cultured on soft substrata suggested that integrin-mediated adhesions would also be reduced under these conditions. We evaluated focal adhesions by staining for activated phosphorylated focal adhesion kinase (pFAK; Figure 6A). We found that cells cultured on soft substrata had significantly fewer pFAK-containing focal adhesions than those cultured on stiff substrata (Figure 6, B and C), consistent with previous reports (Paszek et al., 2005; Wei et al., 2008). However, treatment with MMP3 increased the number of focal adhesions when cells were cultured on stiff, but not on soft, substrata (Figure 6, A–C, and Supplemental Figure S6). Consistent with previous reports indicating a key role for β1-integrins in cellular response to substratum stiffness (Paszek et al., 2005), we found that disrupting focal adhesions by knocking down β1-integrin (Figure 7A) prevented MMP3-induced ROS (Figure 7B) and EMT (Figure 7C). Conversely, expressing an autoclustering β1-integrin mutant (β1V737N; Paszek et al., 2005) promoted membrane localization of YFP-Rac1b (Figure 7D) and allowed Rac1b to interact with NADPH oxidase (Figure 7E). These changes resulted in significant production of ROS and promoted EMT on soft substrata (Figure 7, F and G, and Supplemental Figure S7). These results indicate that integrin-mediated adhesion transduces mechanical signals to control membrane localization and functional activation of Rac1b, which in turn induces NADPH oxidase–mediated production of ROS and EMT.
FIGURE 6:
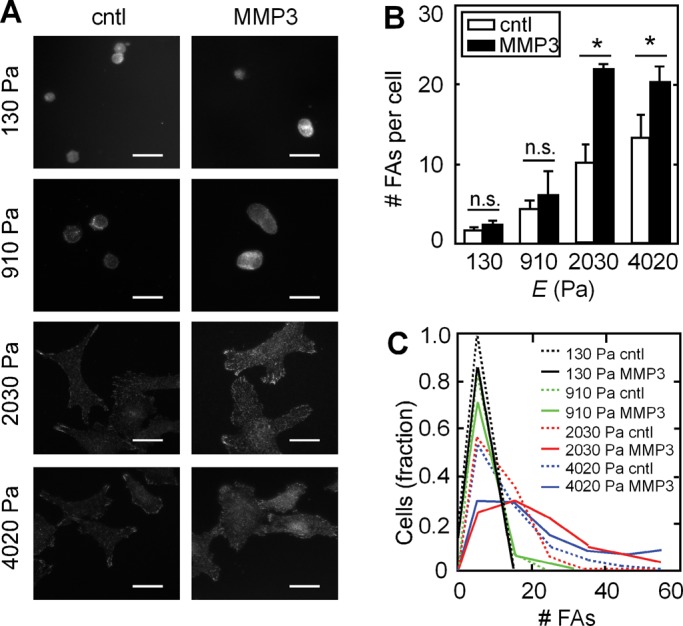
Substratum compliance alters focal adhesion formation. (A) Fluorescence images of cells cultured with or without MMP3 on substrata of various compliances and stained for pFAK Y397. (B) Quantification of the average number of FAs per cell in cells treated with or without MMP3 on substrata of various compliances. (C) Histogram of number of FAs in cells treated with or without MMP3 on substrata of various compliances. *p < 0.05. Scale bars, 25 μm.
FIGURE 7:

Clustering of β1-integrin is required for Rac1b localization to the membrane and MMP3-induced EMT. (A) Depletion of β1 integrin using short hairpin RNA (shRNA) targeting β1 integrin. (B) Fold change in cellular ROS levels in cells transfected with shRNA targeting β1-integrin (β1 KD) or scrambled shRNA (cntl; n = 4; 60 cells/n). (C) Fold change in the transcript levels of keratin-14 and vimentin for control cells or knockdown for β1-integrin (β1 KD; n = 3). (D) Fluorescence images of YFP-Rac1b in cells cultured on soft substrata and simultaneously expressing control β1-integrin or the autoclustering mutant β1V737N. Scale bars, 10 μm. (E) SCp2 cells were cotransfected with Rac1b together with β1V737N and cultured on soft (130 Pa) or stiff (4020 Pa) substrata. Immunoprecipitation (IP) was performed using anti-Rac1b antibody. Whole-cell lysates and IP products were assayed by immunoblot analysis to detect p67phox and Rac1b. (F) Fold change in cellular ROS levels in cells expressing Rac1b and β1V737N and cultured on soft (130 Pa) or stiff (4020 Pa) substrata (n = 4; 60 cells/n). (G) Fold change in the transcript levels of keratin-14 and vimentin for cells simultaneously expressing Rac1b and β1V737N and cultured on soft substrata (n = 4). Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Here we showed that soft microenvironments characteristic of the normal mammary gland protect against pathological insult from MMP3, which induces epithelial dedifferentiation and acquisition of fibrotic attributes in stiffer settings. These findings are congruent with the effects of mechanical stiffness on scattering of Madin–Darby canine kidney epithelial cells (de Rooij et al., 2005) and amplification of fibrosis in the lung (Liu et al., 2010). Although the mechanical compliance of the microenvironment is increasingly recognized as a regulator of cellular phenotype, the molecular basis for its effects on cells is largely undefined. Our data support a model in which substratum compliance regulates MMP3-induced EMT through integrin-mediated modulation of Rac1b localization, NADPH oxidase assembly, and downstream signaling (Figure 8). In rigid microenvironments, Rac1b is recruited to the plasma membrane and interacts with components of the NADPH oxidase complex, which in turn induces ROS production, activation of Snail, and induction of EMT. In tissue-mimetic soft microenvironments, mammary epithelial cells form very few cell–ECM adhesions, Rac1b is not translocated to the membrane, and EMT is prevented. The mechanical properties of the tissue microenvironment thus modulate signaling events controlling epithelial plasticity.
FIGURE 8:

The role of matrix compliance in regulating EMT. When cells encounter a rigid matrix, integrins become activated, which leads to clustering and subsequent recruitment and activation of signaling proteins, including FAK. This force-induced signal promotes localization of Rac1b to the plasma membrane and formation of the NADPH oxidase complex, which results in increased generation of ROS and EMT. In cells cultured on soft matrix, integrins exist in a resting and inactive state and thereby inhibit formation of focal adhesions, the membrane localization of Rac1b, and its interaction with NADPH oxidase. Therefore, soft, compliant matrix inhibits MMP3-induced ROS production and EMT.
Small GTPases act as molecular switches by cycling between GTP-bound and GDP-bound states through nucleotide exchange and hydrolysis. Rac1b contains a 19–amino acid insertion that endows it with accelerated GEF-independent nucleotide exchange and decelerated hydrolysis (Fiegen et al., 2004; Orlichenko et al., 2010); as a self-activating GTPase, Rac1b essentially functions as a naturally occurring, constitutively active variant of Rac1. Here we show that the downstream signaling of Rac1b is not immutable but is determined by the physical properties of the cellular microenvironment. Given that Rac1b is expressed in a wide variety of embryonic epithelia, as well as in tumors (Jordan et al., 1999), this mechanical control suggests a possible mechanism for differential signaling in development and disease. This may be particularly relevant for cancer. Because Rac1b is highly expressed in human breast cancer and particularly so in regions predicted to be stiffened (Figure 5B), our data suggest that therapeutic strategies aimed at reducing the mechanical rigidity of tumors or chemically blocking the association of Rac1b with the plasma membrane may ameliorate some of its pathological effects. The latter strategy is especially intriguing because terpenoid chemotherapeutics disrupt the isoprenylation of Rac1 in rat mammary epithelium in vivo (Ren and Gould, 1998), and statins exhibit antitumor effects for many types of cancer (Sassano and Platanias, 2008).
Studies of mechanotransduction—the mechanisms by which individual cells sense mechanical stimuli such as substratum rigidity and transduce this information into alterations in biochemical signaling and gene expression—have revealed that mechanical cues are transmitted from focal adhesions through direct linkages to the cytoskeleton (Schwartz, 2010; Eyckmans et al., 2011). Our data suggest that information about substratum rigidity may also be transmitted from focal adhesions through their effects on localization of key signaling molecules. Indeed, growing fibroblasts in suspension culture results in a complete loss of adhesion and blocks signaling downstream of Rac1 to its effector PAK (del Pozo et al., 2000, 2002). These previous studies suggested that adhesion (or the lack thereof) acted in a binary capacity to promote membrane localization of Rac1 and thereby control anchorage-dependent cell growth (del Pozo et al., 2004, 2005). Here we propose that the targeting of small GTPases to the membrane may be fine tuned by the compliance of the substratum, and thus may play a larger role in rigidity sensing and control of epithelial phenotype. Indeed, we found that the membrane localization of the Rac1 parent protein is also sensitive to matrix stiffness (Supplemental Figure S6), likely affecting signaling through a large number of downstream effectors. These data suggest a novel mode of mechanotransduction in which information transmitted through cell–ECM adhesions is conveyed to the cell through altered protein localization.
The precise mechanisms by which substratum stiffness regulates the membrane localization of Rac1 and Rac1b remain to be determined. Nonetheless, it is clear that integrin-mediated signaling plays a role, as expressing the autoclustering β1V737N mutant of β1-integrin promoted Rac1b membrane localization and ROS induction when cells were cultured on soft substrata. Our findings are concordant with those of a recent study that suggested that integrin–ligand anchoring plays a role in cellular response to soft polyacrylamide substrata (Trappmann et al., 2012), and future investigation is needed to decouple possible effects on integrin clustering due to ligand anchoring from those due to compliance per se. Regardless, integrin clustering leads to the activation of a number of signaling pathways, including those downstream of Src-p190RhoGAP (Arthur et al., 2000) and integrin-linked kinase (ILK; Legate et al., 2006), which affect cytoskeletal contractility in Rho-dependent and Rho-independent manners, respectively (Deng et al., 2001; Brunton et al., 2004). Artificially inducing actomyosin-mediated contraction in cells on soft substrata promoted the translocation of Rac1b to the plasma membrane and induction of ROS (Supplemental Figure S8), suggesting a likely role for activation of the cell's contractile machinery in the regulation of Rac1b localization. Curiously, the membrane localization and functional activation of Rac1b appear to be independent of both Src and RhoA signaling but highly sensitive to the levels of ILK (Supplemental Figure S8). Although this is the first implication of a possible relationship with Rac1b, ILK has been shown to lead to activation of the Rac1 parent protein (Boulter et al., 2006; Ho and Dagnino, 2012). Given that Rac1b is constitutively activated, it will be interesting to determine how ILK and other integrin-mediated signaling pathways function to control Rac1b localization to the plasma membrane.
Our data also suggest that the mechanical properties of the microenvironment tune the response of cells to signals that induce epithelial plasticity through EMT and a potentially fibrotic response. In effect, more-rigid microenvironments promote the induction of EMT, which may amplify fibrosis and neoplastic progression. EMT can lead to activation of myofibroblastic characteristics, associated with deposition and contraction of ECM proteins, including collagen and fibronectin, resulting in an increase in matrix density and rigidity. This ECM-associated stiffening of the tissue can in turn act to further promote pathological EMT, thus creating a mechanical self-sustaining positive-feedback loop. Our results provide insight into how to uncouple this loop—by normalizing the microenvironment, as perceived by cells, back to that of a nondiseased state—as a potential therapeutic approach to treatment of fibrosis and cancer.
MATERIALS AND METHODS
Cell culture and reagents
SCp2 mouse mammary epithelial cells were cultured and treated with MMP3 as described previously (Radisky et al., 2005), with a 4-d time point for all experiments unless otherwise specified. Stimulation with MMP3 was achieved by treating cells with medium that had been conditioned by cells stably expressing tetracycline-regulated, autoactivated MMP3; MMP3 expression was induced by growth in the absence of tetracycline. Conditioned medium from cells repressed by treatment with tetracycline was used for controls. For gene repression, a 4 mg/ml stock solution of tetracycline (Sigma-Aldrich, St. Louis, MO) in 100% ethanol was diluted 1:800 into culture medium and changed daily. For other experiments, mammary epithelial cells were treated with 25 μM H2O2 for 4 d. The following reagents were used at the concentrations indicated: NADPH oxidase inhibitor DPI, 0.1 μM (Sigma-Aldrich); mevinolin, 0.5 μM (Sigma-Aldrich); TGFβ type I receptor (Alk-5) kinase inhibitor [3-(pyridine-2-yl)-4-(4-quinonyl)]-1H-pyrazole, 0.36 μM (Calbiochem, La Jolla, CA); calyculin A, 1 μM (Calbiochem); Src inhibitor PP2 (Tocris Bioscience, Ellisville, MO); and Rho inhibitor I (Cytoskeleton, Denver, CO).
Synthetic substrata
Polyacrylamide gels with a fixed acrylamide monomer concentration of 5% and bis-acrylamide cross-linker concentrations ranging from 0.01 to 0.35% were prepared using a protocol adapted from Pelham and Wang (1997). Briefly, glass coverslips were treated with 0.1 N NaOH for 5 min, followed by rinsing in Millipore water (Millipore, Billerica, MA) and drying. Coverslips were then soaked in a 2% (vol/vol) solution of aminopropyltrimethoxysilane (Sigma-Aldrich) in acetone for 10 min, rinsed extensively with acetone, and air dried. Next the coverslips were incubated in a solution of 0.5% (vol/vol) glutaraldehyde (Sigma-Aldrich) in phosphate-buffered saline (PBS) for 30 min, followed by extensive rinsing with Millipore water. Acrylamide was mixed with bis-acrylamide, ammonium persulfate (1/200), and N,N,N′,N′-tetramethylethylenediamine (1/2000, TEMED; Bio-Rad, Hercules, CA), and 36 μl was placed onto the activated coverslip surface. The acrylamide solution was then covered by a 32-mm-diameter dichlorodimethylsilane-treated (Sigma-Aldrich) coverslip and allowed to polymerize for 30 min. The top coverslip was carefully removed, and the gel was rinsed with PBS.
Fibronectin is normally present within the mouse mammary gland (Friedman et al., 1984) and by itself does not promote EMT in mammary epithelial cells (Shintani et al., 2006). Accordingly, fibronectin was cross-linked to the gel surfaces through the use of sulfosuccinimidyl-6-(4′-azido-2′-nitrophenyl-amino)-hexanoate (sulfo-SANPAH; Pierce, Rockford, IL) chemistry. Polyacrylamide gels were rinsed in a solution of 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 8.5. The gel surfaces were then treated with a 2 mM solution of sulfo-SANPAH and exposed to a germicidal ultraviolet lamp for 10 min. Gels were rinsed once with 50 mM HEPES, pH 8.5, and then the sulfo-SANPAH treatment was repeated. Gels were rinsed twice with HEPES and then incubated with 0.2 mg/ml human fibronectin (BD Biosciences, San Diego, CA) overnight at 4°C. Before plating of cells, gels were rinsed extensively with PBS, followed by incubation in culture media at 37ºC for 1 h.
Characterization of substrata by enzyme-linked immunosorbent assay
The amount of fibronectin conjugated to the surface of the polyacrylamide gels was measured using an enzyme-linked immunosorbent assay. Fibronectin-conjugated gels were washed extensively in PBS to remove excess fibronectin and then incubated with blocking buffer (1% bovine serum albumin in PBS). The gels were then incubated successively with biotinylated anti-fibronectin, streptavidin-conjugated β-galatosidase (Invitrogen, Carlsbad, CA), and p-nitrophenyl-β-d-galactopyranoside (Sigma-Aldrich). The reaction was stopped with 0.5 M sodium carbonate. The optical density at 405 nm was measured in a plate reader (Promega, Madison, WI) and compared with that for untreated gels. This analysis revealed that substrata had comparable densities of surface-associated ECM protein (Supplemental Figure S1).
Characterization of substrata by rheometry
The viscoelastic properties of the polyacrylamide gels were measured with an Anton Paar MCR 501 rheometer (Anton Paar, Ashland, VA), using 25-mm parallel plates at a gap height of 0.8–1.3 mm. Bulk gels (∼1 mm thick) were prepared by polymerizing acrylamide solution between two dichlorodimethylsilane-treated coverslips. Measurements were performed on gels hydrated with culture media. A Peltier plate was used to maintain the temperature at 37°C, and a humidity chamber was used to prevent dehydration of the sample. All measurements were performed in the linear viscoelastic regime. Values of Young's modulus (E) of the polyacrylamide gels were computed from measured shear modulus, G, using the equation E = 2G(1 + ν), where ν is the Poisson ratio (ν = 0.48 for polyacrylamide; Boudou et al., 2006).
Transfections and adenoviral transductions
Rac1b was cloned from cDNA isolated from SCp2 mouse mammary epithelial cells and fused with YFP using the pEYFP-C1 plasmid (Clontech, Mountain View, CA) as previously described (Radisky et al., 2005). SAAX and myristoylation mutants were generated using the QuikChange II XL site-directed mutagenesis kit (Stratagene, Santa Clara, CA). The autoclustering β1-integrin mutant (β1V737N) was generated using the QuikChange Lightning site-directed mutagenesis kit (Stratagene). Mutations were verified by sequencing. Cells were plated onto polyacrylamide substrata 24 h after transfection with plasmids using FuGENE HD (Roche, Indianapolis, IN). Recombinant adenovirus encoding human Snail (Ad-Snail) was a gift from Paul Wade (National Institute of Environmental Health Sciences, Research Triangle Park, NC; Kajita et al., 2004). Custom recombinant adenoviruses encoding YFP-tagged mouse Rac1b (Ad-Rac1b), its mutants (Ad-Rac1b-myr and Ad-Rac1b-SAAX), and control adenovirus encoding GFP (Ad-GFP) were obtained from Vector BioLabs (Burlingame, CA). High-titer preparations of recombinant adenoviruses were generated using the AdEasy virus purification kit (Stratagene). Cells were transduced at a multiplicity of infection resulting in >99% transduction efficiency.
For knockdown of β1-integrin, gene silencing was accomplished using Mission shRNA as previously described (Orlichenko et al., 2010); five constructs targeting mouse β1-integrin were evaluated, and significant knockdown was observed using clone NM_010578,1-111s1c1.
Quantitative real-time PCR
Total RNA was isolated using either a Qiagen RNeasy Mini Kit or TRIzol reagent (Invitrogen), followed by cDNA synthesis using a Super Script First-Strand Synthesis Kit (Invitrogen). Transcript levels were measured by quantitative real-time PCR using a Bio-Rad Mini-Opticon instrument and SYBR Green chemistry. Amplification was followed by melting curve analysis to verify the presence of a single PCR product. Primers for keratin-14, E-cadherin, αSMA, Rac1b, Snail, vimentin, and 18S rRNA (Supplemental Table S1) were designed using Beacon Designer software (Bio-Rad) and determined to be specific by BLAST and dissociation curve analysis. The expression level of each mRNA was normalized to that of 18S in the same sample.
Coimmunoprecipitation
SCp2 or MCF10A cells expressing GFP or YFP-Rac1b were cultured on soft (130 Pa) or stiff (4020 Pa) polyacrylamide substratum. Whole-cell lysates were prepared using lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.1% NP-40, 1 mM dithiothreitol, 1 mM Na3VO4, 10 mM NaF, and protease inhibitor cocktail) and immunoprecipitated with anti-GFP (Sigma-Aldrich) or anti-Rac1b (Radisky et al., 2005) antibody. The whole-cell lysates and immunoprecipitation products were subject to immunoblotting to detect p67phox (Millipore), p47phox (Millipore), Rac1b (Millipore), and GFP.
Immunoblotting
Samples were lysed using RIPA buffer, mixed with Laemmli sample buffer, boiled at 95°C for 5 min, resolved by SDS–PAGE, and transferred to nitrocellulose. Membranes were blocked in 5% milk and incubated overnight at 4°C in blocking buffer containing antibodies specific for Rac1b (Radisky et al., 2005), Rac1 (Cytoskeleton), Snail (Cell Signaling, Beverly, MA), E-cadherin (Cell Signaling), or actin (Cell Signaling). The signals were visualized using the ECL Plus Western Blotting Detection System (GE Healthcare, Piscataway, NJ).
Immunofluorescence
For staining of keratins, samples were fixed with an ice-cold solution of 1:1 methanol/acetone at −20ºC for 10 min, followed by rinsing with PBS. After blocking for 1 h with 10% goat serum (Sigma-Aldrich) and 0.5% Tween (Sigma-Aldrich), the samples were then incubated for 1 h with rabbit anti-pan keratin (Dako, Carpinteria, CA). Samples were then rinsed and incubated with Alexa 594 goat anti–rabbit immunoglobulin G (IgG; Invitrogen). Nuclei were counterstained with Hoechst 33342 (Invitrogen). Levels of cellular ROS were assessed by staining with 5-(and-6)carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA; Invitrogen) or CellROX Deep Red reagent (Invitrogen). Samples were incubated in the dark with 50 μM carboxy-H2DCFDA or 5 μM CellROX in serum- and phenol red–free media for 30 min. For staining of pFAK, samples were fixed with 4% paraformaldehyde in PBS for 15 min, followed by rinsing with PBS and permeabilization with 0.1% Triton X-100 for 15 min. After blocking for 1 h with 10% goat serum (Sigma-Aldrich), the samples were then incubated at 4°C overnight with rabbit anti-FAK (pY397; Invitrogen). Samples were then rinsed and incubated with Alexa 594 goat anti–rabbit IgG for 1 h.
Microscopy and analysis
Samples were imaged using a 10× (numerical aperture [NA] 0.30), 20× (NA 0.45), or 40× (NA 0.60) air objective on a Nikon Eclipse Ti-U inverted fluorescence microscope (Nikon, Melville, NY) equipped with a Hamamatsu ORCA charge-coupled device camera (Hamamatsu, Hamamatsu, Japan). Confocal images were collected using a Hamamatsu ER camera coupled to a spinning disk confocal microscope (Bio-Rad). Image analysis was performed using ImageJ (National Institutes of Health, Bethesda, MD) and Photoshop (Adobe, San Jose, CA) software. Focal adhesions were quantified using a modification of the Water algorithm (Nelson et al., 2004) as follows. Original images of immunostained cells were filtered to subtract background fluorescence and segmented with a threshold of 0.45 μm2 to quantify the number of focal adhesions per cell. Analysis was performed on at least 130 cells over at least three independent experiments. Statistical analysis was performed using the unpaired Student's t test.
Immunohistochemical analysis
Nonmalignant breast tissue (cancer-free prophylactic mastectomy) and breast cancer biopsies were derived from waste surgical material from deidentified patients and were Formalin fixed and paraffin embedded. Sections from four nonmalignant and four breast cancer samples were evaluated. For immunohistochemistry, tissue sections were deparaffinized by placing them into three changes of xylene and rehydrated in a graded ethanol series. The rehydrated tissue samples were rinsed in water, and sections were subjected to heat antigen retrieval as described by the manufacturer (Dako). Slices were incubated with the appropriate primary antibody (Rac1b, Millipore; Collagen I, Abcam, Cambridge, MA) for 30 min at room temperature. Sections were then rinsed with Tris-buffered saline/Triton X-100 (TBST) wash buffer, and secondary incubation was with Dako DUAL+ horseradish peroxidase for 15 min. Tissue slices were rinsed with TBST wash buffer and then incubated in 3,3′-diaminobenzidine (DAB+; Dako) and counterstained with modified Schmidt's hematoxylin.
Supplementary Material
Acknowledgments
We thank Robert Prud'homme for use of the rheometer, Yibin Kang for use of the luminometer, and Amira Pavlovich and Esther Gomez for technical assistance. This work was supported in part by the National Institutes of Health (CA128660, CA116201, GM083997, and HL110335), Susan G. Komen for the Cure (FAS0703855), the David and Lucile Packard Foundation, the Alfred P. Sloan Foundation, and the Camille and Henry Dreyfus Foundation. C.M.N. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. C.L. was supported by the Lidow Senior Thesis Fund.
Abbreviations used:
- ECM
extracellular matrix
- EMT
epithelial–mesenchymal transition
- FAK
focal adhesion kinase
- ILK
integrin-linked kinase
- MMP
matrix metalloproteinase
- ROS
reactive oxygen species
- SMA
smooth muscle actin
- TGF
transforming growth factor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-02-0166) on August 23, 2012.
REFERENCES
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz J, Xu R, Mori H, Nelson CM, Mroue R, Spencer VA, Brownfield D, Radisky DC, Bustamante C, Bissell MJ. Laminin and biomimetic extracellular elasticity enhance functional differentiation in mammary epithelia. EMBO J. 2008;27:2829–2838. doi: 10.1038/emboj.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S, et al. Post-translational processing of rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J Biol Chem. 1992;267:25709–25713. [PubMed] [Google Scholar]
- Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol. 2000;10:719–722. doi: 10.1016/s0960-9822(00)00537-6. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudou T, Ohayon J, Picart C, Tracqui P. An extended relationship for the characterization of Young's modulus and Poisson's ratio of tunable polyacrylamide gels. Biorheology. 2006;43:721–728. [PubMed] [Google Scholar]
- Boulter E, Grall D, Cagnol S, Van Obberghen-Schilling E. Regulation of cell-matrix adhesion dynamics and Rac-1 by integrin linked kinase. FASEB J. 2006;20:1489–1491. doi: 10.1096/fj.05-4579fje. [DOI] [PubMed] [Google Scholar]
- Brunton VG, MacPherson IR, Frame MC. Cell adhesion receptors, tyrosine kinases and actin modulators: a complex three-way circuitry. Biochim Biophys Acta. 2004;1692:121–144. doi: 10.1016/j.bbamcr.2004.04.010. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol. 2005;171:153–164. doi: 10.1083/jcb.200506152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA. Integrins regulate Rac targeting by internalization of membrane domains. Science. 2004;303:839–842. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Balasubramanian N, Alderson NB, Kiosses WB, Grande-Garcia A, Anderson RG, Schwartz MA. Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat Cell Biol. 2005;7:901–908. doi: 10.1038/ncb1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo MA, Kiosses WB, Alderson NB, Meller N, Hahn KM, Schwartz MA. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol. 2002;4:232–239. doi: 10.1038/ncb759. [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 2000;19:2008–2014. doi: 10.1093/emboj/19.9.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng JT, Van Lierop JE, Sutherland C, Walsh MP. Ca2+-independent smooth muscle contraction. a novel function for integrin-linked kinase. J Biol Chem. 2001;276:16365–16373. doi: 10.1074/jbc.M011634200. [DOI] [PubMed] [Google Scholar]
- Didsbury JR, Uhing RJ, Snyderman R. Isoprenylation of the low molecular mass GTP-binding proteins rac 1 and rac 2: possible role in membrane localization. Biochem Biophys Res Commun. 1990;171:804–812. doi: 10.1016/0006-291x(90)91217-g. [DOI] [PubMed] [Google Scholar]
- Diekmann D, Abo A, Johnston C, Segal AW, Hall A. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science. 1994;265:531–533. doi: 10.1126/science.8036496. [DOI] [PubMed] [Google Scholar]
- Engler A, Bacakova L, Newman C, Hategan A, Griffin M, Discher D. Substrate compliance versus ligand density in cell on gel responses. Biophys J. 2004;86:617–628. doi: 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Eyckmans J, Boudou T, Yu X, Chen CS. A hitchhiker's guide to mechanobiology. Dev Cell. 2011;21:35–47. doi: 10.1016/j.devcel.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 2011;30:4707–4720. doi: 10.1038/onc.2011.181. [DOI] [PubMed] [Google Scholar]
- Fiegen D, Haeusler LC, Blumenstein L, Herbrand U, Dvorsky R, Vetter IR, Ahmadian MR. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem. 2004;279:4743–4749. doi: 10.1074/jbc.M310281200. [DOI] [PubMed] [Google Scholar]
- Friedman R, Gelfand T, Weiss DW, Doljanski F. Patterns of fibronectin deposition in normal and neoplastic fibroblasts and mammary tissue. Int J Tissue React. 1984;6:291–301. [PubMed] [Google Scholar]
- Ghilardi G, Biondi ML, Caputo M, Leviti S, DeMonti M, Guagnellini E, Scorza R. A single nucleotide polymorphism in the matrix metalloproteinase-3 promoter enhances breast cancer susceptibility. Clin Cancer Res. 2002;8:3820–3823. [PubMed] [Google Scholar]
- Guo WH, Frey MT, Burnham NA, Wang YL. Substrate rigidity regulates the formation and maintenance of tissues. Biophys J. 2006;90:2213–2220. doi: 10.1529/biophysj.105.070144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner KJ, Matrisian LM, Jensen RA, Rodgers WH. Expression of most matrix metalloproteinase family members in breast cancer represents a tumor-induced host response. Am J Pathol. 1996;149:273–282. [PMC free article] [PubMed] [Google Scholar]
- Heyworth PG, Knaus UG, Xu X, Uhlinger DJ, Conroy L, Bokoch GM, Curnutte JT. Requirement for posttranslational processing of Rac GTP-binding proteins for activation of human neutrophil NADPH oxidase. Mol Biol Cell. 1993;4:261–269. doi: 10.1091/mbc.4.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho E, Dagnino L. Emerging role of ILK and ELMO2 in the integration of adhesion and migration pathways. Cell Adh Migr. 2012;6:168–172. doi: 10.4161/cam.20399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- Iglesias-De La Cruz MC, Ruiz-Torres P, Alcami J, Diez-Marques L, Ortega-Velazquez R, Chen S, Rodriguez-Puyol M, Ziyadeh FN, Rodriguez-Puyol D. Hydrogen peroxide increases extracellular matrix mRNA through TGF-beta in human mesangial cells. Kidney Int. 2001;59:87–95. doi: 10.1046/j.1523-1755.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan P, Brazao R, Boavida MG, Gespach C, Chastre E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene. 1999;18:6835–6839. doi: 10.1038/sj.onc.1203233. [DOI] [PubMed] [Google Scholar]
- Kajita M, McClinic KN, Wade PA. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol. 2004;24:7559–7566. doi: 10.1128/MCB.24.17.7559-7566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokha R, Werb Z. Mammary gland reprogramming: metalloproteinases couple form with function. Cold Spring Harb Perspect Biol. 2011;3:a004333. doi: 10.1101/cshperspect.a004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Choi GH, Na DC, Ahn EY, Kim GI, Lee JE, Cho JY, Yoo JE, Choi JS, Park YN. Human hepatocellular carcinomas with “Stemness”-related marker expression: keratin 19 expression and a poor prognosis. Hepatology. 2011;54:1707–1717. doi: 10.1002/hep.24559. [DOI] [PubMed] [Google Scholar]
- Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella BT, Erdman RA, Maltese WA. Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J Biol Chem. 1991;266:9786–9794. [PubMed] [Google Scholar]
- Klein EA, Yin L, Kothapalli D, Castagnino P, Byfield FJ, Xu T, Levental I, Hawthorne E, Janmey PA, Assoian RK. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol. 2009;19:1511–1518. doi: 10.1016/j.cub.2009.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanning CC, Daddona JL, Ruiz-Velasco R, Shafer SH, Williams CL. The Rac1 C-terminal polybasic region regulates the nuclear localization and protein degradation of Rac1. J Biol Chem. 2004;279:44197–44210. doi: 10.1074/jbc.M404977200. [DOI] [PubMed] [Google Scholar]
- Lee K, Gjorevski N, Boghaert E, Radisky DC, Nelson CM. Snail1, Snail2, and E47 promote mammary epithelial branching morphogenesis. EMBO J. 2011;30:2662–2674. doi: 10.1038/emboj.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997a;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Srebrow A, Sympson CJ, Terracio N, Werb Z, Bissell MJ. Misregulation of stromelysin-1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin-1-dependent invasive properties. J Biol Chem. 1997b;272:5007–5015. doi: 10.1074/jbc.272.8.5007. [DOI] [PubMed] [Google Scholar]
- Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol. 2011;3:910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui C, Lee K, Nelson CM. Matrix compliance and RhoA direct the differentiation of mammary progenitor cells. Biomech Model Mechanobiol. 2011 doi: 10.1007/s10237-011-0362-7. DOI: 10.1007/s10237-011-0362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos P, Jordan P. Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp Cell Res. 2005;305:292–299. doi: 10.1016/j.yexcr.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Matos P, Jordan P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol Cancer Res. 2008;6:1178–1184. doi: 10.1158/1541-7786.MCR-08-0008. [DOI] [PubMed] [Google Scholar]
- Matos P, Oliveira C, Velho S, Goncalves V, da Costa LT, Moyer MP, Seruca R, Jordan P. B-Raf(V600E) cooperates with alternative spliced Rac1b to sustain colorectal cancer cell survival. Gastroenterology. 2008;135:899–906. doi: 10.1053/j.gastro.2008.05.052. [DOI] [PubMed] [Google Scholar]
- Michaelson D, Abidi W, Guardavaccaro D, Zhou M, Ahearn I, Pagano M, Philips MR. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol. 2008;181:485–496. doi: 10.1083/jcb.200801047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakopoulou L, Giannopoulou I, Gakiopoulou H, Liapis H, Tzonou A, Davaris PS. Matrix metalloproteinase-1 and -3 in breast cancer: correlation with progesterone receptors and other clinicopathologic features. Hum Pathol. 1999;30:436–442. doi: 10.1016/s0046-8177(99)90120-x. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Khauv D, Bissell MJ, Radisky DC. Change in cell shape is required for matrix metalloproteinase-induced epithelial-mesenchymal transition of mammary epithelial cells. J Cell Biochem. 2008;105:25–33. doi: 10.1002/jcb.21821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Pirone DM, Tan JL, Chen CS. Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell. 2004;15:2943–2953. doi: 10.1091/mbc.E03-10-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- Noe V, Fingleton B, Jacobs K, Crawford HC, Vermeulen S, Steelant W, Bruyneel E, Matrisian LM, Mareel M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci. 2001;114:111–118. doi: 10.1242/jcs.114.1.111. [DOI] [PubMed] [Google Scholar]
- Orlichenko L, Geyer R, Yanagisawa M, Khauv D, Radisky ES, Anastasiadis PZ, Radisky DC. The 19-amino acid insertion in the tumor-associated splice isoform Rac1b confers specific binding to p120 catenin. J Biol Chem. 2010;285:19153–19161. doi: 10.1074/jbc.M109.099382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pelham R, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol. 2005;204:198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Gould MN. Modulation of small G protein isoprenylation by anticancer monoterpenes in in situ mammary gland epithelial cells. Carcinogenesis. 1998;19:827–832. doi: 10.1093/carcin/19.5.827. [DOI] [PubMed] [Google Scholar]
- Rudolph-Owen LA, Chan R, Muller WJ, Matrisian LM. The matrix metalloproteinase matrilysin influences early-stage mammary tumorigenesis. Cancer Res. 1998;58:5500–5506. [PubMed] [Google Scholar]
- Sassano A, Platanias LC. Statins in tumor suppression. Cancer Lett. 2008;260:11–19. doi: 10.1016/j.canlet.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000;19:3013–3020. doi: 10.1038/sj.onc.1203621. [DOI] [PubMed] [Google Scholar]
- Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2:a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani Y, Wheelock MJ, Johnson KR. Phosphoinositide-3 kinase-Rac1-c-Jun NH2-terminal kinase signaling mediates collagen I-induced cell scattering and up-regulation of N-cadherin expression in mouse mammary epithelial cells. Mol Biol Cell. 2006;17:2963–2975. doi: 10.1091/mbc.E05-12-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23:9369–9380. doi: 10.1038/sj.onc.1208182. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhouk RS, Chin JR, Unemori EN, Werb Z, Bissell MJ. Proteinases of the mammary gland: developmental regulation in vivo and vectorial secretion in culture. Development. 1991;112:439–449. doi: 10.1242/dev.112.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trappmann B, et al. Extracellular-matrix tethering regulates stem-cell fate. Nat Mater. 2012;11:642–649. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- Wang N, Ostuni E, Whitesides GM, Ingber DE. Micropatterning tractional forces in living cells. Cell Motil Cytoskeleton. 2002;52:97–106. doi: 10.1002/cm.10037. [DOI] [PubMed] [Google Scholar]
- Wei WC, Lin HH, Shen MR, Tang MJ. Mechanosensing machinery for cells under low substratum rigidity. Am J Physiol Cell Physiol. 2008;295:C1579–1589. doi: 10.1152/ajpcell.00223.2008. [DOI] [PubMed] [Google Scholar]
- Wiseman BS, Sternlicht MD, Lund LR, Alexander CM, Mott J, Bissell MJ, Soloway P, Itohara S, Werb Z. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J Cell Biol. 2003;162:1123–1133. doi: 10.1083/jcb.200302090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty JP, Wright JH, Matrisian LM. Matrix metalloproteinases are expressed during ductal and alveolar mammary morphogenesis, and misregulation of stromelysin-1 in transgenic mice induces unscheduled alveolar development. Mol Biol Cell. 1995;6:1287–1303. doi: 10.1091/mbc.6.10.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T, Georges P, Flanagan L, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey P. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.