

Abstract
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is a common human enzymopathy that affects cellular redox status and may lower flux into nonoxidative pathways of glucose metabolism. Oxidative stress may worsen systemic glucose tolerance and cardiometabolic syndrome. We hypothesized that G6PD deficiency exacerbates diet-induced systemic metabolic dysfunction by increasing oxidative stress but in myocardium prevents diet-induced oxidative stress and pathology. WT and G6PD-deficient (G6PDX) mice received a standard high-starch diet, a high-fat/high-sucrose diet to induce obesity (DIO), or a high-fructose diet. After 31 wk, DIO increased adipose and body mass compared with the high-starch diet but to a greater extent in G6PDX than WT mice (24 and 20% lower, respectively). Serum free fatty acids were increased by 77% and triglycerides by 90% in G6PDX mice, but not in WT mice, by DIO and high-fructose intake. G6PD deficiency did not affect glucose tolerance or the increased insulin levels seen in WT mice. There was no diet-induced hypertension or cardiac dysfunction in either mouse strain. However, G6PD deficiency increased aconitase activity by 42% and blunted markers of nonoxidative glucose pathway activation in myocardium, including the hexosamine biosynthetic pathway activation and advanced glycation end product formation. These results reveal a complex interplay between diet-induced metabolic effects and G6PD deficiency, where G6PD deficiency decreases weight gain and hyperinsulinemia with DIO, but elevates serum free fatty acids, without affecting glucose tolerance. On the other hand, it modestly suppressed indexes of glucose flux into nonoxidative pathways in myocardium, suggesting potential protective effects.
Keywords: reactive oxygen species, fructose, diet, nonoxidative glucose pathways
diet-induced obesity (DIO) is increasing in prevalence and is a major risk factor for type II diabetes and heart disease (35). Obesity results in metabolic dysfunction such as increased serum glucose, triglycerides, and free fatty acids, abnormal insulin secretion, and insulin resistance. Cardiovascular problems associated with metabolic dysfunction are highly prevalent in African Americans (36). Glucose-6-phosphate dehydrogenase (G6PD) deficiency, the most common enzyme deficiency in the world, is particularly common among individuals of African descent (7). Less severe forms of G6PD deficiency (10–60% residual activity) are common and are largely clinically undetected (7, 54, 68). G6PD is the rate-determining enzyme of the pentose phosphate pathway and is the main cytoplasmic source of NADPH (15, 27). The effects of G6PD deficiency on the interaction between metabolic dysfunction and the heart are unclear. Clinical studies suggest that G6PD deficiency may reduce the risk of heart disease (12, 42), perhaps due to less NADPH generation and subsequent formation of reactive oxygen species (ROS) (19, 20, 37, 38). The risk of developing heart disease is strongly associated with obesity, metabolic dysfunction, and diabetes (17). However, despite its possible cardioprotective role, G6PD deficiency may increase the risk of diabetes (47, 58, 68, 74). Studies in G6PD deficient (G6PDX) mice found that G6PD deficiency may increase β-cell apoptosis and result in insulin resistance (79). Thus, G6PD deficiency may negatively affect metabolic derangements while protecting against heart disease.
By producing NADPH, G6PD provides reducing equivalents for the antioxidant system (34). G6PD deficiency decreases antioxidant capacity and increases cell death in response to oxidant challenge in cultured cells, (27, 70, 71). Consistent with an increase in oxidative stress, G6PD deficiency increased pancreatic β-cell death and associated metabolic dysfunction in aging mice (79), increased myocardial injury following ischemia/reperfusion (28), and was associated with moderate cardiac dysfunction in older mice (27). Thus, G6PD deficiency may decrease resistance to oxidant stress and result in greater cardiometabolic dysfunction. On the other hand, G6PD provides NADPH required for the generation of superoxide by NADPH oxidase (1, 2, 37, 38), uncoupled nitric oxide synthase (44, 45), and xanthine oxidase (22); thus, deficiency could be protective (18, 19, 21, 60). G6PD activity and NADPH were increased in the liver and myocardium of genetically obese rats, and pharmacological inhibition of G6PD suppressed superoxide generation in vitro (18, 60, 64). Similar findings were seen in myocardium from humans and dogs with heart failure (19, 21). Thus, in contrast to the antioxidant effects of G6PD activity, excessive flux through G6PD may promote the formation of NADPH-dependent ROS, which would be prevented by G6PD deficiency.
High sugar intake and obesity can exert toxic effects by increasing oxidative stress (40, 41, 76, 78, 79). Antioxidant therapy prevents the exacerbation of heart failure caused by high sugar intake (10). Furthermore, inhibition of NADPH oxidase attenuates cardiac dysfunction after myocardial infarction or chronic pressure overload (13, 26, 33) and the adverse effects of obesity on the heart (39). Thus, NADPH-dependent oxidant mechanisms may mediate the adverse effects of obesity on the development of heart failure, while a reduction in G6PD activity may prevent diet-induced ROS formation. Oxidative stress may accelerate flux through nonoxidative glucose pathways through GAPDH inhibition and suppression of glycolytic flux (16, 67). Nonoxidative glucose pathways include the formation of advanced glycation end products (AGEs), the hexosamine biosynthetic pathway (HBP), and (in part) the pentose phosphate shunt. Previous studies have indicated greater oxidative stress and increased flux through these pathways in obesity (48, 51, 52); however, the interaction among these pathways is not well understood, nor is it clear how they are impacted by G6PD deficiency. A decrease in oxidant stress in G6PD-deficient myocardium could decrease flux through other nonoxidative pathways of glucose metabolism, resulting in an overall beneficial effect on cardiac structure and function (16, 21).
In the present investigation, we assessed the effects of G6PD deficiency on diet-induced cardiometabolic defects, oxidant stress, and other nonoxidative glucose pathways. Clinical evidence suggests that G6PD deficiency may lead to metabolic dysfunction while decreasing cardiovascular disease (12, 19, 42, 47, 58, 68, 74). Thus, we hypothesized that G6PD deficiency would exacerbate metabolic dysfunction caused by diet-induced obesity (DIO) or high fructose intake while decreasing diet induced cardiac pathology. We further hypothesized that activation of other nonoxidative glucose pathways would correspond inversely with oxidative stress. Wild-type (WT) and G6PDX mice were subjected to long-term dietary stress using either a high-fat/high-sucrose diet to cause DIO or a high-fructose diet, which were compared with a standard high-starch diet that reflects a commercial diet typically consumed by laboratory mice. The high-fat/high-sucrose and high-fructose diets both induce cardiometabolic stress, but high fructose does so without inducing obesity (9, 24, 41, 66, 67). We envisaged that DIO and high fructose intake would both increase oxidative stress and decrease cardiac and metabolic function compared with a high-starch diet in WT mice. We further predicted that G6PD deficiency would exacerbate systemic diet-induced oxidative stress and metabolic dysfunction but prevent diet-induced cardiac effects.
RESEARCH DESIGN AND METHODS
Experimental design and animal treatment.
All procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals (NIH publication no. 85-23) and were approved by the University of Maryland Institutional Animal Care and Use Committee. G6PDX mice were generated previously on a C3H/HeJ background by Pretsch et al. (50). G6PDX and littermate control (WT) breeding pairs were provided by Dr. Jane Leopold, and genotyping was performed as previously described (28). Mice were fed ad libitum custom manufactured purified diets (Table 1) (Research Diets, New Brunswick, NJ). Sixty female homozygous G6PDX and 60 female WT littermate control mice were weaned at 3 wk of age and subsequently placed on a high-starch diet (Starch), a high-fructose diet (Fructose), or a high-fat/high-sucrose diet intended to induce obesity (DIO) (n = 20/group). Food intake was determined by weighing the food and dividing by the number of mice in the cage. Blood pressure was assessed by tail cuff plethysmography at 11 and 23 wk of age, and heart function was assessed by echocardiography at 12 and 24 wk. Metabolic function was assessed by intraperitoneal glucose tolerance test at 28 wk. A subset of mice (n = 10/group) were euthanized at 12 wk of age, and 31 ± 0.2 wk (28–33 wk) of age for biochemical analysis.
Table 1.
Macronutrient composition of diets
| Fat |
Carbohydrate |
Protein |
|||||
|---|---|---|---|---|---|---|---|
| Diet | Soy Oil | Lard | Cornstarch | Maltodextrin | Fructose | Sucrose | Casein |
| Starch | 6 | 4 | 57 | 12 | 0 | 1 | 20 |
| Fructose | 6 | 4 | 9 | 0 | 60 | 1 | 20 |
| DIO | 6 | 39 | 7 | 10 | 0 | 17 | 20 |
Values are energy contribution to the diet, %total. Diets were matched for vitamin and mineral content. DIO, diet-induced obesity.
Glucose tolerance test.
Glucose tolerance was assessed at 28 wk of age. Animals were fasted overnight for 15 h and injected with glucose (2 g/kg body mass ip). Glucose was measured from blood collected from the tail vein immediately before injection and then at 30, 60, and 120 min following injection.
Echocardiography.
Cardiac function was assessed by echocardiography at 12 and 24 wk of age (Vevo 770 High-Resolution Imaging Systems; Visual Sonics, Toronto, ON, Canada) with a 30-MHz linear array transducer. Mice were anesthetized with 2% isoflurane in oxygen, shaved, and placed on a warming pad. Guided M-mode frames were recorded from the parasternal short axis. Pulse wave Doppler measurements were recorded from the apical view. Left ventricle (LV) absolute wall thickness was calculated as the sum of the thickness of the posterior and anterior walls, and relative wall thickness as the absolute thickness divided by the end-diastolic LV chamber diameter, as previously described in detail (23, 24). LV end-diastolic and end-systolic volumes, stroke volume, and ejection fraction were calculated using standard equations as previously described (23, 24). Myocardial performance index was determined as the sum of the isovolumetric relaxation and contraction times divided by the ejection time.
Tissue harvest.
The terminal procedure was performed from 4 to 6 h into the light phase of the light-dark cycle. Nonfasted animals were anesthetized with 5% isoflurane, the chest was opened, and the heart was dissected in ice-cold phosphate -buffered saline. The left ventricle, liver, and combined soleus and gastrocnemius muscles were frozen in liquid nitrogen. Blood was drawn from the thorax and centrifuged at 1,500 g for 15 min to obtain serum. All samples were stored at −80°C for subsequent biochemical analysis.
Biochemical parameters.
The enzymatic activities of G6PD, aconitase, and citrate synthase (CS) were assayed in tissue homogenates and normalized to wet tissue mass (10). To obtain homogenized tissue, 10–20 mg of tissue was combined with 150 μl of buffer containing 0.1 M Tris·HCl and 15 mM tricarballylic acid, pH 7.8. Tissue was homogenized in a Bullet Blender (Next Advance, Averill Park, NY). All enzyme activities were assayed in duplicate for each sample.
To obtain G6PD activity, 10 μl of LV homogenate (or 10 μl of liver homogenate or 20 μl of hindlimb skeletal muscle homogenate) was added to cuvettes containing 490 μl of G6PD reaction mix [25 mM HEPES-Tris, 0.5 mM NADP+, 3.3 mM MgCl2, 0.5 mM 6-phosphogluconate (6PG), pH 7.8]. The increase in light absorbance over 5 min was measured at 340 nm, after which 5 μl of 5 mM glucose 6-phosphate (G6P) was added, and the increase in light absorbance at 340 nm was measured again. Activity was then determined by multiplying the slope to the molar concentration constant for NADPH (6.7 M−1). To obtain G6PD activity, the slope of the absorbance without G6P was subtracted from the slope of the absorbance with G6P to obtain G6PD activity, because 6PGD, a downstream enzyme from G6PD in the pentose phosphate pathway, also produces NADPH, and the slope with G6P represents the activity of both enzymes.
For aconitase activity, 10 μl of LV homogenate (or 10 μl of liver homogenate or 20 μl of hindlimb skeletal muscle homogenate) was added to cuvettes containing 490 μl of aconitase reaction mix (0.083% chloroform, 1.67 mM sodium citrate, 26.7 mM triethanolamine, 0.5 mM NADP+, 0.5 mM MgCl2, pH 7.4), and the increase in absorbance at 340 nm was measured over 5 min. Activity was then determined by multiplying the slope to the molar concentration constant for NADPH (6.7 M−1).
For CS activity, 0.5 μl of LV homogenate (or 1 μl of liver homogenate or 4 μl of hindlimb skeletal muscle homogenate) was added to cuvettes containing 500 μl of CS reaction mix (0.1 M Tris·HCl, 1.25 mM 5,5′-dithiobis[2-nitrobenzoic acid], pH 8). Then, 25 μl of 50 mM oxaloacetate and 5 mM acetyl-CoA were added to the reaction mixture, and the increase in absorbance at 412 nm was measured over 5 min. Activity was then determined by multiplying the slope to the molar concentration constant for 5,5′-dithiobis[2-nitrobenzoic acid] (13,600 M−1).
Serum glucose, triglycerides, and free fatty acid concentrations were assayed spectrophotometrically, and leptin, insulin, and adiponectin were measured by enzyme-linked immunosorbent assays using commercially available kits according to the manufacturers' instructions (23, 24). Oxidative stress was assessed by measuring aconitase activity as described above (10) and by determining the lipid peroxidation products malondialdehyde (MDA) and 4-hydroxyalkenals (4HA) (Oxford Biomedical Research, Oxford, MI) in tissue homogenates by using spectrophotometric assays. Methylglyoxal (Cell Biolabs, San Diego, CA) and PKC activity (Enzo Life Sciences, Farmingdale, NY) were assayed using enzyme-linked immunosorbent assays. GAPDH activity (Biomedical Research Service Centre, University at Buffalo, Buffalo, NY), caspase-3 activity (BioVision, Mountain View, CA), and sorbitol (BioVision) were assessed spectrophotometrically using commercially available kits according to the manufacturers' instructions.
Total O-GlcNAc expression was evaluated by SDS-PAGE as described previously (51, 52). Briefly, frozen heart and liver tissues were homogenized with modified ice-cold RIPA buffer, and the supernatant was centrifuged twice at 13,000 g for 10 min at 4°C and then stored at −80°C until further use. We employed the Bradford assay for protein quantification, and total O-linked N-acetylglucosamine (O-GlcNAc) expression evaluated by SDS-PAGE (CTD110.6; Santa Cruz Biotechnology, Santa Cruz, CA). Total O-GlcNAcylation expression (per lane) was quantified by densitometric analysis using β-actin (Cell Signaling, Beverly, MA) as a loading control.
Glycogen was assayed by incubating tissue homogenates with and without amyloglucosidase and then determining glucose concentration. Tissue (25–35 mg) was homogenized in 3 M perchloric acid, after which homogenates were incubated for 2 h with and without amyloglucosidase (50 μg/ml in 50 mM sodium acetate, 0.02% BSA, pH 5.5). Following this treatment, samples were centrifuged for 5 min at 2,500 rpm, and 25 μl of the supernatant was used to determine the glucose concentration spectrophotometrically with a commercially available kit (Wako Diagnostics, Richmond, VA).
Liver triglycerides were determined as previously described (24). Briefly, 10–20 mg of tissue sample was homogenized in 1.5 ml of 2:1 chloroform-methanol. After homogenization, 3.5 ml of 2:1 chloroform-methanol was added to samples, and samples were sonicated at 4°C for 1 h and vortexed for 3 min. Vortexed samples were centrifuged at 1,550 g for 15 min, and the supernatant was transferred to new glass vials. Pellets were resuspended in 5 ml of 2:1 chloroform-methanol, vortexed, and centrifuged again, and the supernatants were transferred to the tubes containing the supernatants from the first centrifugation. The supernatants were allowed to dry and were then assayed for triglycerides using a colorimetric assay kit (Wako Diagnostics).
Statistical analysis.
Values are shown as means ± SE. Survival was assessed by Log-Rank Kaplan-Meier analysis. Comparisons between strain and diet grouping were made using a two-way ANOVA. Interaction among the three diets was tested with a Holm-Sidak post hoc test. A Pearson regression analysis was used to assess correlation between parameters. P < 0.05 was considered significant.
RESULTS
Decreased growth in G6PD-deficient mice with an obesogenic diet.
Compared with WT mice, G6PD activity was decreased in G6PDX mice in heart, liver, and skeletal muscle (Fig. 1). Of interest, high fructose intake increased G6PD activity in the heart and liver (Fig. 1, A–C), whereas DIO decreased G6PD activity in the liver and skeletal muscle (Fig. 1, A, B, and F). Similar dietary effects on G6PD activity have been previously characterized (5, 43, 55, 57, 59, 65). Energy intake was greater with the high-fructose diet than the starch diet and was greatest in animals fed the high-fat/high-sugar diet (Table 2). G6PD deficiency had no effect on energy intake. Over the course of the study, there were no differences in survival among groups (Fig. 2).
Fig. 1.

Glucose-6-phosphate dehydrogenase (G6PD) activity. A and B: liver G6PD activity. C and D: left ventricular (LV) G6PD activity. E and F: hindlimb skeletal muscle G6PD activity. Data were obtained using tissue from mice at 12 and 31 wk of age. Values are means ± SE; n = 7–10/group. DIO, diet-induced obesity; G6PDX, G6PD-deficient mice; gww, grams of wet tissue weight; WT, wild type.
Table 2.
Energy intake, morphometric data, and liver content
| WT |
G6PD Deficient |
|||||
|---|---|---|---|---|---|---|
| Parameter | Starch | Fructose | DIO | Starch | Fructose | DIO |
| Duration of the study | ||||||
| Energy intake, kcal·mouse−1·day−1 | 8.33 ± 0.43 | 9.71 ± 0.40† | 10.89 ± 0.29†‡ | 8.22 ± 0.29 | 9.31 ± 0.28† | 10.44 ± 0.26†‡ |
| 12 wk | ||||||
| Tibia length, mm | 19.1 ± 0.2 | 19.5 ± 0.3 | 19.6 ± 0.2 | 19.6 ± 0.2 | 19.6 ± 0.2 | 19.8 ± 0.2 |
| Kidney mass, mg | 135 ± 5 | 147 ± 3 | 162 ± 4†‡ | 134 ± 3 | 154 ± 3† | 160 ± 7† |
| Liver mass, g | 1.05 ± 0.03 | 1.28 ± 0.11† | 1.15 ± 0.03 | 1.07 ± 0.04 | 1.38 ± 0.05† | 1.08 ± 0.04‡ |
| Liver glycogen, mg/gww | 47.8 ± 4.2 | 47.3 ± 8.8 | 36.8 ± 3.8 | 49.6 ± 4.3 | 49.0 ± 6.3 | 27.7 ± 2.8†‡ |
| Liver triglycerides, mg/gww | 9.17 ± 0.76 | 7.53 ± 0.68 | 10.1 ± 0.76 | 7.76 ± 0.65 | 6.88 ± 0.48 | 12.14 ± 1.06†‡ |
| Gonadal fat/body mass, mg/g | 18.0 ± 1.35 | 15.5 ± 1.43 | 30.0 ± 3.53†‡ | 15.3 ± 1.22 | 17.5 ± 2.30 | 33.1 ± 3.00†‡ |
| 31 wk | ||||||
| Tibia length, mm | 19.8 ± 0.1 | 20.1 ± 0.1 | 19.6 ± 0.1 | 19.4 ± 0.3 | 19.5 ± 0.2* | 18.8 ± 0.4* |
| Kidney mass, mg | 162 ± 4 | 177 ± 4 | 169 ± 4 | 155 ± 7 | 163 ± 5 | 153 ± 8 |
| Liver mass, g | 1.42 ± 0.06 | 1.81 ± 0.04† | 1.51 ± 0.05‡ | 1.24 ± 0.06* | 1.80 ± 0.08† | 1.18 ± 0.03†‡ |
| Liver glycogen, mg/gww | 56.4 ± 2.2 | 51.7 ± 4.9 | 38.2 ± 2.9†‡ | 53.5 ± 4.7 | 53.4 ± 5.0 | 38.0 ± 3.5†‡ |
| Liver triglycerides, mg/gww | 12.0 ± 1.3 | 11.0 ± 1.8 | 19.9 ± 1.3†‡ | 8.10 ± 1.05 | 10.8 ± 1.1 | 15.0 ± 2.1*† |
| Gonadal fat/body mass, mg/g | 28.9 ± 3.7 | 39.5 ± 4.1 | 61.0 ± 3.7†‡ | 20.3 ± 4.4 | 27.0 ± 3.8* | 57.5 ± 4.4†‡ |
Values are means ± SE;
P < 0.05 vs. WT within dietary treatment group;
P < 0.05 vs. starch within mouse strain;
P < 0.05 vs. fructose within mouse strain; n = 4 cages/group for energy intake; for all other parameters n = 10/group at 12 wk, n = 7–10/group at 31 wk of age. G6PD, glucose-6-phosphate dehydrogenase; gww, grams of wet tissue weight.
Fig. 2.

Survival. There was no statistically significant difference in overall survival throughout the study (P = 0.181). Survival was assessed by Log Rank Kaplan-Meier analysis; n = 20/group until 12 wk and n = 10/group thereafter.
There were no differences in initial body mass at 3 wk of age when mice were weaned and placed into dietary groups (body mass, 11.1 ± 0.3 g for all groups). Initially, both WT and G6PDX mice experienced greater weight gain in response to DIO than either the high-starch or high-fructose diets (Fig. 3A). However, body mass plateaued after 16 wk in G6PDX mice in the DIO group (Fig. 3, B and C). DIO increased fat pad mass in WT and G6PDX mice but to a lesser extent in absolute terms in G6PDX mice at 31 wk (Fig. 3, D and E). However, when fat pad mass is expressed relative to body mass, there was no difference among groups, suggesting that the difference in body mass was partially due to a lower increase in lean tissue mass in the G6PDX from 16 to 31 wk (Table 2). G6PD deficiency also decreased tibia length compared with WT among high-fructose and DIO groups at 31 wk (Table 2). These results indicate that, despite similar energy intake between G6PDX and WT mice, G6PD deficiency decreased the extent of the increase in both lean and fat tissues in response to an obesogenic diet, suggesting that G6PD deficiency blunted the increase in overall growth.
Fig. 3.
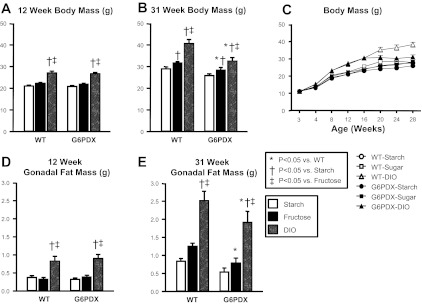
G6PD deficiency results in decreased growth. A: DIO increased 12 wk body mass to the same extent in G6PDX and WT mice. B: DIO increased terminal 31-wk body mass to a lesser extent in G6PDX mice than in WT mice. C: body mass data indicate an increase in body mass over the course of the study, with greater increase in body mass in response to high-fat/high-sugar diet (DIO). Furthermore, data indicate that in G6PDX mice there was a cessation in the increase in body mass around 16 wk. D: DIO increased gonadal fat mass the same extent in G6PDX and WT mice at 12 wk. E: DIO increased gonadal fat mass to a lesser extent in G6PDX mice than in WT mice at 31 wk of age. Values are means ± SE; n = 7–10/group.
Effect of G6PD deficiency on diet-induced metabolic dysfunction.
Normally, attenuating obesity in mice results in fewer metabolic abnormalities and greater metabolic function (24). Thus, we assessed indexes of whether metabolic dysfunction was attenuated by the decrease in obesity in G6PD-deficient mice. Increased serum triglycerides and free fatty acids may be indicative of metabolic syndrome and increased fat mobilization (17). DIO increased serum free fatty acids, and high fructose intake increased serum triglycerides in G6PDX mice (Fig. 4, A and B). Leptin secretion is increased in obesity and corresponds to increased fat mass. Here, we observed an increase in leptin in both WT and G6PDX mice but to a significantly less extent in G6PDX mice (Fig. 4C and Table 3) and corresponding to the extent of fat pad mass increase in response to DIO. Increased insulin secretion is indicative of insulin resistance and a prediabetic state (14). DIO increased serum insulin in WT mice; however, G6PDX mice failed to increase serum insulin in response to DIO (Fig. 4D). Decreased insulin in G6PDX mice may be due to β-cell apoptosis and dysfunction, as previously reported (79). Metabolic dysfunction can lead to renal failure, and G6PD deficiency is associated with increased serum creatinine and albuminuria (75–77). In our study, DIO resulted in a modest but significant increase in serum creatinine in G6PDX mice, suggesting lower creatinine clearance and mild renal dysfunction (Table 3). The high-fructose diet had no effect on serum creatinine.
Fig. 4.
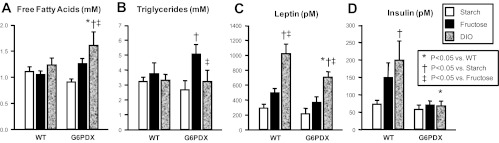
Serum metabolic parameters. A: DIO increased free fatty acids in G6PDX mice but not in WT mice. B: high fructose intake increased triglycerides in G6PDX mice. C: DIO increased leptin in G6PDX and WT mice but to a lesser extent in G6PDX mice. D: DIO increased insulin in WT mice but failed to increase insulin in G6PDX mice. Data were obtained using serum from mice at 31 wk of age. Values are means ± SE; n = 6–10/group.
Table 3.
Serum data
| WT |
G6PD Deficient |
|||||
|---|---|---|---|---|---|---|
| Parameter | Starch | Fructose | DIO | Starch | Fructose | DIO |
| 12 wk | ||||||
| Glucose, mM | 12.8 ± 0.5 | 13.9 ± 0.7 | 13.2 ± 0.4 | 14.0 ± 1.9 | 14.3 ± 0.4 | 13.0 ± 0.5 |
| Triglycerides, mM | 1.91 ± 0.31 | 1.53 ± 0.27 | 2.23 ± 0.34 | 1.95 ± 0.31 | 2.02 ± 0.20 | 1.78 ± 0.17 |
| Free fatty acids, mM | 1.30 ± 0.12 | 1.18 ± 0.09 | 1.53 ± 0.17 | 1.22 ± 0.12 | 1.11 ± 0.11 | 1.50 ± 0.16 |
| Insulin, pM | 33.4 ± 3.2 | 41.5 ± 8.5 | 48.2 ± 13.7 | 33.7 ± 3.4 | 36.2 ± 4.9 | 35.1 ± 3.8 |
| Leptin, pM | 115 ± 22 | 165 ± 57 | 567 ± 70†‡ | 121 ± 20 | 176 ± 20 | 526 ± 87†‡ |
| Adiponectin, mg/ml | 10.0 ± 1.0 | 8.2 ± 1.7 | 16.1 ± 3.1 | 13.6 ± 4.4 | 7.8 ± 1.2 | 12.8 ± 2.7 |
| 31 wk | ||||||
| Glucose, mM | 11.9 ± 0.5 | 13.5 ± 0.6 | 13.4 ± 1.1 | 11.7 ± 0.4 | 13.5 ± 0.7 | 11.8 ± 0.7 |
| Adiponectin, mg/ml | 9.1 ± 1.2 | 11.3 ± 1.5 | 10.4 ± 1.6 | 12.8 ± 3.4 | 9.4 ± 1.7 | 10.3 ± 1.5 |
| Creatinine, μM | 35.1 ± 20.2 | 50.0 ± 21.5 | 50.8 ± 21.5 | 60.0 ± 22.9 | 81.0 ± 21 | 121 ± 25* |
Values are means ± SE;
P < 0.05 vs. WT within dietary treatment group;
P < 0.05 vs. Starch within mouse strain;
P < 0.05 vs. Fructose within mouse strain; n = 6–10/group.
Obesity often corresponds to an increase in liver triglycerides content (hepatic steatosis) (56). Compared with the high-starch diet, DIO but not high fructose intake increased triglycerides in the liver in both WT and G6PDX mice at 31 wk but to a lesser extent in G6PDX mice, consistent with a decrease in body mass in response to a high-fat/high-sugar diet (Table 2). At 12 wk, DIO resulted in increased liver triglycerides only in G6PDX mice. The effects of DIO and G6PD deficiency on liver glycogen was effectively the inverse of what was observed with triglyceride content, with lower glycogen in both WT and G6PDX mice at 31 wk and only in G6PDX mice at 12 wk.
Glucose tolerance has been used to assess metabolic dysfunction and may be decreased in response to obesity and insulin resistance (14). Previous investigators observed a decreased glucose tolerance in male G6PDX mice compared with WT mice (79). Thus, we hypothesized that dietary stress would further exacerbate glucose intolerance in G6PDX mice. However, we found that blood glucose after glucose challenge was identical between WT and G6PDX mice among all diet groups (Fig. 5). DIO decreased glucose tolerance but to the same degree in both G6PDX and WT mice, indicating a similar degree of diet-induced metabolic dysfunction in both strains. There was a trend for high fructose intake to increase glucose tolerance, but this was not statistically significant. Overall, G6PD deficiency had no effect on glucose tolerance.
Fig. 5.

Glucose tolerance. Intraperitoneal glucose tolerance tests indicated decreased glucose tolerance with obesity and no effect of G6PD deficiency. Data were obtained from mice after 25 wk of dietary treatment (28 wk of age). Values are means ± SE; n = 5–10/group.
Cardiovascular effects.
DIO did not affect overall blood pressure and resulted in relatively minor changes in the myocardium. In WT mice, DIO increased LV mass by 11% and atrial mass by 33% at 12 wk, indicative of diet-induced hypertrophy in young WT animals (Table 4). In older animals, DIO increased relative wall thickness by 44% and decreased the chamber volume compared with the high-fructose diet with no effect on overall heart mass, indicating concentric remodeling in WT mice. This dietary effect was not observed in G6PDX mice. Diastolic dysfunction may precede systolic dysfunction; however, we did not observe an increase in myocardial performance index, as would be expected with severe diastolic function (46). More direct indexes of diastolic function were not assessed (e.g., mitral inflow or LV filling pressure). Compared with WT mice, there was significant interaction among mice consuming the high-fructose diet for G6PD deficiency to increase LV mass, diastolic volume, and stroke volume.
Table 4.
Cardiovascular data
| WT |
G6PD Deficient |
|||||
|---|---|---|---|---|---|---|
| Parameter | Starch | Fructose | DIO | Starch | Fructose | DIO |
| 11–12 wk | ||||||
| Cardiovascular function | ||||||
| Systolic blood pressure, mmHg | 117 ± 3 | 114 ± 4 | 114 ± 5 | 113 ± 3 | 113 ± 4 | 118 ± 4 |
| Heart rate, BPM | 573 ± 11 | 553 ± 12 | 563 ± 10 | 543 ± 7 | 540 ± 12 | 565 ± 9 |
| Diastolic volume, μl | 40.5 ± 2.8 | 40.5 ± 2.5 | 42.1 ± 2.0 | 47.0 ± 2.3 | 47.4 ± 2.6* | 46.6 ± 2.5 |
| Systolic volume, μl | 21.4 ± 1.9 | 21.8 ± 1.6 | 21.7 ± 1.7 | 25.6 ± 1.3 | 25.2 ± 1.8 | 24.3 ± 1.6 |
| Stroke volume, μl | 19.2 ± 1.1 | 18.7 ± 1.4 | 20.4 ± 1.0 | 21.4 ± 1.3 | 22.2 ± 1.1* | 22.3 ± 1.3 |
| Ejection fraction % | 48.8 ± 1.9 | 46.3 ± 2.5 | 49.2 ± 2.1 | 45.5 ± 1.3 | 47.9 ± 1.6 | 48.4 ± 1.6 |
| Absolute wall thickness, mm | 1.73 ± 0.08 | 1.77 ± 0.10 | 1.73 ± 0.07 | 1.62 ± 0.08 | 1.61 ± 0.07 | 1.61 ± 0.06 |
| Relative wall thickness | 0.533 ± 0.039 | 0.539 ± 0.040 | 0.523 ± 0.029 | 0.465 ± 0.031 | 0.463 ± 0.030 | 0.464 ± 0.03 |
| Anterior wall at diastole, mm | 0.983 ± 0.080 | 1.04 ± 0.08 | 0.918 ± 0.063 | 0.873 ± 0.078 | 0.855 ± 0.058 | 0.822 ± 0.047 |
| End diastolic diameter, mm | 3.35 ± 0.08 | 3.36 ± 0.07 | 3.41 ± 0.06 | 3.54 ± 0.06* | 3.54 ± 0.07 | 3.52 ± 0.07 |
| Posterior wall at diastole, mm | 0.749 ± 0.027 | 0.729 ± 0.038 | 0.846 ± 0.034‡ | 0.746 ± 0.026 | 0.751 ± 0.041 | 0.788 ± 0.038 |
| Anterior wall at systole, mm | 1.23 ± 0.09 | 1.27 ± 0.08 | 1.17 ± 0.06 | 1.09 ± 0.07 | 1.12 ± 0.06 | 1.08 ± 0.05 |
| End systolic diameter, mm | 2.68 ± 0.09 | 2.72 ± 0.07 | 2.72 ± 0.07 | 2.89 ± 0.05* | 2.85 ± 0.08 | 2.82 ± 0.07 |
| Posterior wall at systole, mm | 0.951 ± 0.040 | 0.891 ± 0.040 | 1.02 ± 0.03 | 0.944 ± 0.022 | 0.953 ± 0.037 | 0.972 ± 0.047 |
| Ejection time, ms | 61.0 ± 1.8 | 55.4 ± 1.8 | 52.6 ± 1.9† | 57.9 ± 1.8 | 55.4 ± 1.8 | 52.3 ± 1.8 |
| IVRT + IVCT, ms | 77.3 ± 4.0 | 67.4 ± 4.0 | 63.6 ± 4.2 | 75.7 ± 4.0 | 64.1 ± 4.0 | 58.4 ± 4.0† |
| Myocardial performance index | 1.25 ± 0.06 | 1.21 ± 0.06 | 1.21 ± 0.06 | 1.31 ± 0.06 | 1.16 ± 0.06 | 1.13 ± 0.06 |
| Heart mass | ||||||
| Left ventricular mass, mg | 59.1 ± 1.4 | 59.6 ± 1.6 | 65.4 ± 1.6†‡ | 63.4 ± 1.3* | 66.6 ± 1.3* | 67.4 ± 1.6 |
| Right ventricular mass, mg | 18.2 ± 0.8 | 16.4 ± 0.8 | 18.9 ± 0.8 | 17.1 ± 1.2 | 17.4 ± 1.2 | 18.4 ± 0.9 |
| Atrial mass, mg | 3.78 ± 0.23 | 4.14 ± 0.29 | 5.02 ± 0.29† | 4.46 ± 0.24 | 4.53 ± 0.33 | 4.57 ± 0.24 |
| 23–24 wk | ||||||
| Cardiovascular function | ||||||
| Systolic blood pressure, mmHg | 96 ± 2 | 94 ± 3 | 99 ± 4 | 100 ± 3 | 100 ± 3 | 97 ± 4 |
| Heart rate, BPM | 634 ± 13 | 617 ± 25 | 652 ± 21 | 564 ± 17* | 594 ± 22 | 638 ± 20† |
| Diastolic volume, μl | 44.8 ± 3.5 | 53.9 ± 3.1 | 39.7 ± 4.8‡ | 50.3 ± 4.1 | 50.0 ± 2.9 | 42.0 ± 3.7 |
| Systolic volume, μl | 25.0 ± 2.7 | 28.9 ± 2.4 | 19.9 ± 3.1‡ | 27.1 ± 2.5 | 27.7 ± 2.6 | 19.4 ± 2.3 |
| Stroke volume, μl | 19.9 ± 1.2 | 25.0 ± 1.0 | 19.8 ± 2.1 | 23.2 ± 2.2 | 22.4 ± 0.9 | 22.6 ± 1.5 |
| Ejection fraction, % | 45.2 ± 2.1 | 46.9 ± 1.7 | 50.7 ± 2.3 | 46.5 ± 2.4 | 45.6 ± 2.4 | 54.6 ± 1.8†‡ |
| Absolute wall thickness, mm | 1.84 ± 0.13 | 1.79 ± 0.08 | 2.21 ± 0.20 | 1.67 ± 0.14 | 1.77 ± 0.16 | 1.84 ± 0.14 |
| Relative wall thickness | 0.538 ± 0.051 | 0.486 ± 0.029 | 0.699 ± 0.099‡ | 0.476 ± 0.059 | 0.494 ± 0.05 | 0.549 ± 0.050 |
| Anterior wall at diastole, mm | 1.06 ± 0.13 | 0.948 ± 0.103 | 1.18 ± 0.13 | 0.874 ± 0.135 | 0.913 ± 0.108 | 0.991 ± 0.123 |
| End diastolic diameter, mm | 3.48 ± 0.09 | 3.71 ± 0.07 | 3.31 ± 0.14‡ | 3.61 ± 0.11 | 3.62 ± 0.07 | 3.40 ± 0.10 |
| Posterior wall at diastole, mm | 0.783 ± 0.040 | 0.842 ± 0.047 | 1.03 ± 0.11 | 0.799 ± 0.061 | 0.854 ± 0.070 | 0.853 ± 0.036 |
| Anterior wall at systole, mm | 1.26 ± 0.13 | 1.19 ± 0.10 | 1.43 ± 0.13 | 1.11 ± 0.13 | 1.12 ± 0.11 | 1.26 ± 0.14 |
| End systolic diameter, mm | 2.85 ± 0.10 | 3.01 ± 0.08 | 2.61 ± 0.13‡ | 2.93 ± 0.10 | 2.96 ± 0.10 | 2.62 ± 0.10 |
| Posterior wall at systole, mm | 0.951 ± 0.044 | 1.04 ± 0.20 | 1.22 ± 0.08† | 1.02 ± 0.05 | 1.08 ± 0.07 | 1.10 ± 0.06 |
| Ejection time, ms | 48.5 ± 1.9 | 47.0 ± 1.9 | 45.5 ± 1.9 | 47.5 ± 2.0 | 49.0 ± 1.9 | 47.2 ± 2.0 |
| IVRT + IVCT, ms | 52.3 ± 3.7 | 52.0 ± 3.7 | 56.8 ± 3.7 | 55.8 ± 3.9 | 55.3 ± 3.7 | 48.3 ± 3.9 |
| Myocardial performance index | 1.08 ± 0.09 | 1.11 ± 0.09 | 1.32 ± 0.09 | 1.16 ± 0.09 | 1.14 ± 0.09 | 1.02 ± 0.09 |
| 31 wk | ||||||
| Heart mass | ||||||
| Left ventricular mass, mg | 75.8 ± 1.7 | 77.4 ± 0.9 | 79.4 ± 2.2 | 72.1 ± 2.8 | 72.7 ± 2.2 | 74.7 ± 3.2 |
| Right ventricular mass, mg | 19.6 ± 0.9 | 21.0 ± 0.8 | 21.8 ± 1.2 | 18.7 ± 1.9 | 19.6 ± 0.9 | 20.8 ± 1.0 |
| Atrial mass, mg | 4.63 ± 0.39 | 4.79 ± 0.40 | 5.20 ± 0.28 | 4.11 ± 0.17 | 4.56 ± 0.14 | 4.60 ± 0.18 |
Values are means ± SE. BPM, beats per minute; IVCT, isovolumetric contraction time; IVRT, isovolumetric relaxation time.
P < 0.05 vs. WT within dietary treatment group;
P < 0.05 vs. starch within mouse strain;
P < 0.05 vs. fructose within mouse strain; n = 19–20/group at 12 wk for blood pressure and echocardiography, n = 9–10/group for all other parameters at 31 wk of age.
G6PD deficiency increased LV mass by 7% at 12 wk (Table 4; P < 0.001 for main effect). This corresponded to a 15% increase in LV end-diastolic volume (P = 0.003 for main effect), a 16% increase in end-systolic volume (P = 0.012 for main effect), an 8% decrease in absolute wall thickness (P = 0.028 for main effect), and a 13% decrease in relative wall thickness (P = 0.012 for main effect) in response to G6PD deficiency, indicating modest LV dilation in young G6PDX mice. These changes corresponded to a 13% increase in stroke volume in young G6PDX mice compared with WT mice (P = 0.008 for main effect), which was independent of any change in blood pressure (Table 4). However, these effects were not observed in older mice at 31 wk. Thus, G6PD deficiency modestly increased myocardial dilation and hypertrophy in young animals but not in later weeks.
Oxidative stress.
The activity of aconitase, a mitochondrial oxidative enzyme, is decreased in response to oxidative stress due to inactivation of its iron-sulfur clusters by superoxide and thus is used as an indicator of mitochondrial oxidative damage (6). MDA and 4HA are lipid peroxidation breakdown products and are also indicative of oxidative damage. Therefore, we assayed aconitase activity and lipid peroxidation products (MDA + 4HA) as indexes of oxidative stress (Fig. 6 and Table 5).
Fig. 6.

Aconitase activity. A: DIO decreased aconitase activity at 31 wk in skeletal muscle of WT mice but not G6PDX mice. B: myocardial aconitase activity was higher in G6PDX mice than in WT mice at 31 wk. Values are means ± SE; n = 6–10/group.
Table 5.
Biochemical data
| WT |
G6PD Deficient |
|||||
|---|---|---|---|---|---|---|
| Parameter | Starch | Fructose | DIO | Starch | Fructose | DIO |
| 12 wk | ||||||
| Liver | ||||||
| MDA + 4HA, pmol/mg protein | 276 ± 21 | 262 ± 20 | 257 ± 20 | 264 ± 21 | 219 ± 20 | 273 ± 20 |
| Aconitase activity, mmol·gww−1·min−1 | 2.99 ± 0.19 | 3.10 ± 0.22 | 3.28 ± 0.28 | 2.73 ± 0.15 | 3.43 ± 0.33 | 2.77 ± 0.29 |
| CS activity, μmol·gww−1·min−1 | 33.8 ± 2.5 | 45.4 ± 4.0 | 32.8 ± 3.8 | 34.3 ± 2.0 | 49.1 ± 6.6† | 36.5 ± 4.4 |
| GAPDH activity, AU/g protein | 132 ± 25 | 183 ± 85 | 632 ± 127‡† | 1010 ± 167* | 437 ± 253† | 742 ± 129 |
| PKC activity, ng·ml−1·min−1 | 1.85 ± 0.02 | 1.73 ± 0.01† | 1.73 ± 0.02† | 1.75 ± 0.02* | 1.76 ± 0.02 | 1.73 ± 0.02 |
| Caspase 3 activity, AU/g | 230 ± 15 | 262 ± 16 | 241 ± 14 | 263 ± 14 | 235 ± 14 | 203 ± 14† |
| Skeletal muscle | ||||||
| MDA +4HA, pmol/mg protein | 2.42 ± 0.62 | 1.39 ± 0.39 | 2.82 ± 0.44 | 2.98 ± 0.81 | 3.18 ± 0.83 | 4.83 ± 1.86* |
| Aconitase activity, mmol·gww−1·min−1 | 1.30 ± 0.14 | 1.45 ± 0.14 | 1.57 ± 0.14 | 1.22 ± 0.14 | 1.26 ± 0.14 | 1.18 ± 0.14 |
| CS activity, μmol·gww−1·min−1 | 14.8 ± 1.1 | 15.5 ± 1.1 | 19.2 ± 1.2 | 14.5 ± 1.1 | 15.1 ± 1.1 | 15.5 ± 1.1 |
| Left ventricle | ||||||
| MDA + 4HA, pmol/mg protein | 1768 ± 479 | 2690 ± 455 | 1623 ± 455 | 1939 ± 455 | 2962 ± 455 | 2306 ± 455 |
| Aconitase activity, mmol·gww−1·min−1 | 10.7 ± 2.0 | 10.2 ± 2.3 | 13.7 ± 2.0 | 11.5 ± 2.0 | 12.7 ± 2.5 | 14.7 ± 1.9 |
| CS activity, μmol·gww−1·min−1 | 157 ± 12 | 150 ± 12 | 198 ± 12 | 173 ± 12 | 186 ± 13 | 176 ± 14 |
| Sorbitol, nmol/ml | 19.6 ± 2.9 | 26.2 ± 2.7 | 34.9 ± 2.9† | 21.6 ± 3.8 | 32.9 ± 2.5† | 21.8 ± 2.9*‡ |
| PKC activity, ng·ml−1·min−1 | 1.93 ± 0.16 | 1.33 ± 0.16† | 2.20 ± 0.20‡ | 1.68 ± 0.16 | 2.23 ± 0.13*† | 2.13 ± 0.20 |
| Caspase 3 activity, AU/g | 138 ± 7 | 134 ± 7 | 142 ± 6 | 140 ± 8 | 147 ± 6 | 158 ± 8 |
| 31 wk | ||||||
| Liver | ||||||
| MDA + 4HA, pmol/mg protein | 328 ± 16 | 252 ± 17† | 336 ± 17‡ | 315 ± 19 | 309 ± 19* | 292 ± 19 |
| Aconitase activity, mmol·gww−1·min−1 | 3.30 ± 0.40 | 3.12 ± 0.40 | 3.15 ± 0.96 | 3.19 ± 0.26 | 2.86 ± 0.22 | 3.08 ± 0.18 |
| CS activity, μmol·gww−1·min−1 | 34.1 ± 5.3 | 39.8 ± 5.5 | 43.4 ± 15.6 | 30.7 ± 3.4 | 42.4 ± 7.8 | 34.9 ± 1.1 |
| GAPDH activity, AU/g protein | 0.087 ± 5 | 1.213 ± 89† | 0.086 ± 4‡ | 0.096 ± 15 | 0.078 ± 7* | 0.087 ± 10 |
| PKC activity, ng·ml−1·min−1 | 1.78 ± 0.04 | 1.75 ± 0.01 | 1.75 ± 0.02 | 1.75 ± 0.03 | 1.91 ± 0.01 | 1.89 ± 0.01 |
| Caspase 3 activity, AU/g | 242 ± 10 | 241 ± 9 | 259 ± 10 | 217 ± 12 | 203 ± 9* | 200 ± 13* |
| Skeletal muscle | ||||||
| MDA + 4HA, pmol/mg protein | 3.32 ± 1.05 | 3.28 ± 1.17 | 5.30 ± 1.05 | 4.84 ± 1.25 | 3.91 ± 1.10 | 2.44 ± 1.25 |
| CS activity, μmol·gww−1·min−1 | 12.5 ± 1.1 | 12.3 ± 1.3 | 11.2 ± 1.3 | 13.7 ± 1.4 | 14.2 ± 1.2 | 14.3 ± 1.4 |
| Left ventricle | ||||||
| MDA + 4HA, pmol/mg protein | 1167 ± 227 | 1299 ± 254 | 920 ± 227 | 1293 ± 271 | 1573 ± 239 | 830 ± 271 |
| CS activity, μmol·gww−1·min−1 | 130 ± 12 | 146 ± 13 | 179 ± 12† | 179 ± 14* | 154 ± 12 | 146 ± 15 |
| Sorbitol, nmol/ml | 26.1 ± 3.1 | 31.1 ± 2.6 | 29.0 ± 2.8 | 27.6 ± 3.1 | 31.8 ± 2.6 | 33.9 ± 2.8 |
| PKC activity, ng·ml−1·min−1 | 2.17 ± 0.26 | 2.61 ± 0.23 | 2.45 ± 0.20 | 1.87 ± 0.23 | 2.73 ± 0.23† | 2.51 ± 0.23 |
| Caspase 3 activity, AU/g | 156 ± 23 | 160 ± 19 | 189 ± 21 | 227 ± 23* | 245 ± 23* | 254 ± 23* |
Values are means ± SE.
P < 0.05 vs. WT within dietary treatment group;
P < 0.05 vs. starch within mouse strain;
P < 0.05 vs. fructose within mouse strain; n = 4–10/group. 4HA, 4-hydroxyalkenals; AU, arbitrary units; CS, citrate synthase activity; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MDA, malondialdehyde; PKC, protein kinase C.
Liver aconitase activity was unaffected by diet or strain; however, compared with the high-starch diet, high fructose intake decreased lipid peroxidation products in the liver of older WT mice but not in that of G6PDX mice (Table 5). This decrease in lipid peroxidation products in response to high fructose corresponded to increased G6PD activity in WT mice (Fig. 1), suggesting that an increase in G6PD activity may decrease hepatic oxidative stress.
Skeletal muscle aconitase activity was decreased by DIO compared with high starch intake in older WT mice, indicative of increased ROS in response to DIO (Fig. 6). However, this effect of diet on decreasing aconitase activity was absent in G6PDX mice. No differences were observed in skeletal muscle citrate synthase activity, indicating that changes in aconitase activity were not due to decreased mitochondrial oxidative capacity (Table 5). At 12 wk, lipid peroxidation products were elevated in skeletal muscle in response to DIO in G6PDX mice compared with WT mice, indicative of increased ROS in obese G6PDX mice at that time point. Thus, there was a temporal change, in which G6PD deficiency increased ROS in response to short-term DIO but prevented oxidative damage in the long term. The preservation of aconitase activity in skeletal muscle of older G6PDX mice may have something to do with the decreased body mass found in these animals at the later time point (Fig. 3).
In the LV, aconitase activity was higher in G6PDX mice than in WT mice among DIO animals at 31 wk, indicative of a decrease in myocardial ROS in G6PDX mice compared with WT mice and similar to the skeletal muscle results (Table 5). We observed this effect despite an increase in myocardial citrate synthase activity by DIO compared with high starch intake in WT mice.
Additional nonoxidative glucose pathways.
G6PD is the rate-determining enzyme of the pentose phosphate pathway, and decreased flux through this enzyme may affect other nonoxidative glucose pathways. High fructose intake and DIO increased O-GlcNAc compared with high starch intake in the liver and myocardium of WT mice after 12 wk, indicating an increase in HBP activation (Fig. 7). After 31 wk, DIO no longer resulted in HBP activation but instead increased methylglyoxal, indicating that DIO increased AGE formation after long-term DIO in WT mice. G6PD deficiency blunted the early diet-induced increase in HBP activation in the liver. In the heart, G6PD deficiency increased baseline O-GlcNAc at the early time point but decreased it later on. Thus, there were dynamic temporal changes in O-GlcNAc in response to G6PD deficiency. Further, methylglyoxal was not increased by DIO in G6PDX hearts at 31 wk, suggesting that G6PD deficiency decreases AGE formation.
Fig. 7.
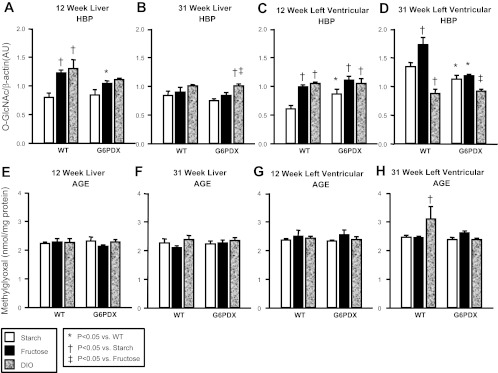
Additional nonoxidative glucose pathways. A–D: hexosamine biosynthetic pathway (HBP) activation as assessed by O-GlcNAc levels in liver and LV tissue at 12 and 31 wk of age. E–H: advanced glycation end product (AGE) formation as assessed by methylglyoxal concentration in liver and LV tissue at 12 and 31 wk of age. Values are means ± SE; n = 4–10/group.
DISCUSSION
The novel findings of this study are that G6PD deficiency attenuated the acceleration in growth usually observed with a high-fat/high-sugar diet while increasing diet-induced metabolic abnormalities (increased serum triglycerides and free fatty acids) and preventing the increase in insulin levels normally associated with greater fat mass. On the other hand, G6PD deficiency did not affect glucose intolerance with any of the diets, indicating that the adverse effects of G6PD on overall metabolism were modest. Overall, our results suggest that G6PD deficiency decreases weight gain in response to an obesogenic diet and predisposes to metabolic abnormalities. In terms of myocardial oxidative stress and LV remodeling, G6PD deficiency preserved myocardial aconitase activity in response to DIO compared with WT mice, suggesting that G6PD deficiency prevents diet-induced myocardial oxidative stress. G6PD deficiency also decreased diet-induced flux through other nonoxidative glucose pathways, suggesting that G6PD deficiency provides cardioprotection in the face of diet-induced metabolic stress. Overall, G6PD deficiency adversely affects systemic metabolism while providing some beneficial cardiac effects.
Effects of G6PD on obesity development.
The results indicate that G6PD deficiency decreased weight gain in response to a high-fat/high-sugar diet. Other investigators saw a similar effect in response to treatment with DHEA, an inhibitor of G6PD, in genetically obese rats and with growth of cell in culture (11, 71). NADPH produced by the pentose phosphate shunt is in high demand for fatty acid synthesis in adipose tissue, and it is plausible that a decrease in G6PD activity may decrease fatty acid synthesis and thereby decrease obesity (32). G6PD knockdown attenuates adipocyte differentiation (49). Normally a decrease in body mass confers beneficial cardiometabolic effects, but a decrease in adipocyte differentiation and prevention of normal fat mass expansion may result in increased circulating lipids and ectopic fat deposition (69). In our study, G6PD-deficient animals showed increased circulating free fatty acids in response to DIO, which corresponded to low insulin levels, suggesting that the decrease in insulin stimulated lipid storage and protein synthesis with G6PD deficiency.
Effect of G6PD on systemic metabolic profile.
Our results suggest a complex effect of G6PD deficiency on diet-induced abnormalities. Although there was no effect in the systemic metabolic profile with the control high-starch diet, G6PD-deficient mice had an increase in triglycerides in response to high fructose intake and an increase in free fatty acids in response to DIO. G6PD deficiency prevented an increase in insulin but had no effect on glucose tolerance. Furthermore, G6PD deficiency increased serum creatinine in response to DIO compared with WT mice at the later time point, indicating impaired renal function. The increase in free fatty acids in G6PDX mice with DIO suggests greater mobilization of fatty acids from adipose tissue, which corresponded to a decrease in fat and body mass and lower insulin levels compared with WT mice with DIO. The glycemic response to intraperitoneal glucose injection was similar between WT and G6PDX mice despite lower body and adipose mass in deficient mice, consistent with the idea that insulin secretion was reduced. Elevated insulin is indicative of insulin resistance and a “prediabetic” state; thus, our findings suggest that G6PD deficiency may protect against development of obesity-induced diabetes. Alternatively, this may reflect impaired insulin secretion, as recent studies showed that G6PDX mice have increased β-cell apoptosis and dysfunction and thus may be unable to increase insulin secretion in response to glucose stimulation (79).
G6PD and oxidative stress.
High fructose intake and DIO-induced metabolic dysfunction both lead to increased cardiac oxidative stress (10, 18, 40). Fructose bypasses a regulatory step in the liver and is metabolized immediately to result in a transient increase in circulating triglycerides. DIO can lead to increased adipose mobilization and free fatty acid release (41, 67). In either case, when circulating lipids are overabundant, they can be taken up by the heart and inhibit glycolysis to increase flux through nonoxidative pathways that increase superoxide formation (9, 16, 48). In either case, it is predicted that flux through G6PD would be increased (as a nonoxidative glucose metabolism pathway) to possibly increase superoxide formation and that G6PD deficiency would dampen this effect. However, because G6PD is required to fuel the antioxidant glutathione pathway, the effect of G6PD on overall oxidative stress induced by either high fructose intake or DIO is difficult to predict (21, 77). We hypothesized that dietary stress (DIO or high fructose intake) would increase oxidative stress in both WT and G6PDX mice,; however, this was not consistently evident (41). The high-fructose diet decreased markers of lipid peroxidation (MDA + 4HA) in the liver of WT mice. Compared with the mice used in other studies (41), the C3H/HeJ strain employed here may cope better with high fructose and thus did not have increased oxidative stress. Suppression of aconitase activity is an established marker of oxidative damage in the mitochondria (6). Since G6PD is a cytoplasmic enzyme, we would not expect it to affect mitochondrial ROS. However, in the skeletal muscle of WT mice we saw a decrease in aconitase activity in response to DIO that was not observed in G6PDX mice. This suggests that DIO increases oxidative damage in the mitochondria, which is prevented by G6PD deficiency. G6PD deficiency could trigger a compensatory mechanism(s) that enhances mitochondrial antioxidant capacity in skeletal muscle. The mechanism(s) responsible for this phenomenon remains to be elucidated.
Effect of G6PD on NonOxidative glucose metabolism.
We hypothesized that G6PD deficiency decreases activation of the HBP by preventing diet-induced oxidative stress in the heart. Our data indicate that with long-term feeding G6PD deficiency decreased cardiac HBP activation and prevented AGE formation. This corresponded to a decrease in cardiac oxidative stress in the heart. Initially, there was basal cardiac HBP activation in response to G6PD deficiency, which occurred before any evidence of oxidative stress. The initial increase in HBP may have occurred because G6PD normally consumes a portion of the glucose within a cell and that glucose was free to proceed through other nonoxidative pathways. In WT mice, the HBP was upregulated in the short term by DIO and high fructose intake in the liver and myocardium as expected. In response to long-term feeding, activation of the HBP in the liver and heart in response to DIO was downregulated (likely by feedback inhibition). The long-term downregulation of DIO -induced HBP activation corresponded to induction of cardiac AGE formation. Thus, there was a switch from early activation of the HBP to later AGE formation in the heart in WT mice, which was prevented by G6PD deficiency.
Cardiac specific effects.
On the basis of previous results from our laboratory and others' in WT mice (9, 10), we hypothesized that both DIO and the high-fructose diet would accelerate generation of ROS, which would be associated with an increase in cardiac mass and myocardial dysfunction. Moreover, this pathology would be prevented by G6PD deficiency as a result of less NADPH and superoxide formation (25, 60). Unexpectedly, both DIO and high fructose intake caused little or no cardiac hypertrophy or dysfunction, and the effects of G6PD deficiency were minimal. At 12 wk there was a slight increase in LV mass in response to DIO that was blunted by G6PD deficiency; however, there were no effects after 31 wk of treatment. Our hypothesis that G6PD deficiency would decrease in the generation ROS in the heart was based on the previous observation that superoxide production is decreased by pharmacological G6PD inhibition in the myocardium of genetically obese Zucker rats (60). Here, we found that aconitase activity was higher in the myocardium of G6PDX mice with DIO compared with WT mice with DIO (Fig. 6), suggesting less oxidative stress in mitochondria. In WT mice, the high-fructose diet had few cardiometabolic effects. In the liver and myocardium, fructose increased G6PD activity, modestly increased body mass, and had minimal effects on the metabolic profile or cardiac function and mass. Our results are consistent with previous reports showing that high sugar intake did not exert adverse cardiometabolic effects (23, 29, 30, 41), but in contrast to other studies that found adverse cardiometabolic effects (8–10, 41, 61, 62). The minimal effects observed in the present study may heve been due to the strain of mice and the use of female mice. Clearly, the interactive effects of diet, obesity, G6PD activity, ROS generation, and subsequent oxidative damage and cardiac structure and function are not clear, and further investigation is needed.
Study limitations and clinical relevance.
We used homozygous mutant female mice in the current experiment, which is important to keep in mind when comparing our results to previous work. The g6pdx gene is X-linked, and G6PD deficiency is detected more often in males, because females carry a heterozygous or homozygous mutation rather than simply a heterozygous mutation as in males (7). However, due to X-chromosome inactivation, heterozygous females can still be affected by G6PD deficiency, as there are mosaics of deficient and nondeficient cells (3, 4). We used homozygous mutant female mice to avoid the issue of X-chromosome inactivation. Previous studies in male G6PDX and WT mice found differences in glucose tolerance (79). Investigations assessing glucose tolerance following an intraperitoneal injection using different mouse strains found a peak in blood glucose at ∼15 min following injection (63, 73). We employed the same protocol as Zhang et al. (79), who reported differences between G6PDX and WT mice at 30 min; however, we did not replicate these findings in females (79). On the other hand, renal defects were previously observed in both male and female G6PD-deficient mice (77). Overall, it appears that the pathology induced by G6PDX mice may be worse in males than in females, although direct perspective comparison has not been done.
We used a mixed-background mouse strain (C3H/HeJ), which has not been previously been characterized in terms of DIO or fructose intake. This strain appears to be more resistant to diet-induced cardiometabolic defects than other strains, such as the commonly used C57BL/6J mice, which develop severe dyslipidemia and metabolic dysfunction (31, 41, 53, 72). Toye et al. (72) compared C3H/HeH mice with C57BL/6J mice, and found three genetic loci that may contribute to a defect in β-cell glucose metabolism in C57BL/6J mice; however, a more extensive comparison between strains has not been performed. The diet-induced metabolic dysfunction in our study was relatively modest, as was seen in the increased serum insulin and decreased glucose tolerance without being elevated in serum glucose, free fatty acids, or triglycerides in WT mice. We did not assess superoxide production, and our markers of ROS (increased MDA + 4HA and decreased aconitase activity) reflect lipid damage and oxidative stress rather than the generation of ROS. We found little evidence for induction of oxidative stress by DIO or the high-fructose diet compared with the high-starch diet. Specifically, in WT mice there was no increase in MDA + 4HA or decrease in aconitase activity in the liver or heart in response to DIO or fructose intake. It would be of interest in future studies to determine the effects of G6PD deficiency in a more severe diabetic state or more prolonged dietary interventions.
Although we saw some modest cardiac hypertrophy at 12 wk in response to DIO in WT mice, these effects were relatively minor, there was no contractile dysfunction, and these effects did not persist at the later time point. Thus, we were unable to determine whether G6PD deficiency affects diet-induced contractile dysfunction. Future studies should further assess the effects of G6PD deficiency on diet-induced contractile dysfunction and cardiac remodeling. It may be possible to better address cardiac and metabolic dysfunction if G6PDX mice were back-crossed onto a more sensitive mouse strain (e.g., C57BL/6J mice) (31, 41, 53, 72). Finally, others have reported that G6PD deficiency may prevent an increase in blood pressure, and it would be of interest to see if this is the case in a model of hypertension (37).
In summary, our results suggest that G6PD deficiency decreases weight gain in response to an obesogenic diet and predisposes to metabolic abnormalities. In the heart, G6PD deficiency may decrease diet-induced oxidative stress and flux through other nonoxidative glucose pathways, thus providing cardioprotection. Our results are consistent with clinical observation that G6PD-deficient people have less cardiovascular disease despite increasing diabetes and kidney dysfunction (12, 42, 47, 58, 74, 75) and suggest that further clinical investigation into the interaction between G6PD deficiency and obesity is warranted.
GRANTS
This work was supported by the National Institutes of Health Grant nos. HL-074237 and HL-072751 (to W. C. Stanley), and R03 TW-008932 from the Fogarty International Center (to M. F. Essop). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Fogarty International Center of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: P.A.H., M.F.E., and W.C.S. conception and design of research; P.A.H., R.F.M., R.F.R.J., B.H.B., K.A.O., J.W.C., K.C.S., and G.A. performed experiments; P.A.H., R.F.M., C.P.K., R.F.R.J., B.H.B., K.A.O., J.W.C., K.C.S., G.A., M.F.E., and W.C.S. analyzed data; P.A.H., R.F.M., C.P.K., M.F.E., and W.C.S. interpreted results of experiments; P.A.H. and M.F.E. prepared figures; P.A.H. and M.F.E. drafted manuscript; P.A.H., K.A.O., M.F.E., and W.C.S. edited and revised manuscript; P.A.H., M.F.E., and W.C.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Jane Leopold for providing us with breeding pairs of G6PDX and WT littermate mice.
REFERENCES
- 1. Babior BM. NADPH oxidase: an update. Blood 93: 1464– 1476, 1999 [PubMed] [Google Scholar]
- 2. Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105: 293– 296, 2002 [DOI] [PubMed] [Google Scholar]
- 3. Beutler E. G6PD deficiency. Blood 84: 3613– 3636, 1994 [PubMed] [Google Scholar]
- 4. Beutler E, Yeh M, Fairbanks VF. The normal human female as a mosaic of X-chromosome activity: studies using the gene for G-6-PD-deficiency as a marker. Proc Natl Acad Sci USA 48: 9– 16, 1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brooks SP, Lampi BJ. Time course of enzyme changes after a switch from a high-fat to a low-fat diet. Comp Biochem Physiol B Biochem Mol Biol 118: 359– 365, 1997 [DOI] [PubMed] [Google Scholar]
- 6. Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One 4: e7095, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 371: 64– 74, 2008 [DOI] [PubMed] [Google Scholar]
- 8. Chess DJ, Lei B, Hoit BD, Azimzadeh AM, Stanley WC. Deleterious effects of sugar and protective effects of starch on cardiac remodeling, contractile dysfunction, and mortality in response to pressure overload. Am J Physiol Heart Circ Physiol 293: H1853– H1860, 2007 [DOI] [PubMed] [Google Scholar]
- 9. Chess DJ, Stanley WC. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc Res 79: 269– 278, 2008 [DOI] [PubMed] [Google Scholar]
- 10. Chess DJ, Xu W, Khairallah R, O'Shea KM, Kop WJ, Azimzadeh AM, Stanley WC. The antioxidant tempol attenuates pressure overload-Induced cardiac hypertrophy and contractile dysfunction in mice fed a high-fructose diet. Am J Physiol Heart Circ Physiol 295: H2223– H2230, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cleary MP, Zisk JF. Anti-obesity effect of two different levels of dehydroepiandrosterone in lean and obese middle-aged female Zucker rats. Int J Obes 10: 193– 204, 1986 [PubMed] [Google Scholar]
- 12. Cocco P, Todde P, Fornera S, Manca MB, Manca P, Sias AR. Mortality in a cohort of men expressing the glucose-6-phosphate dehydrogenase deficiency. Blood 91: 706– 709, 1998 [PubMed] [Google Scholar]
- 13. Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, Sorrentino S, Manes C, Schieffer B, Drexler H, Landmesser U. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res 100: 894– 903, 2007 [DOI] [PubMed] [Google Scholar]
- 14. Ferrannini E, Gastaldelli A, Iozzo P. Pathophysiology of prediabetes. Med Clin North Am 95: 327– 339, vii–viii, 2011 [DOI] [PubMed] [Google Scholar]
- 15. Frederiks WM, Kummerlin IP, Bosch KS, Vreeling-Sindelarova H, Jonker A, Van Noorden CJ. NADPH production by the pentose phosphate pathway in the zona fasciculata of rat adrenal gland. J Histochem Cytochem 55: 975–980, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 107: 1058– 1070, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grundy SM, Brewer HB, Jr, Cleeman JI, Smith SC, Jr, Lenfant C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation 109: 433– 438, 2004 [DOI] [PubMed] [Google Scholar]
- 18. Gupte RS, Floyd BC, Kozicky M, George S, Ungvari ZI, Neito V, Wolin MS, Gupte SA. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med 47: 219– 228, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupte RS, Vijay V, Marks B, Levine RJ, Sabbah HN, Wolin MS, Recchia FA, Gupte SA. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail 13: 497– 506, 2007 [DOI] [PubMed] [Google Scholar]
- 20. Gupte SA. Glucose-6-phosphate dehydrogenase: a novel therapeutic target in cardiovascular diseases. Curr Opin Investig Drugs 9: 993– 1000, 2008 [PubMed] [Google Scholar]
- 21. Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol 41: 340– 349, 2006 [DOI] [PubMed] [Google Scholar]
- 22. Hayashi K, Kimata H, Obata K, Matsushita A, Fukata A, Hashimoto K, Noda A, Iwase M, Koike Y, Yokota M, Nagata K. Xanthine oxidase inhibition improves left ventricular dysfunction in dilated cardiomyopathic hamsters. J Card Fail 14: 238– 244, 2008 [DOI] [PubMed] [Google Scholar]
- 23. Hecker PA, Galvao TF, O'Shea KM, Brown BH, Henderson R, Jr, Riggle H, Gupte SA, Stanley WC. High-sugar intake does not exacerbate metabolic abnormalities or cardiac dysfunction in genetic cardiomyopathy. Nutrition 28: 520– 526, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hecker PA, O'Shea KM, Galvao TF, Brown BH, Stanley WC. Role of adiponectin in the development of high fat diet-induced metabolic abnormalities in mice. Horm Metab Res 43: 100– 105, 2011 [DOI] [PubMed] [Google Scholar]
- 25. Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41: 2164– 2171, 2003 [DOI] [PubMed] [Google Scholar]
- 26. Infanger DW, Cao X, Butler SD, Burmeister MA, Zhou Y, Stupinski JA, Sharma RV, Davisson RL. Silencing nox4 in the paraventricular nucleus improves myocardial infarction-induced cardiac dysfunction by attenuating sympathoexcitation and periinfarct apoptosis. Circ Res 106: 1763– 1774, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jain M, Brenner DA, Cui L, Lim CC, Wang B, Pimentel DR, Koh S, Sawyer DB, Leopold JA, Handy DE, Loscalzo J, Apstein CS, Liao R. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res 93: e9– e16, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Jain M, Cui L, Brenner DA, Wang B, Handy DE, Leopold JA, Loscalzo J, Apstein CS, Liao R. Increased myocardial dysfunction after ischemia-reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation 109: 898– 903, 2004 [DOI] [PubMed] [Google Scholar]
- 29. Jordan JE, Simandle SA, Tulbert CD, Busija DW, Miller AW. Fructose-fed rats are protected against ischemia/reperfusion injury. J Pharmacol Exp Ther 307: 1007– 1011, 2003 [DOI] [PubMed] [Google Scholar]
- 30. Joyeux-Faure M, Rossini E, Ribuot C, Faure P. Fructose-fed rat hearts are protected against ischemia-reperfusion injury. Exp Biol Med (Maywood) 231: 456– 462, 2006 [DOI] [PubMed] [Google Scholar]
- 31. Kanasaki K, Koya D. Biology of obesity: lessons from animal models of obesity. J Biomed Biotechnol 2011: 197636, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Katz J, Landau BR, Bartsch GE. The pentose cycle, triose phosphate isomerization, and lipogenesis in rat adipose tissue. J Biol Chem 241: 727– 740, 1966 [PubMed] [Google Scholar]
- 33. Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107: 15565– 15570, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leopold JA, Loscalzo J. Oxidative enzymopathies and vascular disease. Arterioscler Thromb Vasc Biol 25: 1332– 1340, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Lewis CE, McTigue KM, Burke LE, Poirier P, Eckel RH, Howard BV, Allison DB, Kumanyika S, Pi-Sunyer FX. Mortality, health outcomes, and body mass index in the overweight range: a science advisory from the American Heart Association. Circulation 119: 3263– 3271, 2009 [DOI] [PubMed] [Google Scholar]
- 36. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De SG, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 121: e46– e215, 2010 [DOI] [PubMed] [Google Scholar]
- 37. Matsui R, Xu S, Maitland KA, Hayes A, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation 112: 257– 263, 2005 [DOI] [PubMed] [Google Scholar]
- 38. Matsui R, Xu S, Maitland KA, Mastroianni R, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6-phosphate dehydrogenase deficiency decreases vascular superoxide and atherosclerotic lesions in apolipoprotein E-/- mice. Arterioscler Thromb Vasc Biol 26: 910– 916, 2006 [DOI] [PubMed] [Google Scholar]
- 39. Matsushima S, Kinugawa S, Yokota T, Inoue N, Ohta Y, Hamaguchi S, Tsutsui H. Increased myocardial NAD(P)H oxidase-derived superoxide causes the exacerbation of postinfarct heart failure in type 2 diabetes. Am J Physiol Heart Circ Physiol 297: H409– H416, 2009 [DOI] [PubMed] [Google Scholar]
- 40. Mellor K, Ritchie RH, Meredith G, Woodman OL, Morris MJ, Delbridge LM. High-fructose diet elevates myocardial superoxide generation in mice in the absence of cardiac hypertrophy. Nutrition 26: 842– 848, 2010 [DOI] [PubMed] [Google Scholar]
- 41. Mellor KM, Ritchie RH, Davidoff AJ, Delbridge LM. Elevated dietary sugar and the heart: experimental models and myocardial remodeling. Can J Physiol Pharmacol 88: 525– 540, 2010 [DOI] [PubMed] [Google Scholar]
- 42. Meloni L, Manca MR, Loddo I, Cioglia G, Cocco P, Schwartz A, Muntoni S, Muntoni S. Glucose-6-phosphate dehydrogenase deficiency protects against coronary heart disease. J Inherit Metab Dis 31: 412– 417, 2008 [DOI] [PubMed] [Google Scholar]
- 43. mir-Ahmady B, Salati LM. Regulation of the processing of glucose-6-phosphate dehydrogenase mRNA by nutritional status. J Biol Chem 276: 10514– 10523, 2001 [DOI] [PubMed] [Google Scholar]
- 44. Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol 26: 2439– 2444, 2006 [DOI] [PubMed] [Google Scholar]
- 45. Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB, Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC, Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation 117: 2626– 2636, 2008. 18474817 [Google Scholar]
- 46. Morgan EE, Faulx MD, McElfresh TA, Kung TA, Zawaneh MS, Stanley WC, Chandler MP, Hoit BD. Validation of echocardiographic methods for assessing left ventricular dysfunction in rats with myocardial infarction. Am J Physiol Heart Circ Physiol 287: H2049– H2053, 2004 [DOI] [PubMed] [Google Scholar]
- 47. Niazi GA. Glucose-6-phosphate dehydrogenase deficiency and diabetes mellitus. Int J Hematol 54: 295– 298, 1991 [PubMed] [Google Scholar]
- 48. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404: 787– 790, 2000 [DOI] [PubMed] [Google Scholar]
- 49. Park J, Rho HK, Kim KH, Choe SS, Lee YS, Kim JB. Overexpression of glucose-6-phosphate dehydrogenase is associated with lipid dysregulation and insulin resistance in obesity. Mol Cell Biol 25: 5146– 5157, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pretsch W, Charles DJ, Merkle S. X-linked glucose-6-phosphate dehydrogenase deficiency in Mus musculus. Biochem Genet 26: 89– 103, 1988 [DOI] [PubMed] [Google Scholar]
- 51. Rajamani U, Essop MF. Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am J Physiol Cell Physiol 299: C139– C147, 2010 [DOI] [PubMed] [Google Scholar]
- 52. Rajamani U, Joseph D, Roux S, Essop MF. The hexosamine biosynthetic pathway can mediate myocardial apoptosis in a rat model of diet-induced insulin resistance. Acta Physiol (Oxf) 202: 151– 157, 2011 [DOI] [PubMed] [Google Scholar]
- 53. Rebuffe-Scrive M, Surwit R, Feinglos M, Kuhn C, Rodin J. Regional fat distribution and metabolism in a new mouse model (C57BL/6J) of non-insulin-dependent diabetes mellitus. Metabolism 42: 1405– 1409, 1993 [DOI] [PubMed] [Google Scholar]
- 54. Reclos GJ, Hatzidakis CJ, Schulpis KH. Glucose-6-phosphate dehydrogenase deficiency neonatal screening: preliminary evidence that a high percentage of partially deficient female neonates are missed during routine screening. J Med Screen 7: 46– 51, 2000 [DOI] [PubMed] [Google Scholar]
- 55. Risse S, Schulke B, Dargel D, Fischer K. [Behavior of some parameters of lipid and energy metabolism. 2. Activity of citrate synthase, ATP citrate lyase, fatty acid synthase, and glucose-6-phosphate dehydrogenase in liver of growing rats on diets differing in fat content]. Nahrung 20: 613– 618, 1976 [DOI] [PubMed] [Google Scholar]
- 56. Roden M. Mechanisms of disease: hepatic steatosis in type 2 diabetes—pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab 2: 335– 348, 2006 [DOI] [PubMed] [Google Scholar]
- 57. ROSSIF [Decrease of glucose-6-phosphate dehydrogenase activity prevented by choline in the liver of rats on high fat diet]. Boll Soc Ital Biol Sper 35: 1400– 1402, 1959 [PubMed] [Google Scholar]
- 58. Saeed TK, Hamamy HA, Alwan AA. Association of glucose-6-phosphate dehydrogenase deficiency with diabetes mellitus. Diabet Med 2: 110– 112, 1985 [DOI] [PubMed] [Google Scholar]
- 59. Salati LM, mir-Ahmady B. Dietary regulation of expression of glucose-6-phosphate dehydrogenase. Annu Rev Nutr 21: 121– 140, 2001 [DOI] [PubMed] [Google Scholar]
- 60. Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS, Gupte SA. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol 297: H153– H162, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sharma N, Okere IC, Barrows BR, Lei B, Duda MK, Yuan CL, Previs SF, Sharov VG, Azimzadeh AM, Ernsberger P, Hoit BD, Sabbah H, Stanley WC. High-sugar diets increase cardiac dysfunction and mortality in hypertension compared with low-carbohydrate or high-starch diets. J Hypertens 26: 1402– 1410, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sharma N, Okere IC, Duda MK, Johnson J, Yuan CL, Chandler MP, Ernsberger P, Hoit BD, Stanley WC. High fructose diet increases mortality in hypertensive rats compared to a complex carbohydrate or high fat diet. Am J Hypertens 20: 403– 409, 2007 [DOI] [PubMed] [Google Scholar]
- 63. Sloan C, Tuinei J, Nemetz K, Frandsen J, Soto J, Wride N, Sempokuya T, Alegria L, Bugger H, Abel ED. Central leptin signaling is required to normalize myocardial fatty acid oxidation rates in caloric-restricted ob/ob mice. Diabetes 60: 1424– 1434, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spencer NY, Yan Z, Boudreau RL, Zhang Y, Luo M, Li Q, Tian X, Shah AM, Davisson RL, Davidson B, Banfi B, Engelhardt JF. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and NADPH oxidase-4. J Biol Chem 286: 8977– 8987, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Spolarics Z. A carbohydrate-rich diet stimulates glucose-6-phosphate dehydrogenase expression in rat hepatic sinusoidal endothelial cells. J Nutr 129: 105– 108, 1999 [DOI] [PubMed] [Google Scholar]
- 66. Stanley WC, Dabkowski ER, Ribeiro RF, Jr, O'Connell KA. Dietary fat and heart failure: moving from lipotoxicity to lipoprotection. Circ Res 110: 764– 776, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093– 1129, 2005 [DOI] [PubMed] [Google Scholar]
- 68. Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 64: 362– 369, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Taubes G. Insulin resistance. Prosperity's plague. Science 325: 256– 260, 2009 [DOI] [PubMed] [Google Scholar]
- 70. Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol Cell Physiol 276: C1121– C1131, 1999 [DOI] [PubMed] [Google Scholar]
- 71. Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem 273: 10609– 10617, 1998 [DOI] [PubMed] [Google Scholar]
- 72. Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, Quarterman J, Freeman HC, Ashcroft FM, Cox RD. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48: 675– 686, 2005 [DOI] [PubMed] [Google Scholar]
- 73. Vyas AK, Koster JC, Tzekov A, Hruz PW. Effects of the HIV protease inhibitor ritonavir on GLUT4 knock-out mice. J Biol Chem 285: 36395– 36400, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wan GH, Tsai SC, Chiu DT. Decreased blood activity of glucose-6-phosphate dehydrogenase associates with increased risk for diabetes mellitus. Endocrine 19: 191– 195, 2002 [DOI] [PubMed] [Google Scholar]
- 75. Wiesenfeld SL, Petrakis NL, Sams BJ, Collen MF, Cutler JL. Elevated blood pressure, pulse rate and serum creatinine in Negro males deficient in glucose-6-phosphate dehydrogenase. N Engl J Med 282: 1001– 1002, 1970 [DOI] [PubMed] [Google Scholar]
- 76. Xu Y, Osborne BW, Stanton RC. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am J Physiol Renal Physiol 289: F1040– F1047, 2005 [DOI] [PubMed] [Google Scholar]
- 77. Xu Y, Zhang Z, Hu J, Stillman IE, Leopold JA, Handy DE, Loscalzo J, Stanton RC. Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J 24: 609– 616, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yokota T, Kinugawa S, Hirabayashi K, Matsushima S, Inoue N, Ohta Y, Hamaguchi S, Sobirin MA, Ono T, Suga T, Kuroda S, Tanaka S, Terasaki F, Okita K, Tsutsui H. Oxidative stress in skeletal muscle impairs mitochondrial respiration and limits exercise capacity in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 297: H1069– H1077, 2009 [DOI] [PubMed] [Google Scholar]
- 79. Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J, Guo L, Kulkarni RN, Loscalzo J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J 24: 1497– 1505, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]