

Abstract
Background
Pacific white shrimp (Litopenaeus vannamei), the major species of farmed shrimps in the world, has been attracting extensive studies, which require more and more genome background knowledge. The now available transcriptome data of L. vannamei are insufficient for research requirements, and have not been adequately assembled and annotated.
Methodology/Principal Findings
This is the first study that used a next-generation high-throughput DNA sequencing technique, the Solexa/Illumina GA II method, to analyze the transcriptome from whole bodies of L. vannamei larvae. More than 2.4 Gb of raw data were generated, and 109,169 unigenes with a mean length of 396 bp were assembled using the SOAP denovo software. 73,505 unigenes (>200 bp) with good quality sequences were selected and subjected to annotation analysis, among which 37.80% can be matched in NCBI Nr database, 37.3% matched in Swissprot, and 44.1% matched in TrEMBL. Using BLAST and BLAST2Go softwares, 11,153 unigenes were classified into 25 Clusters of Orthologous Groups of proteins (COG) categories, 8171 unigenes were assigned into 51 Gene ontology (GO) functional groups, and 18,154 unigenes were divided into 220 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. To primarily verify part of the results of assembly and annotations, 12 assembled unigenes that are homologous to many embryo development-related genes were chosen and subjected to RT-PCR for electrophoresis and Sanger sequencing analyses, and to real-time PCR for expression profile analyses during embryo development.
Conclusions/Significance
The L. vannamei transcriptome analyzed using the next-generation sequencing technique enriches the information of L. vannamei genes, which will facilitate our understanding of the genome background of crustaceans, and promote the studies on L. vannamei.
Introduction
Pacific white shrimp, Litopenaeus vannamei, formerly Penaeus vannamei, belongs to the Penaeidae family of decapod crustaceans, is the major species of farmed shrimps in the world [1]. Because of its great economic value and important evolutionary status, more and more studies have been focused on breeding, growth, development, immunity, genetics and evolution of L. vannamei in recent twenty years [2]–[6]. The L. vannamei genome with higher recombination rates than other genomes of closely related penaeid prawns is predicted to be 2.0 Gigabases and has not been sequenced up to now [7], [8]. The absence of a fully sequenced and assembled genome hindered the studies on L. vannamei, including determination of gene functions and regulations, and establishment of novel genetic manipulation technologies.
Many transcriptome studies of L. vannamei have been carried out and a large number of expressed sequence tags (ESTs) were obtained using cDNA library and Sanger sequencing methods. By May 2012, 162,993 ESTs from many organs and tissues have been released on Genbank. These data have been used for cloning functional genes, selecting genetic molecular markers, and designing cDNA microarrays [2], [9]–[11]. However, because of the limitations of the traditional methods used for ESTs sequencing, the now available transcriptome data of L. vannamei are still insufficient for research requirements relative to the size of its genome. Moreover, the now available EST sequences have not been systematically analyzed. Up to now, only 7968 unigenes have been assembled and annotated, largely limiting the use of the ESTs sequence data.
The next-generation high-throughput DNA sequencing techniques, such as Solexa/Illumina (Illumina), 454 (Roche) and SOLiD (ABI), have been developed and growing rapidly in recent years [12]. They can sequence millions of DNA fragments simultaneously and provide Gigabases of data with high fidelity in a single machine run, greatly improving work efficiency and increasing data output [13]. The enormous advantages of these technologies make them admirably suited for genomics research, such as de novo and re-sequencing of genome, mRNA and microRNA [14]–[18]. Especially in transcriptome analysis, the usage of the next-generation sequencing techniques make it no longer necessary to establish cDNA libraries with bacteria cells as carriers, which could introduce DNA fragments deletion during the cloning process [19].
In this study, we analyzed the transcriptome of whole bodies of L. vannamei larvae using Solexa/Illumina high-throughout sequencing method, providing over 2.4 Gb data of raw sequences, which were assembled into 109,169 unigenes. We further annotated the unigenes by matching against Nr, Swissprot, Clusters of Orthologous Groups of proteins (COG), Gene ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Part of the results of unigene assemblies and annotations were primarily verified by RT-PCR, gel electrophoresis, Sanger sequencing, and real-time PCR. The assembled and annotated unigenes provide useful information for the studies on genomes and functional genes of L. vannamei and crustaceans.
Methods
Breeding of L. vannamei
Newly hatched L. vannamei larvae from a relatively high-WSSV-resistant family [20], obtained from Hengxing (Evergreen) shrimp farm in Zhanjiang, China, were fed with larvae of Chirocephalus diaphanus and bred in seawater of pH 8.0 and 30 g kg−1 salinity in a 3 m3 indoor tank with recirculating and filtering units at 28°C.
Illumina Sequencing
One hundred L. vannamei larvae at 20 days post spawning were fasted for 24h to avoid contamination from the feed Chirocephalus diaphanous larvae and then sampled. Total RNA was extracted using the SV Total RNA Isolation System (Promega) according to the manufacturer’s protocol. The total RNA concentration, quality and integrity were determined using a NanoDrop 1000 spectrophotometer and an Agilent 2100 Bioanalyzer. The total RNA with absorbance 260/280 nm Ratio of ∼2.0 was chosen and treated with DNase I prior to library construction. Poly-(A) mRNA was then purified using oligo(dT) magnetic beads and fragmented by treatment with divalent cations and heat, followed by reverse-transcribing into cDNA using reverse transcriptase and random hexamer-primers. The second-strand cDNA was synthesized using RNaseH and DNA polymerase I. The double-stranded cDNA was end-repaired using T4 DNA polymerase, Klenow fragment, and T4 polynucleotide kinase followed by a single (A) base addition using Klenow 3′ to 5′ exo-polymerase, and further ligated with an Illumina PE adapter oligo mix using T4 DNA ligase. Adaptor-modified fragments with length of 200±25 bp were selected by gel purification and subjected to PCR amplification as templates. After 15 cycles of PCR amplification the libraries were sequenced on an Illumina sequencing platform (GAII) and the raw reads were generated using Solexa GA pipeline 1.6.
Transcriptome De novo Assembly and Analysis
Fig. 1 shows the workflow of transcriptome assembly and analysis. The raw reads of Illumina sequencing were preprocessed by removing adaptor sequences, low-quality reads (reads with ambiguous bases N), and duplication sequences, and were then assembled using the SOAP denovo software (http://soap.genomics.org.cn/soapdenovo.html) with the default settings. Firstly, the clean reads are combined by SOAP denovo based on sequence overlap to form longer fragments without N, which are called contigs, and then the reads are mapped back to contigs. Next, scaffolds were made using SOAP denovo by connecting the contigs with N to present unknown sequences between each two contigs in a same transcript. Scaffolds’ gaps can be filled by paired-end reads of sequencing to get sequences with least Ns and cannot be extended on either end. Such sequences are defined as unigenes, and the following analysis are based on them [15], [21], [22].
Figure 1. Workflow diagram for transcriptome sequencing, assembly and analysis.

After ruling out short-length sequences and low-quality sequences containing more than 10% ambiguous ‘N’ nucleotides or consecutive 14 ‘N’ nucleotides, unigenes with a minimum length of 200 bp were selected, submitted to NCBI Transcriptome Shotgun Assembly (TSA) database (http://www.ncbi.nlm.nih.gov/genbank/TSA.html, accession number: JP355723-JP376614 and JP382831-JP435443), and subjected to annotation analysis. The coding regions (CDSs) of the assembled >200 bp unigenes were determined by matching sequences against NCBI Nr (non-redundant, http://www.ncbi.nlm.nih.gov/), UniProtKB (including Swiss-prot and TrEMBL, http://www.uniprot.org/), KEGG, and COG databases in turn using the BLASTx algorithm of the BLAST software (ftp://ftp.ncbi.nih.gov/blast/executables/release/2.2.23/) with a significant threshold of E value<0.00001. Unigenes (>200 bp) that cannot be aligned to Nr and Swiss-prot databases were scanned by ESTScan (http://www.ch.embnet.org/software/ESTScan2.html) to determine their CDSs [23].
To compare the assembled unigenes with L.vannamei ESTs available from Genbank (http://www.ncbi.nlm.nih.gov/nucest/?term=Litopenaeus vannamei), EST sequences were firstly clustered by TIGR Gene Indices clustering tools (TGICL) [24] with minimum overlap length of 60 bp, minimum identity of 94% for the overlap, and maximum mismatched overhang of 30 bp, and then combined using Phrap (http://bozeman.mbt.washington.edu/phrap.docs/phrap.html) with minmatch 35, minscore 35 and repeat stringency 0.95. Comparison between assembled unigenes and assembled ESTs was performed using BLASTn algorithm and E-value threshold of 0.00001.
Based on Nr annotation, we used BLAST2GO program (http://www.BLAST2go.org/) to get GO annotation of unigenes [25]. GO functional classification for all unigenes was performed using WEGO software (http://wego.genomics.org.cn/cgi-bin/wego/index.pl) [26]. KEGG metabolic pathway annotation and COG classification of unigenes were determined by BLASTx searching against KEGG database and COG database, respectively [27]–[30].
RT-PCR and Real-time PCR Assays
12 annotated unigenes that may relate to embryo development were selected to be analyzed using real-time PCR and RT-PCR, and their specific primers were listed in Table 1. For real-time PCR assays, Parent prawns mated and laid eggs in a 3 m3 tank at 28°C with seawater of pH 8.0 and 30 g kg−1 salinity. The timing of sampling was according to the embryogenesis stages, which were determined by microscopic examination and reference to previous studies [31], [32]. 60 fertilized eggs per sample were collected at 0, 140, 215, 275, 480, 600 and 660 minutes post-spawning (mps), respectively. The spawning and sampling experiments were repeated for three biological replicates. Total RNA of each sample was isolated with TRIzol (Invitrogen, USA) and subjected to DNase I treatment (Promega, USA) according to the manufacturer’s protocols. The cDNA was synthesized with SuperScript II RT (Invitrogen, USA), and quantitative real-time PCR was triply performed using the LightCycle 480 System (Roche, Germany) with a volume of 10 µl contained 1 µl of 1∶10 cDNA diluted with ddH2O, 5 µl of 2×SYBRGreen Master Mix (Toyobo, Japan), and 250 nM of each primer. The cycling parameters were 95°C for 2 min to activate the polymerase, followed by 40 cycles of 95°C for 15 s, 62°C for 1 min, and 70°C for 1 s. Cycling ended at 95°C with 5°C/s calefactive velocity to create the melting curve. Fluorescence measurements were taken at 70°C for 1 s during each cycle. Expression levels of each gene were normalized to 18S ribosomal RNA (18S rRNA, GenBank accession number: AF186250, 18S-rRNA-qF and 18S-rRNA-qR primers: 5′-CTGCGACGCTAGAGGTGAAATTC-3′ and 5′- AGGTTAGAACTAGGGCGGTATCTG-3′). Data were calculated with the relative quantification method described by Muller et al. [33], and subjected to statistical analysis.
Table 1. Analyzed genes and their specific primers.
| Unigene | similarity | Size (bp) | RT-PCR Primer sequence (5′–3′) | Realtime-PCR Primer sequence (5′–3′) |
| 98112 | abdominal-A[Strigamia maritima] | 586 | F: TCGTCGTGGACAGCCGTTTGR: CGGTTCTCGTGCCTGGTGTC | F: CGTCTGTGGTCGCCGTCATR: CAGAAGTATGTACCCCTACGTGAGT |
| 10296 | abdominal-B(Strigamia maritime) | 1049 | F: GGCTAGGGGCAGAGGGGTGACAGGAR: CAGCGAGGGAGGAGGACTAGACAG | F: GGCATGACTCCGTGGTCATCR: CCACGTACTATAACATTGCGGAC |
| 54158 | homeotic antennapedia protein [Culex quinquefasciatus] | 216 | F: CAGCTCGAGGGTCTGGTACCGCGTGR: GGAGGCGAAGGAGCCGAAG | F: TGGTACCGCGTGTAGGACGTTCR: CTCAGATCTACCCGTGGATGAAGAG |
| 16317 | Wnt6 [Monodelphis domestica] | 555 | F: CCATCGTGATGCTCCACAACAACR: GCTATCATCAATTATTCCGTAATC | F: ATGTCGCTTCAAGTTCTGCTGTGR: CCGGACGCTATCATCAATTATTCC |
| 18019 | Wnt10 [Tribolium castaneum] | 408 | F:CTGGCACCGAACCGGGTGTTGTCGR:TCTGGGACCACGTGCTCGACCC | F: GTGATGGCGTGACCGAAGGR: CGGAAGGCAGTTTGCGAATC |
| 105360 | beta-catenin [Parhyale hawaiensis] | 979 | F:GACTTTGGTTGCAGCATCTGAGAGGR: CAACCAGGCTGCCTTCACCAGC | F: GCAGCAGGAGGTTGTGGAGTGR: CGCCAGGGTTTATTAGCCATTTTC |
| 92779 | Pumilio(Apis mellifera) | 468 | F: GTTTTGTCGTCATCGAGGCCGTR: AAAAGCTGTGGGGAGTGGAAG | F: CTGAGTTGCAGGTTGGCCATGR: TCAAGATGGTCGAGTACGTGTTG |
| 99210 | pumilio homolog 2-like (Saccoglossus kowalevskii) | 619 | F: CTGGCGGATCCTCATCCTTATTCR: GCTGAATAACACGGCAACCATAGG | F: GGCCACGTTTTGCCTTTAGCR: ATCCAGTTCACGCACGATGTCT |
| 95206 | Dorsal (Litopenaeus vannamei) | 514 | F: AGTTACGAGAGGAGATTAGAGTGGR: AGTCTAGAGGCAAATACTGGAATG | F: TTCCAGACCGGGTTTTCTCATCR: TTCCCCTTTTCTGATCCCTCG |
| 99694 | Spalt (Tribolium castaneum) | 635 | F: GCTGTCCCCCGAGGCCCTCAGR: TTGAGAAGCCTCGGTCGCAGATG | F: CCCACGCCGTCGGACTACTR: GACGGAGCCGCTGATATTGTG |
| 10400 | extra sex combs (Schistocerca americana) | 567 | F: GCATTTTGTTGGTCATGGAAATGCR: TCCAAAGCAAACCTCATAAACCAG | F: TTTCAACCCAAGCAGAGCAGTGR: CCAACGCACACAATCCACATAGT |
| 20337 | HIRA (Takifugu rubripes) | 4036 | F: CAAAGATGGCGGAGGAAGCGTCR: GCTTGCTAACTTTCATGAGTTTCAC | F: GGAGGCTTGGCTTTTGTTAGGTR: CACTTTGATGACAGGGAGGCAG |
For RT-PCR assays, the 12 unigenes were amplified by LA Taq DNA polymerase (TaKaRa, Japan) using cDNA from L. vannamei larvae at 20 days as template. The PCR products were analyzed using agarose gel electrophoresis, and subcloned into the PMD19-T vector (TaKaRa, Japan) for Sanger sequencing.
Results
Illumina Sequencing and Sequence Assembly
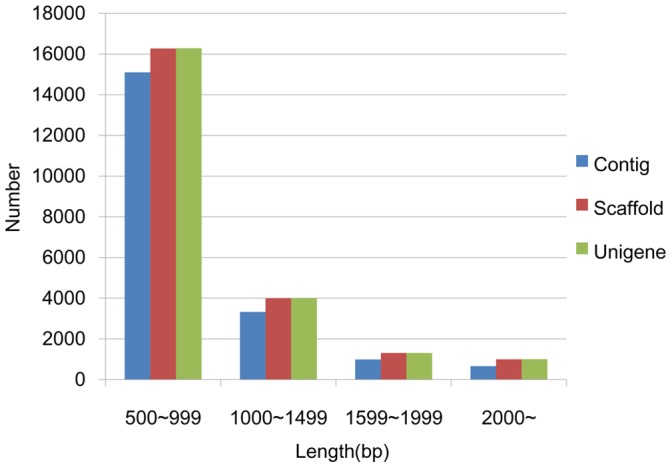
Sequences of mRNA pooled from one hundred whole bodies of L. vannamei larvae were analyzed on an Illumina GAII platform. Up to 27,394,946 reads (accumulated length of 2,465,545,140 bp with a GC percentage of 47.89%) were obtained and assembled into 882,339 contigs (>75 bp, mean length of 127 bp and an N50 length of 90 bp), of which 79.64% (702,760) were with length of 75–100 bp, 11.34% (100,065) with 100–200 bp, 3.93% (34,655) with 200–300 bp, 1.80% (15,914) with 300–400 bp, 1.01% (8,951) with 400–500 bp, and 2.27% (20,054) with >500 bp. A total of 162,342 scaffolds (>100 bp), with a mean length of 306 bp and an N50 of 399 bp, were generated by assembling and paired end-joining of these contigs. About 86.09% (139,764) of scaffolds were 100–500 bp long, 10.02% (16,271) were 500–1000 bp, 2.46% (3,993) were 1000–1500 bp, 0.81% (1,311) were 1500–2000 bp, and 0.62% (1,003) were >2000 bp. Most of the assembled scaffolds (93.77% of the total) were with gaps of 0–5% (N/length). By clustering and gap-filling, these scaffolds were further assembled into 109,169 unigenes (http://marine.sysu.edu.cn/Public/Uploads/files/4f7ff9d500edc5.rar), including 1622 clusters and 107,547 singletons, with a mean length of 396 bp and an N50 of 478 bp, and 20.71% of which were >500 bp long. 105,206 unigenes (96.37% of the total) contained gaps within 5% in length. Summary statistics of L. vannamei transcriptome assembly was shown in Table 2, and the size distributions (>500 bp) of Contigs, Scaffolds, and Unigenes were showed in Fig. 2.
Table 2. Summary statistics of L. vannamei transcriptome sequencing and assembly.
| Summary | Number of total nucleotides(nt) | Number of mean length of total reads | Number of mean length of total reads(bp) |
| Output statistics of sequencing | 2,465,545,140 | 27,394,946 | 90 |
| Assembly | Contig | Scaffold | Unigene |
| Length distribution | |||
| 75–99 | 702,730 | − | − |
| 100–499 | 159585 | 139,764 | 86,564 |
| 500–999 | 15098 | 16,271 | 16,284 |
| 1000–1499 | 3,331 | 3,993 | 4,007 |
| 1500–1999 | 992 | 1,311 | 1,308 |
| ≥2000 | 663 | 1,003 | 1,006 |
| Total No. | 882,399 | 162,342 | 109,169 |
| Length statistics(bp) | |||
| Mean length | 127 | 306 | 396 |
| N50 | 90 | 399 | 478 |
| Total length(Mb) | 112 | 49.6 | 43.2 |
Figure 2. The size distributions (>500 bp) of Contigs, Scaffolds, and Unigenes.
To assess the abundance and coverage of the transcriptome data, we matched the assembled unigenes against the known EST library on Genbank. The 162,926 ESTs downloaded from NCBI were clustered and assembled using TGICL and Phrap, and 25,813 assembled EST-unigenes with mean length of 681 bp and N50 length of 756 bp were generated (http://marine.sysu.edu.cn/Download/download/id/68). Comparisons between transcriptome unigenes and EST-unigenes were performed using BLASTn algorithm. Results were shown in Fig. 3 as a Venn chart and further detailed in Table S1. 60.1% (15,519 out of 25,813) of the EST-unigenes can be matched in the transcriptome unigenes library, whereas only 14.2% (15,519 out of 109,169) of the transcriptome unigene sequences can be found in the ESTs library.
Figure 3. Venn chart for comparisons between assembled transcriptome unigenes and assembled EST-unigenes.

Annotation of Unigenes
After ruling out short-length or low-quality sequences, 73,505 unigenes with a minimum length of 200 bp were selected and subjected to annotation analysis by matching sequences against Nr, Swiss-prot and TrEMBL databases using BLASTx searching with an E value<0.00001. 27,789 unigenes (37.80% of the total) can be matched in Nr database (Table S2), 27,424 (37.3% of the total) matched in Swissprot (Table S3), and 32,439 (44.1% of the total) matched in TrEMBL (Table S4). For main species distribution matched against Nr database, 91.9% of the matched unigenes showed similarities with Homo sapiens, followed by Drosophila melanogaster (84.2%), Mus musculus (72.37%), Danio rerio (47.94%), Rattus norvegicus (43.17%), Tribolium castaneum (43.14%), Drosophila mojavensis (38.50%), Harpegnathos saltator (37.42%), Apis mellifera (37.09%), Anopheles gambiae str. PEST (36.62%), Camponotus floridanus (35.97%), Drosophila virilis (34.95%), Drosophila willistoni (33.31%), Drosophila grimshawi (32.79%), and Drosophila yakuba (32.62%).
The remaining unmatched unigenes (>200 bp) were further analyzed using ESTscan and the predicted CDSs were translated into peptide sequences. CDSs of 11,886 unigenes (16.17% of the total) were successfully predicted by ESTscan.
COG, GO and KEGG Classification
The assembled unigene sequences were subjected to BLAST searching against GO, COG and KEGG databases, and the summary statistics of BLAST assignment was shown in Table 3.
Table 3. Summary statistics of L. vannamei transcriptome blast assignment.
| Species | Number of Unigenes | Number of Nr annotations | Number of Swiss-prot annotations | Number of TrEMBL annotations | ESTscan prediction | Number of COG hits | Number of GO mapped | Number of KEGG hits |
| Litopenaeus vannamei | 109,169 | 27,789 | 27,424 | 32,439 | 11,886 | 11,153 | 45,601 | 18,154 |
COG is a database where orthologous gene products were classified. Every protein in COG is assumed to be evolved from an ancestor protein, and the whole database is built on coding proteins with complete genome as well as system evolution relationships of bacteria, algae and eukaryotes [27], [29]. Phylogenetic classifications of the predicted CDSs of unigenes were analyzed by searching against COG database to predict and classify possible functions of the unigenes (Fig. 4). Possible functions of 11,153 unigenes were classified and subdivided into 25 COG categories (Table S5), among which the cluster for ‘General function prediction only’ represents the largest group (2002, 17.95% of the matched unigenes) followed by ‘Translation, ribosomal structure and biogenesis’ (929, 8.33%) and ‘Posttranslational modification, protein turnover, chaperones’ (830, 7.44%). The following categories: ‘extracellular structures’ (6, 0.05%), ‘nuclear structure’ (9, 0.08%) and ‘RNA processing and modification’ (71, 0.64%), represent the smallest groups.
Figure 4. COG Classification of the unigenes.

Possible functions of 11153 unigenes were classified and subdivided into 25 COG categories.
GO is an international standardized gene functional classification system which offers a dynamic-updated controlled vocabulary and a strictly defined concept to comprehensively describe properties of genes and their products in any organism [25], [26]. Based on the results of Nr annotation, the Gene Ontology (GO) annotations of unigenes were generated using the BLAST2GO program, and the GO functional classifications were performed using WEGO software to understand the distribution of gene functions of L. vannamei from the macro-level (Fig. 5). 45,601 GO term annotations corresponding to 8171 unigenes were produced and assigned into 51 functional groups and three categories, among which 22,268 were assigned in biological process category, 15,403 in cellular component category and 7930 in molecular function category (Table S6). Among the biological process category, ‘cellular process’ (17.65%) and ‘metabolic process’ (14.45%) biological regulation were most highly represented, and other unigenes were categorized into other 25 important biological process, including ‘biological regulation’ (7.53%), ‘multicellular organismal process’ (7.02%), ‘localization’(6.75%), ‘developmental process’ (6.55%), ‘regulation of biological process’ (6.35%), ‘cellular component organization or biogenesis’ (5.64%), and so on. 11 GO functional groups were assigned into the cellular component category, among which ‘cell’ (32.88%) and ‘cell part’ (29.66%) were most highly represented. Similarly, 13 GO functional groups were assigned into the molecular function category, among which ‘catalytic activity’ (42.86%) and ‘binding’ (40.86%) were most highly represented.
Figure 5. GO categories of the unigenes.

8171 unigenes were assigned 45601 GO annotations, which were divided into three categories: biological processes, cellular components, and molecular functions.
The KEGG pathway database records networks of molecular interactions in the cells and variants of them specific to particular organisms. Pathway-based analysis helps us to further learn biological functions of genes [27], [34], [35]. To systematically analyze their inner-cell metabolic pathways and complicated biological behaviors, we classified the unigenes into biological pathways by mapping the annotated CDS sequences to the reference canonical pathways in the KEGG database (Fig. 6). 18,154 unigenes were consequently assigned to 220 KEGG pathways (Table S7), among which 2285 members assigned to ‘metabolic pathways’, followed by ‘Spliceosome’ (1007 members), ‘Regulation of actin cytoskeleton’ (926 members), ‘Amoebiasis’ (848 members), ‘Pathways in cancer’ (843 members), ‘Vibrio cholerae infection’ (748 members), ‘Adherens junction’ (610 members), ‘Focal adhesion’ (576 members), ‘Dilated cardiomyopathy’ (546 members), ‘MAPK signaling pathway’ (545 members), ‘Hypertrophic cardiomyopathy (HCM)’ (539 members), ‘Tight junction’ (507 members), and ‘Pathogenic Escherichia coli infection’ (500 members).
Figure 6. KEGG Classification of the unigenes.18154 unigenes were assigned into 220 KEGG pathways.

The top 10 most abundant KEGG pathways are shown.
RT-PCR and Real-time RT-PCR Assays
To primarily verify the results of assemblies and annotations, 12 assembled unigenes that are homologous to many embryo development-related genes, including three Hox family transcriptional regulators abdominal-A, abdominal-B, and homeotic antennapedia (Antp), three Wnt signaling pathway genes Wnt6, Wnt10 and beta-catenin, two Puf Family RNA-binding translational regulators pumilio (PUM) and pumilio homolog 2 (PUM2), a NF-κB family gene Dorsal, a Zn-finger transcription factor Spalt, a Polycomb group (PcG) gene extra sex combs (esc), and histone cell cycle regulation defective homolog A (HIRA), were chosen and subjected to RT-PCR and real-time PCR analyses (Table 1). Full lengths of these unigenes were amplified by RT-PCR using specific primers designed based on the assembly results. The amplicons were analyzed using agarose gel electrophoresis (Fig. 7) and Sanger sequencing (Table S8), which confirmed their lengths and sequences, suggesting the faithful assembly of transcriptome data.
Figure 7. RT-PCR analyses of the amplicons of 12 unigenes related to shrimp embryo development.
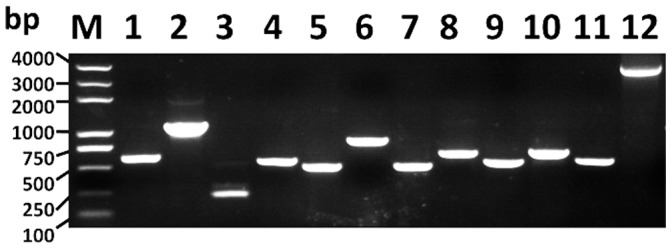
Amplicons: 1:Unigene98112 (similar to Abdominal-A, 580 bp); 2:Unigene10209 (similar to Abdominal-B, 1036 bp); 3:Unigene54158 (similar to homeotic antennapedia, 216 bp); 4:Unigene16317 (similar to Wnt6, 528 bp); 5:Unigene18019 (similar to Wnt10, 394 bp); 6: Unigene105360 (similar to beta-catenin, 979 bp); 7:Unigene92779 (similar to pumilio, 449 bp); 8:Unigene99210 (similar to pumilio homolog 2, 615 bp); 9: Unigene95206 (similar to Dorsal, 507 bp); 10:Unigene99694 (similar to Spalt, 620 bp); 11:Unigene10400 (similar to gene extra sex combs, 555 bp); 12:Unigene20337 (similar to HIRA, 3941 bp).
The sampled L. vannamei embryos were examined under microscope to determine their embryogenesis stages (Fig. 8). To verify their annotations, expression profiles of the 12 unigenes during embryonic development were further detected using real-time RT-PCR (Fig. 9). The Strigamia maritima abdominal-A homolog unigene98122 demonstrated a periodic expression profile with the first peak of 10.14-fold increase at 215 mps and the second peak of 18.54-fold increase at 480 mps, while the Strigamia maritima abdominal-B homolog unigene10296 and Culex quinquefasciatus Antp homolog unigene54158 showed similar expression profiles with peaks at 215 mps of 6.01-fold and 11.52-fold increase levels, respectively. The Wnt6 homolog unigene16317 peaked at 215 to 275 mps with 4.50–4.04-fold, followed by a sharp decrease at 480 mps, and the Wnt10 homolog unigene18019 kept increasing after spawning and reached a peak at 600 mps with 7.89-flod increase. The unigene105360, similar to beta-catenin, the key molecule of the Wnt pathway, peaked at 215 mps with 2.38-fold and returned to baseline levels after 480 mps. The expression of the Apis mellifera PUM homolog unigene92779 was up-regulated during 0–215 mps, and then fell to a low level at 480 mps. The Saccoglossus kowalevskii PUM2 homolog unigene99210 exhibited a 5.49-fold increase at 215 mps, and then returned to the basal level at 275 mps where it remained unchanged. The expression of the unigene95206, the dorsal gene of Litopenaeus vannamei, was up-regulated periodically, with the first peak of 6.15-fold at 215 mps and the second peak of 6.82-fold at 660 mps. The expression of unigene99694, similar to the spalt protein of Tribolium castaneum, was down-regulated during 0–600 mps, with two valleys at 215 mps and 600 mps, and then up-regulated at 660 mps. The unigene10400 (homologous to the esc gene of Schistocerca Americana), and unigene20337 (homologous to the HIRA gene of Takifugu rubripes) showed similar expression profiles, which up-regulated and peaked at 215 mps and fell to a low value at 480 mps.
Figure 8. Embryonic development of L. vannamei at 28°C, pH 8.0 and 30 g kg.

−1 salinity. The sampled L. vannamei embryos were checked under the inverted microscope to confirm their embryogenesis stages. A: Newly Spawned Egg (0 mps); B: Blastula Stage (140 mps); C: Gastrula Stage (215 mps); D: Antennal Limb Bud Stage (275 mps); E: Biramous Antenna and Mandible (480 mps); F: Hatching Nauplius (600 mps); G: Newly Hatched Nauplius (660 mps). Abbreviations: mps, minutes post spawning. Scale bars: 100 µm.
Figure 9. Real-time PCR analyses of the expression profiles of 12 assembled unigenes during embryos development.

Experiments were performed in triplicate and repeated three times with similar results. Bars display mean+s.d., and statistical analysis was performed using Student’s T test and the P values were provided (**, P<0.01; *, P<0.05).
Discussion
Many members of crustacean are of great economic value and important evolutionary status. Up to now, the Daphnia pulex genome is the only one sequenced in the subphylum Crustacea, phylum Arthropod [36]. Many transcriptomes of crustaceans have been analyzed using traditional Sanger sequencing and cDNA microarray method, including Eriocheir sinensis [37], Portunus pelagicus [38], Petrolisthes cinctipes [39], Penaeus monodon [40], Penaeus japonicas [41], Daphnia magna [42], Daphnia pulex [43], and so on. In recent years, the next generation sequencing methods have also been applied to analyze transcriptomes of crustaceans, such as Balanus Amphitrite [44], Euphausia superb [45], Macrobrachium rosenbergii [46] and Parhyale hawaiensis [47] using 454 sequencing, and Eriocheir sinensis [48], [49] using Illumina sequencing. Comparing with the traditional methods, the next-generation high-throughput DNA sequencing techniques provide more ideal methods for transcriptome analyses with high efficiency, low cost and high data output. The development of DNA sequencing technology will facilitate the studies on crustaceans’ gene background.
In this study, using the Illumina sequencing method to analyze the trancriptome of L. vannamei, more than 2.4 Gb of raw data were generated, and 882,339 contigs (>75 bp) were assembled, largely enriching the transcriptome data of L. vannamei and prompting the genome studies of crustaceans. The former studies on L. vannamei transcriptome were performed using traditional cDNA library and Sanger sequencing methods with RNA from many organs such as muscle, blood and hepatopancreas. In our study, RNA used for transcriptome analysis was exacted from whole bodies of L. vannamei larvae, covering all tissues of the species, which could include fuller transcriptional genes of L. vannamei. We compared our transcriptome data with L. vannamei EST sequences obtained from NCBI and showed that more than half of the EST sequences (60.1%) can be matched in the transcriptome data, whereas up to 85.8% of the transcriptome unigenes can not be found in the ESTs library. It suggests the transcriptome data provide abundant information besides the now available ESTs sequences.
Although providing much more data throughout than traditional Sanger sequencing method, the reading lengths of the raw data of the Illumina GAII system are quite short. Up to 79.64% of the obtained contigs are less than 100 bp. The SOAP denovo method, a relative mature technique based on the short oligonucleotide analysis package (SOAP) algorithm, was adopted to process the sequencing data, and 73,505 unigenes (>200 bp) with good quality sequences were assembled and subjected to annotation analyses. There are more unigenes showed similarities to H. sapiens than other arthropods species such as T. castaneum, D. mojavensis, and H. saltator, which are phylogenetically closer to L. vannamei than human. It is maybe because the now available information on gene background of crustaceans and arthropods is limited, and human genes have been much better studied than other species, providing sufficient gene sequences and annotations for comparison analyses. Only 31.83% of the total analyzed unigenes (84.2% of the Nr database-matched) showed similarities to D. melanogaster, a well studied model animal in the Insecta class, phylum Arthropod, maybe because the genome size of L. vannamei is almost 12 times more than that of D. melanogaster [50] and there might be somewhat different between their gene backgrounds. Further investigation should be required to determine whether protein sequences in crustaceans may have divergence from other animals. With more genes from crustaceans being studied and more gene background information being available, unigenes of L. vannamei obtained in this study will be further annotated. Moreover, since it has been reported that there are limitations of the next-generation sequence de novo assembly, which could cause missing of the duplicated sequences [51], the possibilities of missing of the encoding sequences in the assembled unigenes might not be excluded. It might also lead to miss-matching of the L. vannamei unigenes to protein databases of other species. With the development of sequencing methods and short-read assembly algorithms, further analyses will be performed to rearrange the now available L. vannamei transcriptome data and improve their annotations.
Possible functions of the assembled unigenes were analyzed by matching to GO, COG and KEGG databases. Although only a small part of the assembled unigenes were functional annotated, the results of these three databases searching help us learn more about biological features of L. vannamei. For example, 748 unigenes can be classified into the ‘Vibrio cholerae infection’ KEGG pathway, such as protein disulfide isomerase (PDI, unigene15355), ADP ribosylation factor (ARF, Unigene87975) and transport protein Sec61 (Unigene100471), which have been reported that involve in Vibrio infection in arthropods [52]–[55]. It indicated that as an animal living in water, L. vannamei may always deal with the challenges from bacteria in water, and may have evolved complicated systems and signal pathways against infection. Interestingly, although no cancer has ever been reported in Arthropoda animals, our results showed that 843 unigenes can be classified into ‘Pathways in cancer’ KEGG pathway. Cancer is a disease of aberrant multicellularity, and its hallmarks are thought to be intimately associated with those of metazoan multicellularity [56], [57]. It has been reported that many ‘multicellularity’ genes of Amphimedon queenslandica were also implicated in cancer, suggesting the remote origin of cancer and oncogenes [56], [57]. The KEGG ‘Pathways in cancer’ annotations of these L. vannamei unigenes in this study also provided supports for the theme that cancer origin may be related to evolution of multicellularity. Further studies should be performed to confirm this hypothesis.
The 12 unigenes subjected to RT-PCR and real-time PCR analyses were chosen based on the results of annotations, and the primer-pairs were designed based on the sequences of the unigenes assembled with contigs. The products of RT-PCR were sequenced using Sanger sequencing method, which confirmed the sequences of these unigenes, suggesting the faithful results of Illumina sequencing and assemblies. Furthermore, as real-time PCR showed that the expression profiles of these assembled unigenes regularly varied following the developmental stages of embryos, we think the qualities of the annotations of the unigenes may be enough for the requirements of studies on functional genes of L. vannamei.
With the development of sequencing techniques, the data of nucleic sequences boosted every day. As more data will be obtained from other species, the assembled unigenes in this study will be further annotated and analyzed. The data of the annotated unigenes are worthy of deeper mining and further analyzing. It will facilitate our understanding of the genome background of crustaceans, and promote the studies on genetics, functional genes, and gene regulations of L. vannamei.
Supporting Information
Comparisons between assembled transcriptome unigenes and the EST-unigenes which were assembled form EST sequences available from Genbank using TGICL and Phrap softwares.
(ZIP)
BLASTX searching of the Unigenes against NCBI Nr database (E value<0.00001).
(ZIP)
BLASTX searching of the Unigenes against Swissprot database (E value<0.00001).
(ZIP)
BLASTX searching of the Unigenes against TrEMBL database (E value<0.00001).
(ZIP)
COG Classification of the unigenes.
(ZIP)
GO categories of the unigenes.
(ZIP)
KEGG Classification of the unigenes.
(ZIP)
Sanger sequencing of the 12 analyzed unigenes.
(ZIP)
Funding Statement
This research was supported by National Natural Science Foundation of China under grant No. U1131002 (http://www.nsfc.gov.cn/Portal0/default106.htm); National High Technology Research and Development Program of China (973 Program) 2012CB114401 (http://www.973.gov.cn/Default_3.aspx); China Agriculture Research System (47); Special Fund for Agro-scientific Research in the Public Interest 201103034 (http://english.agri.gov.cn/); Foundation from Science and Technology Bureau of Guangdong Province 2011A020102002 and 2009A020102002 (http://www.gdstc.gov.cn/eng/mission.html); Foundation from Administration of Ocean and Fisheries of Guangdong Province A201101B02 (http://www.gdofa.gov.cn/index.php/English/); and the Open Project of the State Key Laboratory of Biocontrol (SKLBC09K04) (http://sklbc.sysu.edu.cn/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Rendon Rodriguez S, Macias Regalado E, Calderon Perez JA, Nunez Pasten A, Solis Ibarra R (2007) Comparison of some reproductive characteristics of farmed and wild white shrimp males Litopenaeus vannamei (Decapoda: Penaeidae). Rev Biol Trop 55: 199–206. [DOI] [PubMed] [Google Scholar]
- 2. Ciobanu DC, Bastiaansen JW, Magrin J, Rocha JL, Jiang DH, et al. (2010) A major SNP resource for dissection of phenotypic and genetic variation in Pacific white shrimp (Litopenaeus vannamei). Anim Genet 41: 39–47. [DOI] [PubMed] [Google Scholar]
- 3. Freitas PD, Calgaro MR, Galetti Jr PM (2007) Genetic diversity within and between broodstocks of the white shrimp Litopenaeus vannamei (Boone, 1931) (Decapoda, Penaeidae) and its implication for the gene pool conservation. Braz J Biol 67: 939–943. [DOI] [PubMed] [Google Scholar]
- 4. Gorbach DM, Hu ZL, Du ZQ, Rothschild MF (2010) Mining ESTs to determine the usefulness of SNPs across shrimp species. Anim Biotechnol 21: 100–103. [DOI] [PubMed] [Google Scholar]
- 5. Liu KF, Chiu CH, Shiu YL, Cheng W, Liu CH (2010) Effects of the probiotic, Bacillus subtilis E20, on the survival, development, stress tolerance, and immune status of white shrimp, Litopenaeus vannamei larvae. Fish Shellfish Immunol 28: 837–844. [DOI] [PubMed] [Google Scholar]
- 6. Shen X, Ren J, Cui Z, Sha Z, Wang B, et al. (2007) The complete mitochondrial genomes of two common shrimps (Litopenaeus vannamei and Fenneropenaeus chinensis) and their phylogenomic considerations. Gene 403: 98–109. [DOI] [PubMed] [Google Scholar]
- 7. Zhang L, Yang C, Zhang Y, Li L, Zhang X, et al. (2007) A genetic linkage map of Pacific white shrimp (Litopenaeus vannamei): sex-linked microsatellite markers and high recombination rates. Genetica 131: 37–49. [DOI] [PubMed] [Google Scholar]
- 8. Zhang X, Zhang Y, Scheuring C, Zhang HB, Huan P, et al. (2010) Construction and characterization of a bacterial artificial chromosome (BAC) library of Pacific white shrimp, Litopenaeus vannamei. Mar Biotechnol (NY) 12: 141–149. [DOI] [PubMed] [Google Scholar]
- 9. Veloso A, Warr GW, Browdy CL, Chapman RW (2011) The transcriptomic response to viral infection of two strains of shrimp (Litopenaeus vannamei). Dev Comp Immunol 35: 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gorbach DM, Hu ZL, Du ZQ, Rothschild MF (2009) SNP discovery in Litopenaeus vannamei with a new computational pipeline. Anim Genet 40: 106–109. [DOI] [PubMed] [Google Scholar]
- 11. Huang XD, Yin ZX, Jia XT, Liang JP, Ai HS, et al. (2010) Identification and functional study of a shrimp Dorsal homologue. Dev Comp Immunol 34: 107–113. [DOI] [PubMed] [Google Scholar]
- 12. Metzker ML (2010) Sequencing technologies - the next generation. Nat Rev Genet 11: 31–46. [DOI] [PubMed] [Google Scholar]
- 13. Voelkerding KV, Dames SA, Durtschi JD (2009) Next-generation sequencing: from basic research to diagnostics. Clin Chem 55: 641–658. [DOI] [PubMed] [Google Scholar]
- 14. Morozova O, Marra MA (2008) Applications of next-generation sequencing technologies in functional genomics. Genomics 92: 255–264. [DOI] [PubMed] [Google Scholar]
- 15. Surget-Groba Y, Montoya-Burgos JI (2010) Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res 20: 1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Git A, Dvinge H, Salmon-Divon M, Osborne M, Kutter C, et al. (2010) Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 16: 991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hashimoto S, Qu W, Ahsan B, Ogoshi K, Sasaki A, et al. (2009) High-resolution analysis of the 5′-end transcriptome using a next generation DNA sequencer. PLoS One 4: e4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li R, Fan W, Tian G, Zhu H, He L, et al. (2010) The sequence and de novo assembly of the giant panda genome. Nature 463: 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wall PK, Leebens-Mack J, Chanderbali AS, Barakat A, Wolcott E, et al. (2009) Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics 10: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao ZY, Yin ZX, Weng SP, Guan HJ, Li SD, et al. (2007) Profiling of differentially expressed genes in hepatopancreas of white spot syndrome virus-resistant shrimp (Litopenaeus vannamei) by suppression subtractive hybridisation. Fish Shellfish Immunol 22: 520–534. [DOI] [PubMed] [Google Scholar]
- 21. Hao da C, Ge G, Xiao P, Zhang Y, Yang L (2011) The first insight into the tissue specific taxus transcriptome via Illumina second generation sequencing. PLoS One 6: e21220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qin YF, Fang HM, Tian QN, Bao ZX, Lu P, et al. (2011) Transcriptome profiling and digital gene expression by deep-sequencing in normal/regenerative tissues of planarian Dugesia japonica. Genomics 97: 364–371. [DOI] [PubMed] [Google Scholar]
- 23.Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol: 138–148. [PubMed]
- 24. Pertea G, Huang X, Liang F, Antonescu V, Sultana R, et al. (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19: 651–652. [DOI] [PubMed] [Google Scholar]
- 25. Conesa A, Gotz S (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008: 619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ye J, Fang L, Zheng H, Zhang Y, Chen J, et al. (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34: W293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, et al. (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28: 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, et al. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29: 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang-di P, Jian-min Z, Wen-guo P, Shi-qiang H, Tian-li T, et al. (2002) The embryonic development of Penaeus vannamei and effect of temperature and salinity on embryonic development. Journal of Shanghai Fisheries University 4: 310–316. [Google Scholar]
- 32. Ronquillo JD, Saisho T, McKinley RS (2006) Early developmental stages of the green tiger prawn,Penaeus semisulcatus de Haan (Crustacea, Decapoda, Penaeidae). Hydrobiologia 560: 175–196. [Google Scholar]
- 33.Muller PY, Janovjak H, Miserez AR, Dobbie Z (2002) Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32: 1372–1374, 1376, 1378–1379. [PubMed]
- 34. Wixon J, Kell D (2000) The Kyoto encyclopedia of genes and genomes–KEGG. Yeast 17: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Altermann E, Klaenhammer TR (2005) PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics 6: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, et al. (2011) The ecoresponsive genome of Daphnia pulex. Science 331: 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang W, Wan H, Jiang H, Zhao Y, Zhang X, et al. (2011) A transcriptome analysis of mitten crab testes (Eriocheir sinensis). Genet Mol Biol 34: 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuballa AV, Holton TA, Paterson B, Elizur A (2011) Moult cycle specific differential gene expression profiling of the crab Portunus pelagicus. BMC Genomics 12: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tagmount A, Wang M, Lindquist E, Tanaka Y, Teranishi KS, et al. (2010) The porcelain crab transcriptome and PCAD, the porcelain crab microarray and sequence database. PLoS One 5: e9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pongsomboon S, Tang S, Boonda S, Aoki T, Hirono I, et al. (2011) A cDNA microarray approach for analyzing transcriptional changes in Penaeus monodon after infection by pathogens. Fish Shellfish Immunol 30: 439–446. [DOI] [PubMed] [Google Scholar]
- 41. Pongsomboon S, Tang S, Boonda S, Aoki T, Hirono I, et al. (2008) Differentially expressed genes in Penaeus monodon hemocytes following infection with yellow head virus. BMB Rep 41: 670–677. [DOI] [PubMed] [Google Scholar]
- 42. Watanabe H, Kobayashi K, Kato Y, Oda S, Abe R, et al. (2008) Transcriptome profiling in crustaceans as a tool for ecotoxicogenomics: Daphnia magna DNA microarray. Cell Biol Toxicol 24: 641–647. [DOI] [PubMed] [Google Scholar]
- 43. Gard AL, Lenz PH, Shaw JR, Christie AE (2009) Identification of putative peptide paracrines/hormones in the water flea Daphnia pulex (Crustacea; Branchiopoda; Cladocera) using transcriptomics and immunohistochemistry. Gen Comp Endocrinol 160: 271–287. [DOI] [PubMed] [Google Scholar]
- 44. De Gregoris TB, Rupp O, Klages S, Knaust F, Bekel T, et al. (2011) Deep sequencing of naupliar-, cyprid- and adult-specific normalised Expressed Sequence Tag (EST) libraries of the acorn barnacle Balanus amphitrite. Biofouling 27: 367–374. [DOI] [PubMed] [Google Scholar]
- 45. Clark MS, Thorne MA, Toullec JY, Meng Y, Guan LL, et al. (2011) Antarctic krill 454 pyrosequencing reveals chaperone and stress transcriptome. PLoS One 6: e15919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jung H, Lyons RE, Dinh H, Hurwood DA, McWilliam S, et al. (2011) Transcriptomics of a giant freshwater prawn (Macrobrachium rosenbergii): de novo assembly, annotation and marker discovery. PLoS One 6: e27938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zeng V, Villanueva KE, Ewen-Campen BS, Alwes F, Browne WE, et al. (2011) De novo assembly and characterization of a maternal and developmental transcriptome for the emerging model crustacean Parhyale hawaiensis. BMC Genomics 12: 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He L, Wang Q, Jin X, Wang Y, Chen L, et al. (2012) Transcriptome profiling of testis during sexual maturation stages in Eriocheir sinensis using Illumina sequencing. PLoS One 7: e33735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ou J, Meng Q, Li Y, Xiu Y, Du J, et al. (2012) Identification and comparative analysis of the Eriocheir sinensis microRNA transcriptome response to Spiroplasma eriocheiris infection using a deep sequencing approach. Fish Shellfish Immunol 32: 345–352. [DOI] [PubMed] [Google Scholar]
- 50. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. (2000) The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. [DOI] [PubMed] [Google Scholar]
- 51. Alkan C, Sajjadian S, Eichler EE (2011) Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vargas-Albores F, Martinez-Martinez A, Aguilar-Campos J, Jimenez-Vega F (2009) The expression of protein disulfide isomerase from Litopenaeus vannamei hemocytes is regulated by bacterial inoculation. Comp Biochem Physiol Part D Genomics Proteomics 4: 141–146. [DOI] [PubMed] [Google Scholar]
- 53. Ma J, Zhang M, Ruan L, Shi H, Xu X (2010) Characterization of two novel ADP ribosylation factors from the shrimp Marsupenaeus japonicus. Fish Shellfish Immunol 29: 956–962. [DOI] [PubMed] [Google Scholar]
- 54. Zhang Y, Chen J, Tang X, Wang F, Jiang L, et al. (2010) Transcriptome analysis of the venom glands of the Chinese wolf spider Lycosa singoriensis. Zoology (Jena) 113: 10–18. [DOI] [PubMed] [Google Scholar]
- 55. Blow NS, Salomon RN, Garrity K, Reveillaud I, Kopin A, et al. (2005) Vibrio cholerae infection of Drosophila melanogaster mimics the human disease cholera. PLoS Pathog 1: e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 57. Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier ME, et al. (2010) The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466: 720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparisons between assembled transcriptome unigenes and the EST-unigenes which were assembled form EST sequences available from Genbank using TGICL and Phrap softwares.
(ZIP)
BLASTX searching of the Unigenes against NCBI Nr database (E value<0.00001).
(ZIP)
BLASTX searching of the Unigenes against Swissprot database (E value<0.00001).
(ZIP)
BLASTX searching of the Unigenes against TrEMBL database (E value<0.00001).
(ZIP)
COG Classification of the unigenes.
(ZIP)
GO categories of the unigenes.
(ZIP)
KEGG Classification of the unigenes.
(ZIP)
Sanger sequencing of the 12 analyzed unigenes.
(ZIP)