

Abstract
MicroRNAs (miRNAs) were recently reported to play an important role in the pathogenesis of pulmonary arterial hypertension (PAH), but it is not clear which miRNAs are important or what pathways are involved in the process. Because hypoxia is an important stimulus for human pulmonary artery smooth muscle cell (HPASMC) proliferation and PAH, we performed miRNA microarray assays in hypoxia-treated and control HPASMC. We found that miR-210 is the predominant miRNA induced by hypoxia in HPASMC. Induction of miR-210 was also observed in whole lungs of mice with chronic hypoxia-induced PAH. We found that transcriptional induction of miR-210 in HPASMC is hypoxia-inducible factor-1α dependent. Inhibition of miR-210 in HPASMC caused a significant decrease in cell number due to increased apoptosis. We found that miR-210 appears to mediate its antiapoptotic effects via the regulation of transcription factor E2F3, a direct target of miR-210. Our results have identified miR-210 as a hypoxia-inducible miRNA both in vitro and in vivo, which inhibits pulmonary vascular smooth muscle cell apoptosis in hypoxia by specifically repressing E2F3 expression.
Keywords: pulmonary arterial hypertension, microRNA, E2F3, human pulmonary artery smooth muscle cells
pulmonary arterial hypertension (PAH) is a devastating, life-threatening disorder characterized by the obstructive remodeling of pulmonary arteries, leading to increased vascular resistance, subsequent right heart dysfunction, and ultimately death (40). Although a number of cellular mechanisms have been proposed, the specific ones responsible for the development of PAH remain largely unknown (41). Pulmonary vascular remodeling that involves abnormal vascular cell proliferation, survival, and migration is a key feature of PAH pathology (28). Chronic hypoxia is an important stimulus for vascular remodeling in patients with PAH (15, 43). By stimulating the release of various mitogens, hypoxia can induce the differentiation of mature, nonproliferative pulmonary artery smooth muscle cells (PASMC) into proliferative PASMC and subsequently cause vascular remodeling (8, 14). However, the cellular and molecular mechanisms involved in these responses are still not completely understood.
MicroRNAs (miRNAs) are small, noncoding, single-stranded RNA molecules of about 21–23 nucleotides in length that posttranscriptionally downregulate gene expression by interacting with the 3′-untranslated region (UTR) of specific mRNA targets (2). In humans, there are currently 1,527 miRNAs listed in the Sanger miRNA registry miRBase 18.0 (22). Bioinformatics prediction suggests that ∼30% of human genes are regulated by miRNAs (33). Emerging data in various organisms indicate that miRNAs function in diverse biological processes, such as embryo development, cell proliferation, apoptosis, regulation of exocytosis and various diseases (1, 10, 37, 38, 44). It has been reported that miR-17–5p and miR-20a play a role in the regulation of BMPR2, a key determinant in most cases of idiopathic familial pulmonary hypertension (5). A recent study demonstrated the role of miRNAs in rat models of PAH induced by chronic hypoxia and monocrotaline (7). Differential expression of miRNA under hypoxia has been studied in a number of cell types including tumor cell lines, endothelial cells, and smooth muscle cells, and miR-210 has been identified as a hypoxia-sensitive marker in many types of cells (11, 19, 26, 29, 39). However, the main function of miR-210 in human PASMC (HPASMC) in hypoxia is not known. Here we demonstrate that miR-210 exerts an antiapoptotic effect in HPASMC in hypoxia and may contribute to the development of PAH.
MATERIALS AND METHODS
Cell culture.
Primary HPASMC were maintained in SmGM-2 BulletKit media (Lonza, Switzerland) with 5% FBS, 0.5 ng/ml human recombinant epidermal growth factor, 2 ng/ml human recombinant fibroblast growth factor, 5 μg/ml insulin, and 50 μg/ml gentamicin in a humidified incubator at 37°C with a constant supply of 5% CO2. Cells were subcultured at subconfluence and used for experiments between passages 6 and 8. For hypoxia experiments, cells were placed in a special hypoxia incubator infused with a gas mixture of 5% CO2 and nitrogen to obtain 3% oxygen concentration. Oxygen concentration was monitored continuously (Forma 3130; Thermo Scientific, Rockford, IL).
Animal model.
All mice experimental protocols described were reviewed and approved by the Animal Care and Use Committee at the University of Illinois. The mice were cared for in accordance with the University of Illinois at Chicago animal care policy following the Guide for the Care and Use of Laboratory Animals. Male C57BL/6 mice (2 mo old) were randomly divided into five groups. Group 1 (n = 4), group 2 (n = 3), group 3 (n = 3), and group 4 (n = 3) were exposed to hypoxia for 2, 7, 14, and 21 days, respectively, in an indigenously designed transparent Plexiglas chamber flushed with 10% oxygen (balance nitrogen) at 0.5–3.0 l/min (FiO2 0.10). The CO2 from the chamber was removed daily by absorption with sodalime (Amsorb Plus). Group 5 (n = 3) served as the normoxic control and was placed in the same chamber open to room air for 21 days. After exposure to hypoxia, mice were weighed and then anesthetized for measurement of hematocrit. Subsequently they were euthanized to determine the masses of right ventricle, left ventricle, and interventricular septum and lungs recovered for analysis.
MicroRNA array profiling.
MicroRNA microarray was performed at Exiqon (Vedbaek, Denmark). Briefly, HPASMC at passage 7 were incubated in normoxia or 3% oxygen for 24 h and 48 h (three experiments each in normoxia and hypoxia). The cells were collected and total RNA extracted using the miRNeasy kit (Qiagen, Hilden, Germany) followed by DNase treatment to eliminate genomic DNA contamination and then quantified using the NanoDrop ND-2000 Spectrophotometer (Nano-Drop Technologies, Wilmington, DE). Total RNA (900 ng) from each sample and reference was labeled with Hy3 and Hy5 fluorescent label, respectively, using the miRCURY LNA Array power labeling kit (Exiqon). The Hy3-labeled samples and a Hy5-labeled reference RNA sample were mixed pairwise and hybridized to the miRCURY LNA array version 11.0 (Exiqon), which contains capture probes targeting all human miRNAs registered in the miRBASE version 14.0 at the Sanger Institute. The hybridization was performed according to the miRCURY LNA array manual using a Tecan HS4800 hybridization station (Tecan, Austria). After hybridization, the microarray slides were scanned and stored in an ozone-free environment (ozone level below 2.0 ppb) to prevent potential bleaching of the fluorescent dyes. The miRCURY LNA array microarray slides were scanned using the Agilent G2565BA Microarray Scanner System (Agilent Technologies, Palo Alto, CA), and the image analysis was carried out using the ImaGene 8.0 software (BioDiscovery, Hawthorne, CA). The quantified signals were background corrected (Normexp with offset value 10; Ritchie et al., 2007) and normalized using the global Lowess (Locally Weighted Scatterplot Smoothing) regression algorithm.
Quantification of miRNA and mRNA expression levels.
miR-210 expression was evaluated using miRCURY LNA Universal RT microRNA PCR system (Exiqon) as specified in their protocol. Real-time qPCR was performed using Power SYBR Fast PCR Master Mix and Step-one plus real-time PCR machine (Applied Biosystems, Foster City, CA). The U6 small nuclear RNA for human or snoRNA-234 for mice was used as an internal control. The transcription levels of hypoxia-inducible factor (HIF)-1α, HIF-2α, α-smooth muscle actin (SMA), SM22, Calponin, and house keeping control (GAPDH) were quantified using SYBR green-I Master PCR Mix with specific real-time PCR primer sets (Forward/reverse primer sequences, for HIF-1α: 5′-TGAACATAAAGTCTGCAACATGGA-3′ /5′-TGAGGTTGGTTACTGTTGGTATCATATA-3′ ; for HIF-2α, 5′-TGCTCCCACGGCCTGTAC-3′/5′-TTGTCACACCTATGGCATATCACA-3′; for α-SMA, 5′-GCAGCCCAGCCAAGCACTGT-3′/5′-AGCCGGCCTTACAGAGCCCA-3′; for Calponin, 5′-GCAACTTCATCAAGGCCATCACCA-3′/5′-TCGAATTTCCGCTCCTGCTTCTCT-3′; for GAPDH, 5′-TTGCCATCAATGACCCCTTCA-3′/5′-CGCCCCACTTGATTTTGGA-3′; SM22, 5′-TTGAAGGCAAAGACATGGCAGCAG-3′/5′-TCCACGGTAGTGCCCATCATTCTT-3′). All reactions were performed in duplicate. Expression levels of miRNA and mRNA were quantified employing the 2 (−ΔΔCt) relative quantification method.
HIF-1 stabilization.
To stabilize endogenous HIF-1 proteins, adenovirus expressing green fluorescent protein (GFP)-tagged oxygen-dependent degradation domains (ODDD: HIF-1α amino acid 531–575) were used to infect HPASMC at MOI = 1:100 according to previously reported methodology (47). The mutated ODDD (ODDDmut: P564A) was used as negative control.
Transfection of siRNA or miRNA knockdown probes.
The siRNAs used to silence human E2F3 were ON-TARGETplus SMARTpool siRNA (siE2F3) obtained from Dharmacon (Lafayette, CO). ON-TARGETplus SMARTpool Nontargeting siRNA were used as control (siCon). The siRNAs directly against human HIF-1α and HIF-2α (siHIF1α and siHIF2α) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). To knock down endogenous miR-210, anti-210 (miRCURY LNA microRNA Power inhibitor) and negative miRNA power inhibitor control (anti-Con) were used (obtained from Exiqon). In brief, cells at 80% confluence after overnight culture on 60-mm Petri dishes were transfected with 80 nM of siRNAs or miRNA inhibitors by Lipofectamine 2000 (Invitrogen). After 6 h of transfection, fresh medium was replaced, and the cells were cultured for 24 h and then exposed to hypoxia for 48 h.
Lentivirus-based miR-210 overexpression.
For miRNA overexpression experiments, we used a lentiviral vector to overexpress miR-210 in HPASMCs. The primary miR-210 was amplified from human genomic DNA with the forward primer 5′- cacctcgag CTGAAGTTGGGCCGAGAG -3′ and reverse primer 5′- gagaattc GTATCTGGCCCAGCCTCA -3′. The PCR product size was 525 bp. After double digestion with XhoI-EcoRI restriction enzymes, the PCR products were cloned into pLVX-Puro vector (Clontech, Mountain View, CA). To monitor the transduction efficiency, the open reading frame of EGFP with stop codon was inserted between CMV promoter and pri-miR-210, named as pLVX/EGFP-miR210. pLVX/EGFP vector without miR-210 was also constructed by deleting miR-210 and used as miRNA negative control. High-titer lentivirus was generated by using a Lenti-X HT Packaging system in 293T cells according to the procedure described by the manufacturer (Clontech). Lentiviral supernatants (100 μl) produced by the transfected packaging cells were then used to infect HPASMCs cultured on 60-mm dishes along with Polybrene (4 μg/ml). To ensure complete transduction, cells were selected with 1.5 μg/ml puromycin for 3–4 days after 2 days of infection.
Apoptosis assay.
HPASMC were transfected overnight and reseeded at 3 × 103 cells per well in 96-well plates with triplicate well for each transfection. After 24 h of attachment, cells were grown in the starvation medium with 0.1% of FBS and then exposed for 2 days of hypoxia before performing an apoptosis assay with Apo-ONE homogeneous kit (Promega, Madison, WI). Caspase-3/7 activity was expressed per number of total viable cells. Apoptosis was also studied using a fluorescence-activated cell sorter (FACS). HPASMCs plated on 60-mm dishes at 70% confluence were transfected with anti-miRNA or siRNA with corresponding controls using Lipofectamine 2000 transfection reagent. One day after transfection, cells were starved for 24 h in SmGM-2 with 0.1% FBS and then exposed to hypoxia. After 2 days of hypoxia treatment, cells were collected, washed with PBS, and then stained with Annexin V-FITC and propidium iodide (PI) reagents in binding buffer using Annexin V-FITC-PI cell apoptosis detection kit (Beyotime Institute of Biotechnology, Beijing, China) and analyzed with an ACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). The results were analyzed using FACStation software.
Cell counting.
The same amount of transfected cells was seeded in six-well plates (∼5,000 cells per well) in SmGM-2 and incubated under hypoxic conditions for 48 h. Adherent cells were detached by trypsin and viable cells were counted using the TC10 Automated Cell Counter (Bio-Rad, Hercules, CA). All experiments were performed in triplicate.
EdU assay.
The transfected cells in 24-well plates were cultured in growth medium for 1 day and then starved for another day in SmGM-2 with 0.1% FBS before exposing them to hypoxia. After 24-h hypoxia treatment, 10 μM of EdU solution was applied to cells for another 24 h in hypoxia. EdU assay was performed using EdU DNA proliferation in vitro detection kit (Guangzhou RiboBio, Guangzhou, China) according to the manufacturer's instructions. Cells were examined using a fluorescent-inverted Olympus IX71 microscope. The proliferation rate was defined as the amount of cells with EdU staining divided by the amount of cells stained with DAPI.
Western blot.
HPASMC were collected and dissolved in mRIPA Mammalian Protein Extraction lysis buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN). Same amount of proteins were resolved on SDS-PAGE and transferred onto nitrocellular membranes. The membranes were blocked with 5% (wt/vol) nonfat milk, washed in Tris-buffered saline-Tween-20 solution, and incubated with primary antibody at 4°C overnight. After being rinsed, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Bio-Rad) at dilutions of 1:10,000 for 1 h at room temperature, and immunoreactive bands were then visualized with Pierce SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). The relative band intensities were quantified by densitometry using NIH ImageJ software (National Institutes of Health) and normalized with image densities of β-actin or β-tubulin that were used as loading controls. The primary antibodies used for this study included rabbit polyclonal anti-human PCNA (1:1,000 dilution; Proteintech Group, Chicago, IL), mouse monoclonal anti-human α-SMA (1:1,000 dilution; Sigma, St. Louis, MO), mouse monoclonal anti-human SM22 (1:3,000 dilution; Sigma); mouse monoclonal anti-human Calponin (1:6,000 dilution; Sigma); rabbit polyclonal anti-human β-tubulin (1:1,000 dilution; Sigma), and mouse monoclonal anti-human E2F3 (1:1,000 dilution; Millipore, Billerica, MA).
miR-210 target validation by UTR luciferase reporter assay.
To construct the E2F3 3′-UTR which contained miR-210 binding site (188–194), two primers were designed: E2F3 3′-UTR forward: 5′-AATTAATAAACAAATTGTCTAAACGCACAGTTGCAGG-3′ and reverse: 5′-CTAGCCTGCAACTGTGCGTTTAGACAATTTGTTTATT-3′. These two primers were annealed to form a double-stranded fragment with EcoRI-XbaI sticky ends at both sides. Then the fragment was directly inserted into a modified pGL3 control vector with corresponding sites (pGL3-E2F3UTR construct). The construct pGL3-E2F3UTR was confirmed by DNA sequence analysis. 4 × 104 HPASMCs were seeded onto 12-well plates, and, after the cells reached ∼80–90% confluence, each well of cells was transfected with 300 ng of 3′-UTR reporter vectors, 1,200 ng of primary miR-210 expressing plasmid (pENTR-EGFP-miR210), or mutated miR-210 (pENTR-EGFP-miR 210m) plasmid and 100 ng of phRL-TK (Promega) with 4 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The pENTR-EGFP without miRNA sequences was used as the control for miR-210 overexpression. The mutant construct of pENTR-EGFP-miR 210m with four mutated residues in the region of miR-210 seeding sequences was generated by site-directed mutagenesis using PCR with primers 5′-CTGCCCCAGACCCACTCACGGTGTGACAGCGGCTGA-3′ (forward) and 5′-TCAGCCGCTGTCACACCGTGAGTGGGTCTGGGGCAG-3′ (reverse), and the construct was confirmed by DNA sequence analysis. Two days after transfection, the cells were harvested and luciferase activities determined using the Dual-Luciferase Reporter Assay System (Promega).
RESULTS
miRNA expression profile in hypoxic HPASMC.
We have previously characterized the role of hypoxia-induced miR-21 in HPASMC proliferation (42). To further explore the miRNAs that may be involved in smooth muscle cell growth or phenotype modulation in response to hypoxia, we conducted miRNA expression profiling using LNA-based miRNA array with HPASMC grown under normoxia (21% oxygen) or hypoxia (3% oxygen). Among 1,265 human miRNAs included in microarray, most of them did not show any significant change in hypoxia. Only five miRNAs (miR-210, miR-637, miR-183*, miR-665, and miR-516a-5p) showed >1.5-fold induction, for at least one time point, in response to hypoxia (Fig. 1, A and B). Consistent with previous reports (6, 11, 17), miR-210 demonstrated the most pronounced increase induced by hypoxia in HPASMC. No miRNAs were significantly downregulated based on our analysis after multiple test correction of P values.
Fig. 1.
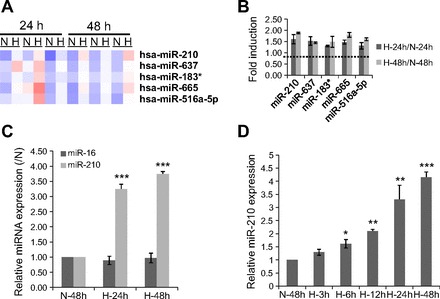
microRNA-210 (miR-210) induction by hypoxia in human pulmonary artery smooth muscle cells (HPASMC). HPASMC were exposed to normoxia (N) or hypoxia (H) for 24 and 48 h and then subjected to microarray analysis. A: cluster analysis of 5 upregulated miRNAs by hypoxia with >1.5-fold induction at least in 1 time point. B: average fold changes related to their normoxic control calculated from 3 independent experiments at each time point. C: expression levels of miR-210 and miR-16 measured by qPCR from the same RNA samples used for microarray analysis. U6 was selected as a reference. The value is shown relative to their normoxic control. D: HPASMC were cultured in normoxia 48 h as control or hypoxia for 3, 6, 12, 24, and 48 h, and the induction of miR-210 was analyzed by qPCR. The value is shown relative to normoxia control for 48 h. Means ± SE of 3 independent experiments are shown (*P < 0.05, **P < 0.01, ***P < 0.001).
Using quantitative real-time RT-PCR (qPCR), we determined the expression levels of miR-210 in the same RNA samples used for the array analysis. Compared with the array data, qPCR showed a higher induction of miR-210 expression by hypoxia (Fig. 1C). To exclude the possibility that this change in miRNA recovery was an artifact, we also determined the expression of miR-16, which is not regulated by hypoxia (based on our own microarray results as well as other reports; Refs. 4 and 11). This miRNA did not show any significant change in expression in hypoxia when assayed by qPCR (Fig. 1C).
To further explore the regulation of miR-210 by hypoxia, we examined the time course of miR-210 induction by hypoxia in HPASMCs. As shown in Fig. 1D, miR-210 increased 1.5-fold as early as 6 h after hypoxic exposure and showed a progressive increase in expression, becoming maximal at the latest (48 h) time point (4.16 ± 0.19, P < 0.001).
miR-210 induction in chronic hypoxia-exposed mice.
In vitro cell-based induction of miR-210 by hypoxia has been found in many different cell types; however, little is known about its regulation in hypoxia-exposed animal models. Because chronic hypoxia is a well-established stimulus for induction of PAH in experimental rodent models, we wished to know whether miR-210 is induced during the development of PAH. Compared with the mice maintained in normoxia, mice exposed to hypoxia (10% oxygen) exhibited a gradual increase in their hematocrit values with the 3-wk hypoxia-exposed mice exhibiting a significantly greater increase than the 2-day hypoxia-exposed mice (Fig. 2A). Right ventricle/left ventricle + septum mass ratios were also significantly increased after 2- and 3-wk exposure to hypoxia compared with mice maintained under normoxia (Fig. 2B), consistent with the development of PAH. The expression level of miR-210 (plus snoRNA-234 for normalization) was analyzed by qPCR using total RNA extracted from normoxic and hypoxia-exposed mouse lungs. Interestingly, a slight upregulation of miR-210 was observed in mouse lungs after 2 days of hypoxic exposure, which significantly increased at 1 wk (2.8-fold change compared with normoxia control), reached a maximum at 2 wk with 3.1-fold change, and subsequently decreased slightly after 3 wk of hypoxia (Fig. 2C). These data demonstrate for the first time that miR-210 is induced by hypoxia, not only in cells, but also in vivo in remodeled lung tissues.
Fig. 2.
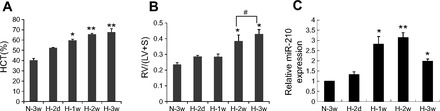
Induction of miR-210 by chronic hypoxia in mice lungs. Mice were exposed to hypoxia (10% oxygen) for indicated times (H-2d: hypoxia 2 day, H-1w: hypoxia 1 wk, H-2w: hypoxia 2 wk, H-3w: hypoxia 3 wk) or remained in normoxia throughout (N-3w: normoxia 3 wk). A and B: hematocrit (HCT) and mass ratio of right ventricle (RV) to left ventricle and septum (LV+S) of different experimental groups are shown. Each bar represents mean ± SE. *P < 0.05, **P < 0.01 vs. normoxic control. C: expression of miR-210 in whole lung tissue was measured by qPCR. The value is shown relative to the normoxic control mice. The data are from 3 mice in each group with the exception of H-2d group, where 4 mice were included. *P < 0.05, **P < 0.01 vs. normoxic control, #not significantly different from H-2w group.
miR-210 expression is regulated by HIF-1α in HPASMC.
Hypoxia-induced miR-210 expression has been reported to be associated with HIF-1α and HIF-2α activity in several cancer cell lines (39, 46). To examine the regulation of miR-210 by the HIF pathway in HPASMC, we transfected HPASMC with a modified HIF-1α-ODDD-wt adenoviral vector, which expresses a GFP-tagged HIF-1α ODDD domain (amino acids from 531 to 575). It is anticipated that the domain will compete with endogenous HIF-1/2α for pVHL recognition and stabilize endogenous HIF-1/2α. HIF-1α-ODDD-mut with a mutation of proline 564 to alanine, which cannot be recognized by the VHL complex for degradation, was used as negative control. As shown in Fig. 3A, overexpression of wild-type ODDD but not mutated ODDD significantly upregulated miR-210 expression in HPASMC under normal culture conditions, indicating that the transient stabilization of HIF-1/2α is sufficient to induce miR-210 expression. To determine the role of HIF-1α vs. HIF-2α in the induction of miR-210 expression, specific siRNA against HIF-1α, HIF-2α, or scrambled control siRNAs was transfected into HPASMC. qPCR confirmed that both of these two siRNAs specifically knocked down their corresponding target gene expression (Fig. 3B). The cells transfected with siRNA control and siRNA against HIF-2α had strong induction of miR-210 expression in hypoxia as detected by qPCR. However, this induction was dramatically reduced in cells that were transfected with siRNAs against HIF-1α (Fig. 3C), indicating that hypoxia-induced miR-210 expression is mainly controlled by HIF-1α in HPASMCs.
Fig. 3.
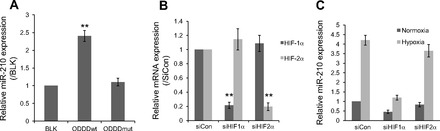
Induction of miR-210 by hypoxia is regulated by hypoxia-inducible factor (HIF)-1α. A: HPASMCs cultured under normoxic conditions were infected with adenovirus expressing green fluorescent protein (GFP)-tagged oxygen-dependent degradation domains (ODDD) or mutated ODDD (ODDDmut). Blank (BLK) control without viral infection was also included. miR-210 expression in normoxic HPASMC was examined by qPCR after 2 days of infection. U6 was selected as a reference. The data shown as means ± SE (n = 3) were relative to BLK control **P < 0.01. B and C: HPASMC were transfected with 80 nM of siRNAs against HIF-1α, HIF-2α, or scramble sequences (siCon). 1 day after transfection, cells were exposed to normoxia or hypoxia for 2 days. The silencing efficiencies of HIF-1α and HIF-2α were measured by qPCR. The value was relative to control siRNA using 18s rRNA as reference (B). miR-210 expression was then assayed by qPCR in transfected HPASMC with or without exposure to 3% O2 for 2 days. The value was relative to normoxia control siRNA using U6 as reference (C). **P < 0.01.
Knockdown of miR-210 induces cell death without affecting HPASMC phenotype.
To understand the role of miR-210 induction in hypoxia, we first investigated the effect of miR-210 inhibition on HPASMC differentiation by measuring the mRNA levels of three smooth muscle cell contractile protein genes (α-SMA, SM22, and Calponin) and their upstream regulator, myocardin. With the transfection of 80 nM of LNA-based miR-210 inhibitor, endogenous miR-210 in hypoxic HPASMC was considerably depleted (over 90% compared with inhibitor control) (Fig. 4A). However, it had no effect on the mRNA expression of the smooth muscle cell-specific markers, α-SMA, SM22, Calponin, as well as myocardin (Fig. 4B). Further experimental evidence by Western blot showed that the protein level of α-SMA was not affected by the loss of miR-210 (Fig. 4C). These results indicate that miRNA-210 may not be necessary to maintain the contractile smooth muscle cell phenotype. Interestingly, we observed a significant decrease in cell number in the miR-210 knockdown group compared with control (Fig. 4D). The decrease in cell number could have been caused by cell death or inhibition of cell proliferation. Therefore, we determined whether the cell proliferation marker PCNA was affected by the loss of miR-210. As shown in Fig. 4C, the protein level of PCNA was not significantly different between anti-210 and anti-Con groups. EdU incorporation further confirmed that the proliferating rate of HPASMC in hypoxia was not significantly changed by the loss of miR-210 compared with knockdown control (Fig. 4E). Therefore, we evaluated the impact of miR-210 inhibition on cell apoptotic response by caspase-3/7 activity assay under hypoxic conditions. We reproducibly noticed that the knockdown of miR-210 in hypoxia-treated HPASMC induced apoptosis by increasing caspase activity when compared with the knockdown control (Fig. 4F), indicating that hypoxia-induced miR-210 may function as an inhibitor of apoptosis in HPASMC. To confirm this antiapoptotic effect, we utilized a second assay of cell apoptosis involving FACS analysis. As shown in Fig. 4G, knockdown of miR-210 resulted in a significant increase in the proportion of FITC-annexin V-positive cells (Fig. 4G, top, right and bottom, right) and a significant decrease of FITC-annexin V-negative/PI-negative cells (Fig. 4G, bottom, left), which further indicated that the loss of miR-210 may lead to cellular apoptosis during hypoxia.
Fig. 4.
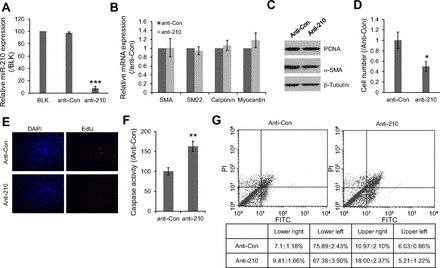
Functions of hypoxia-induced miR-210 in HPASMC. A: expression level of miR-210 in hypoxic HPASMC after transfection with either miR-210 knockdown probe or control probe (anti-Con) were investigated by qPCR. The values are shown relative to the value obtained from blank control. B: mRNA expression levels of α-smooth muscle actin (SMA), SM22, Calponin, and myocardin were examined by qPCR from the same samples mentioned above. 18S rRNA was used as reference. Data was shown relative to control probe. C: protein levels of PCNA and SMA were also examined in the treated cells. β-Tubulin was used as an internal control. D: effect of miR-210 knockdown on cell growth was evaluated by counting the cell number. The values are shown relative to the value obtained with control probe. E: effect of miR-210 knockdown on cell proliferation was also evaluated by EdU incorporation assay. F: induction of apoptosis due to the loss of miR-210 in hypoxic HPASMC was evaluated by measuring Caspase-3/7 activities. G: effects of miR-210 knockdown on cell apoptosis at hypoxia condition were also investigated by analyzing cell death with FACScan flow cytometer. Cells were stained with FITC-conjugated annexin V and PI. All the values are shown relative to the value obtained with control probe. *P < 0.05, **P < 0.01, ***P < 0.001.
miR-210 directly targets E2F3 in HPASMC.
Recently, it was reported that E2F3, a potential target of miR-210, plays an important role in mediating DNA damage-induced apoptosis (34). To determine whether E2F3 is regulated by miR-210 in HPASMC, we first examined the expression level of E2F3 before and after hypoxia treatment. As shown in Fig. 5A, E2F3 protein was significantly decreased by ∼40% in HPASMC after 48-h hypoxia treatment (P < 0.05). To determine whether E2F3 is the target of miR-210 in vivo, we measured E2F3 protein levels in lung tissues of mice with chronic hypoxia-induced PAH. Compared with normoxic control mice, the expression of E2F3 protein was significantly reduced after exposure to hypoxia for 2 wk and further decreased at the 3 wk time point (Fig. 5B). These observations indicate that downregulation of E2F3 might be caused by hypoxia-induced miR-210 in HPASMC and mouse lungs. To further confirm this, we constructed a truncated 3′-UTR of E2F3 containing miR-210 binding site (188–194) into pGL3 vector and performed reporter assays. Figure 5C shows the predicted conserved binding sequence of miR-210 to the human E2F3 3′-UTR. When the miR-210 expression plasmid was cotransfected into the cells with E2F3 3′-UTR luciferase reporter construct, reporter luciferase activity was repressed about 55% compared with cotransfection with control plasmid without miR-210 insert (Fig. 5D). However, the repression was completely abolished when a mutant form of miR-210-expressing plasmid was cotransfected. To further validate our results, HPASMC overexpressing miR-210 were generated by transfection with a lentiviral vector bearing the primary miR-210 sequence under the control of CMV promoter. Figure 5E shows over 10-fold increase of miR-210 expression in miR-210 overexpressing HPASMCs. Consistent with above 3′-UTR dual-luciferase assay, cells with miR-210 overexpression resulted in ∼55% decrease in E2F3 protein expression (Fig. 5F). Correspondingly, knockdown of miR-210 significantly induced E2F3 protein expression (Fig. 5G). Based on these studies, we conclude that E2F3 is the direct target of miR-210 in HPASMCs.
Fig. 5.
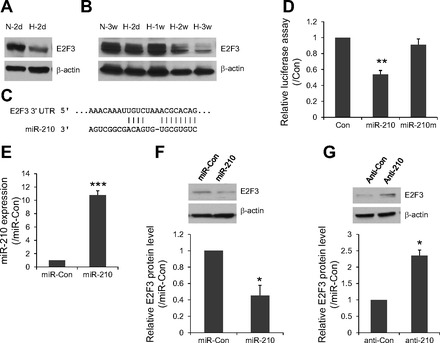
E2F3 expression is regulated by miR-210 in HPASMC. A and B: expression level of E2F3 in hypoxia-treated HPASMC (A) and mice lungs (B) was measured by Western blot. β-Actin was used as a loading control. C: mature miR-210 sequence and its potential binding site in the 3′-untranslated region (3′-UTR) of human E2F3. D: pGL3 luciferase expression vector containing the truncated E2F3 3′UTR was cotransfected into HPASMCs with either miR-210 overexpression vector, mutated miR-210 (miR-210m) vector, or EGFP control vector. phRL-TK vector was also included to normalize the transfection efficiency. 2 days after transfection, the cells were collected for dual-luciferase assay. The value was shown as normalized luciferase activity relative to EGFP control vector (n = 3; **P < 0.01). E: HPASMC were transduced with a lentiviral vector bearing the miR-210 pri-miRNA sequence under the control of CMV promoter. These cells expressed almost 10-fold more mature miR-210 than control miR-Con transduced cells (***P < 0.001; n = 3). F: Western blot analysis showed that miR-210 overexpression resulted in reduced E2F3 protein expression compared with miRNA control vector. G: Western blot analysis showed that miR-210 inhibition caused increased E2F3 protein expression compared with anti-Con probe. Western blot data was quantitated to better view the difference between different treatment groups. Data are represented as means ± SE (n = 3); *P < 0.05, **P < 0.01, ***P < 0.001.
E2F3 downregulation is necessary for miR-210-mediated antiapoptotic effect.
To investigate whether downregulation of E2F3 is an integral part of miR-210-mediated antiapoptosis effect in HPASMC, we silenced the expression of endogenous E2F3 with siRNAs and then performed cell apoptosis assay under hypoxic conditions. Efficient inhibition of E2F3 was confirmed by Western blot analysis (Fig. 6A). The proapoptotic response was interrogated by both caspase-3/7 assay and FACS analysis as described before. We observed that partial loss of E2F3 induced a significant decrease in cellular apoptosis when compared with nontargeting siRNA (Fig. 6, B–C), which is consistent with the above miR-210 knockdown data. These results indicate that hypoxia-induced expression of miR-210 serves, in part, to inhibit apoptosis through its direct target, E2F3, in HPASMC.
Fig. 6.
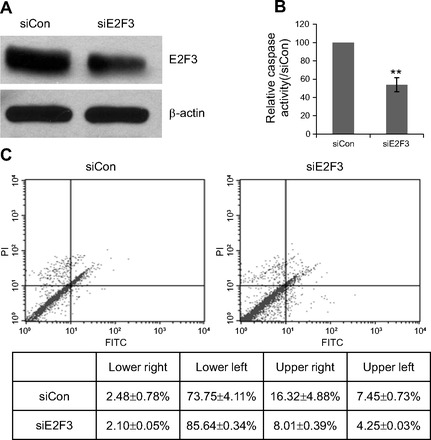
Silencing of E2F3 results in decreased apoptotic activity in HPASMC exposed to hypoxia. A: representative Western blot showed successful silencing of E2F3 in HPASMCs. β-Actin was used as protein loading control. B: effect of E2F3 knockdown on activity of executioner caspases 3 and 7. Same number of HPASMC transfected with siE2F3 or siCon were cultured in starvation medium for 2 days at hypoxic condition. Caspase 3/7 activity was determined using Apo-ONE Homogeneous Caspase-3/7 Assay and normalized by number of viable cells (determined by MTS assay). The value was shown as percentage of control from four replicates. **P < 0.01. C: effect of E2F3 knockdown cell apoptosis was also investigated by analyzing cell death with FACScan flow cytometer. Cells were stained with FITC-conjugated annexin V and propidium iodide (PI).
DISCUSSION
In this study, we show that hypoxia-induced upregulation of miR-210 plays an antiapoptotic role in HPASMC via the transcription factor E2F3. This antiapoptotic effect may contribute to PASMC proliferation, leading to vascular remodeling in hypoxia-induced PAH, as we have demonstrated that lungs of mice exposed to chronic hypoxia exhibit upregulation of miR-210 with a concomitant decrease in E2F3 protein levels.
From the global miRNA profiling, we first identified a small subset of hypoxia-sensitive miRNAs (>1.5-fold change), including miR-210, miR-637, miR-183*, miR-655, and miR-516a-5p. Our results on the upregulation of miR-210 are in agreement with previous reports and were confirmed by qPCR. However, it seems that the sensitivity of LNA-based miRNA microarray seemingly is much less than qPCR. Using the same RNA samples, only 1.9-fold induction of miR-210 was observed by LNA-based miRNA microarray, whereas the induction of miR-210 was noted to be 4.3-fold during qPCR-based analysis of miRNA expression. We focused this study on miR-210 although other miRNAs are also likely to be important in the control of gene expression in hypoxia. Of the ones we identified as responsive to hypoxia, miR-637, which has been reported as a hypoxia-induced miRNA in head and neck squamous cell carcinoma (23), may be of importance. According to the human genomic sequences, miR-637 is located within an the intron of DAPK3 gene. Because the host gene of DAPK3 was upregulated by hypoxia according to data by Ning et al. (36), miR-637 may play an important role in the control of gene expression in HPASMC.
miR-210 is the only miRNA consistently upregulated in all published studies, in both normal and transformed hypoxic cells (6, 13, 18, 20, 21, 27, 39, 46). Our results showing a predominant induction of miR-210 by hypoxia is consistent with reports involving other cell types such as breast adenocarcinoma cells (6, 19), pancreatic cancer cells (27), endothelial cells (19), and mouse HL-1 cardiomyocytes (25). This strong induction of miR-210 in all these different cell types is due to the highly conserved structure of the hypoxia response element in the miR-210 promoter (25). The regulation of miR-210 by HIF proteins has been discussed to some extent (4, 9, 11, 21, 27). Mostly, it is believed that miR-210 is HIF-1α dependent. However, Zhang et al. (46) have reported that HIF-2α could also regulate miR-210 expression. The data we present here provide evidence that HIF-1α, but not HIF-2α, induces miR-210 expression in hypoxia-treated HPASMC. Prior studies showing miR-210 as a hypoxia-induced gene have been performed in cells in vitro, with no data from in vivo studies. A recent study profiled miRNA signatures in models of pulmonary arterial hypertension in rats induced by both chronic hypoxia and monocrotaline and found increased expression of miR-451 and miR-30c in both models; however, miR-210 was not identified through microarray due to its low intensities from hybridization (absolute reading <50). We also found that the hybridization signal of miR-210 was too low to distinguish its expression between normoxic and chronic hypoxia-treated mouse lungs when we performed miRNA microarray analysis (unpublished data). However, a good correlation was clearly noted between miR-210 regulation and chronic hypoxia-induced PAH in mouse lungs when qPCR was used to quantify miR-210 expression levels. This is the first report of upregulation of miR-210 in chronic hypoxia-induced PAH in an in vivo model. A slight decrease of miR-210 after 3 wk of hypoxia treatment indicates that miR-210 may contribute to the early vascular and tissue remodeling event and further development of PAH.
The function of miR-210 includes almost every aspect of hypoxia-related biology, such as angiogenesis, apoptosis, proliferation, differentiation, cell cycle regulation, DNA damage repair, mitochondrial metabolism, and tumor growth (27). Early studies have shown that miR-210 promotes osteoblastic differentiation by inhibiting the expression of a type 1B receptor of activin A (AcvR1b) (35). Hypoxia-induced miR-210 is also associated with K562 differentiation (3). Our data from HPASMC show that loss of miR-210 does not cause any change in α-SMA, SM22, and Calponin expression, indicating that miR-210 may not be involved in the regulation of smooth muscle contractile proteins (32). Interestingly, we found that HPASMC treated with miR-210 inhibitor had fewer cell numbers when compared with cells treated with control inhibitor. This could have been caused by inhibition of proliferation and/or promotion of cell death. To differentiate between these two possibilities, we measured the expression of PCNA and EdU incorporation for cell proliferation assay and caspase activity and annexin V/PI staining for apoptosis assay. We did not observe a significant change in both PCNA protein and EdU incorporation rate after miR-210 knockdown, but caspase activity and the proportion of annexin V-positive cells were increased significantly, indicating miR-210 may have antiapoptotic effects by protecting HPASMCs from cell death during hypoxia. Cheng et al. first described that blockade of miR-210 with antisense inhibitor leads to an increased apoptotic response in HeLa cells (12). Similar results were further documented in colon and breast cancer cells by Kulshreshtha (31). Fasanaro et al. (17) provided evidence that miR-210 blockade in the presence of hypoxia decreases capillary-like formation, EC migration, and EC survival and induces apoptosis. Mechanistically, Kim et al. (30) revealed that miR-210 directly antagonizes an apoptotic component, CASP8AP2. A recent paper described that miR-210 can improve angiogenesis, inhibit apoptosis, and improve cardiac function in a murine model of myocardial infarction and therefore may be developed as a novel therapy for treatment of ischemic heart disease. In contrast, miR-210 was also reported to increase apoptosis in cancer cells and pulmonary arterial endothelial cells (9, 26, 39).
Although more than ten miR-210 targets involved in cellular processes have been identified, including cell-cycle regulator E2F3 (21, 39), homeobox proteins(26), the iron sulfur cluster assembly proteins ISCU1/2 (9), and the subunit D of succinate dehydrogenase complex (39), none of these targets have yet been verified in PAH. Among these, E2F3 is a potential target that has been functionally involved in cell apoptosis (34). However, little is known about E2F3 gene regulation in HPASMC. To determine whether miR-210 regulates E2F3 in HPASMC, we examined the expression pattern of E2F3 protein after hypoxia treatment. We found that E2F3 was downregulated, not only in hypoxia-treated HPASMC, but also in chronic hypoxia-treated mouse lungs. Further experiments in HPASMC showed that overexpression of miR-210 caused the decrease of E2F3, whereas silencing of miR-210 induced the expression of E2F3. Dual luciferase reporter assay confirmed that miR-210 binds to E2F3 3′-UTR directly. More importantly, we found that loss of E2F3 caused the inhibition of apoptosis, which is consistent with the miR-210 functional data in HPASMC. Based on all these results, we surmise that E2F3 is the direct target by which miR-210 plays an important role in PAH.
The E2F family includes both “activating E2Fs” that are potent transcriptional activators, which can drive cell cycle progression by inducing the expression of proliferation-associated genes, whereas the “repressive E2Fs” impede cell growth by repressing these genes (34). In addition to their role in proliferation, E2Fs also influence other biological processes, such as differentiation, the DNA damage response, and apoptosis (16, 24, 34, 48). The role of E2F3 could be completely different in different cells. As shown during myeloid development, E2F3 has no effect on the proliferation of early myeloid progenitors. However, with the development of myeloid cells, E2F3 has totally different roles: first repressing the survival of CD11b+ myeloid progenitors and then promoting the proliferation of CD11b+ macrophages (45). A very recent paper (34) revealed that E2F3 is required for DNA damage-induced apoptosis. We provide evidence that E2F3, a known target of miR-210, emerges as an important mediator of apoptosis in hypoxia-exposed HPASMC. However, the cellular mechanisms for E2F3-mediated apoptosis in HPASMC still remain to be elucidated.
In conclusion, we have confirmed the expression of hypoxia-inducible miRNA-210 in vitro and in vivo and its transcriptional regulation by the HIF pathway in HPASMC. We have shown that E2F3 is the direct target of miR-210 in HPASMC. The functional connection of miR-210 and E2F3 has been demonstrated in the context of cell apoptosis. We hypothesize that miR-210 acts as a homeostatic rheostat and that a balanced level of miR-210 in cells is essential for cell survival. Future studies could be focused on in vivo studies by either conditionally knocking out or overexpressing miR-210 to see whether it is involved in vascular remodeling and the development of hypoxia-induced PAH.
GRANTS
This study was supported by National Natural Science Foundation of China No. 81170047 (to D. Gou), National Basic Research Program of China (973 Program, 2012CB124701); Shenzhen Municipal Basic Research Program No.JC201006010725A (to D. Gou); Shenzhen Pengcheng Distinguished Professor Funding No.000233 (to D. Gou) and National Heart, Lung, and Blood Institute Grants R01 HL075187 (to J. U. Raj) and R01 HL059435 (to J. U. Raj).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.G. and J.U.R. conception and design of research; D.G., R.R., X.P., L.Y., K.K., J.S., Z.W., and G.Z. performed experiments; D.G., R.R., X.P., L.Y., J.S., and Z.W. analyzed data; D.G., R.R., K.K., J.S., Z.W., G.Z., and J.U.R. interpreted results of experiments; D.G., R.R., X.P., K.K., J.S., and Z.W. prepared figures; D.G. drafted manuscript; D.G., R.R., X.P., L.Y., K.K., J.S., G.Z., and J.U.R. edited and revised manuscript; D.G. and J.U.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Tingting Wen, Qiyuan Zhou, and Laura Bach for helpful writing, discussions, and technical assistance.
REFERENCES
- 1. Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, Yfantis HG, Stephens RM, Croce CM. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res 68: 6162–6170, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, function. Cell 116: 281–297, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Bianchi N, Zuccato C, Lampronti I, Borgatti M, Gambari R. Expression of miR-210 during erythroid differentiation and induction of gamma-globin gene expression. BMB Rep 42: 493–499, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Biswas S, Roy S, Banerjee J, Hussain SR, Khanna S, Meenakshisundaram G, Kuppusamy P, Friedman A, Sen CK. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc Natl Acad Sci USA 107: 6976–6981, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 104: 1184–1191, 2009 [DOI] [PubMed] [Google Scholar]
- 6. Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, Harris AL, Gleadle JM, Ragoussis J. Hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res 14: 1340–1348, 2008 [DOI] [PubMed] [Google Scholar]
- 7. Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R, Denby L, Dempsie Y, Long L, Morrell NW, Baker AH. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol 30: 716–723, 2010 [DOI] [PubMed] [Google Scholar]
- 8. Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol 44: 14–30, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab 10: 273–284, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38: 228–233, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 29: 4362–4368, 2010 [DOI] [PubMed] [Google Scholar]
- 12. Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 33: 1290–1297, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Corn PG. Hypoxic regulation of miR-210: shrinking targets expand HIF-1's influence. Cancer Biol Ther 7: 265–267, 2008 [DOI] [PubMed] [Google Scholar]
- 14. Davie NJ, Crossno JT, Jr, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, Carpenter TC, Brunetti JA, McNiece IK, Stenmark KR. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol 286: L668–L678, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Dumas JP, Bardou M, Goirand F, Dumas M. Hypoxic pulmonary vasoconstriction. Gen Pharmacol 33: 289–297, 1999 [DOI] [PubMed] [Google Scholar]
- 16. Ebelt H, Hufnagel N, Neuhaus P, Neuhaus H, Gajawada P, Simm A, Muller-Werdan U, Werdan K, Braun T. Divergent siblings: E2F2 and E2F4 but not E2F1 and E2F3 induce DNA synthesis in cardiomyocytes without activation of apoptosis. Circ Res 96: 509–517, 2005 [DOI] [PubMed] [Google Scholar]
- 17. Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem 283: 15878–15883, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fasanaro P, Greco S, Lorenzi M, Pescatori M, Brioschi M, Kulshreshtha R, Banfi C, Stubbs A, Calin GA, Ivan M, Capogrossi MC, Martelli F. An integrated approach for experimental target identification of hypoxia-induced miR-210. J Biol Chem 284: 35134–35143, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Favaro E, Ramachandran A, McCormick R, Gee H, Blancher C, Crosby M, Devlin C, Blick C, Buffa F, Li JL, Vojnovic B, Pires das Neves R, Glazer P, Iborra F, Ivan M, Ragoussis J, Harris AL. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One 5: e10345, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gee HE, Camps C, Buffa FM, Patiar S, Winter SC, Betts G, Homer J, Corbridge R, Cox G, West CM, Ragoussis J, Harris AL. Hsa-miR-210 is a marker of tumor hypoxia and a prognostic factor in head and neck cancer. Cancer 116: 2148–2158, 2010 [DOI] [PubMed] [Google Scholar]
- 21. Giannakakis A, Sandaltzopoulos R, Greshock J, Liang S, Huang J, Hasegawa K, Li C, O'Brien-Jenkins A, Katsaros D, Weber BL, Simon C, Coukos G, Zhang L. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol Ther 7: 255–264, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34: D140–D144, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hebert C, Norris K, Scheper MA, Nikitakis N, Sauk JJ. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol Cancer 6: 5, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hong S, Paulson QX, Johnson DG. E2F1 and E2F3 activate ATM through distinct mechanisms to promote E1A-induced apoptosis. Cell Cycle 7: 391–400, 2008 [DOI] [PubMed] [Google Scholar]
- 25. Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 122: S124–S131, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, Story M, Le QT, Giaccia AJ. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell 35: 856–867, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang X, Le QT, Giaccia AJ. MiR-210–micromanager of the hypoxia pathway. Trends Mol Med 16: 230–237, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004 [DOI] [PubMed] [Google Scholar]
- 29. Ivan M, Harris AL, Martelli F, Kulshreshtha R. Hypoxia response and microRNAs: no longer two separate worlds. J Cell Mol Med 12: 1426–1431, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim HW, Haider HK, Jiang S, Ashraf M. Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J Biol Chem 284: 33161–33168, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M. A microRNA signature of hypoxia. Mol Cell Biol 27: 1859–1867, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lagna G, Ku MM, Nguyen PH, Neuman NA, Davis BN, Hata A. Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J Biol Chem 282: 37244–37255, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Martinez LA, Goluszko E, Chen HZ, Leone G, Post S, Lozano G, Chen Z, Chauchereau A. E2F3 is a mediator of DNA damage-induced apoptosis. Mol Cell Biol 30: 524–536, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mizuno Y, Tokuzawa Y, Ninomiya Y, Yagi K, Yatsuka-Kanesaki Y, Suda T, Fukuda T, Katagiri T, Kondoh Y, Amemiya T, Tashiro H, Okazaki Y. miR-210 promotes osteoblastic differentiation through inhibition of AcvR1b. FEBS Lett 583: 2263–2268, 2009 [DOI] [PubMed] [Google Scholar]
- 36. Ning W, Chu TJ, Li CJ, Choi AM, Peters DG. Genome-wide analysis of the endothelial transcriptome under short-term chronic hypoxia. Physiol Genomics 18: 70–78, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Plasterk RH. Micro RNAs in animal development. Cell 124: 877–881, 2006 [DOI] [PubMed] [Google Scholar]
- 38. Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432: 226–230, 2004 [DOI] [PubMed] [Google Scholar]
- 39. Puissegur MP, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, Maurin T, Lebrigand K, Cardinaud B, Hofman V, Fourre S, Magnone V, Ricci JE, Pouyssegur J, Gounon P, Hofman P, Barbry P, Mari B. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ 18: 465–478, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rabinovitch M. Pathobiology of pulmonary hypertension. Annu Rev Pathol 2: 369–399, 2007 [DOI] [PubMed] [Google Scholar]
- 40a. Ritchie ME, Silver J, Oshlack A, Holmes M, Diyagama D, Holloway A, Smyth GK. A comparison of background correction methods for two-colour microarrays. Bioinformatics 23: 2700–2707, 2007 [DOI] [PubMed] [Google Scholar]
- 41. Rubin LJ. Pulmonary arterial hypertension. Proc Am Thorac Soc 3: 111–115, 2006 [DOI] [PubMed] [Google Scholar]
- 42. Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 299: L861–L871, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006 [DOI] [PubMed] [Google Scholar]
- 44. Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell 126: 767–774, 2006 [DOI] [PubMed] [Google Scholar]
- 45. Trikha P, Sharma N, Opavsky R, Reyes A, Pena C, Ostrowski MC, Roussel MF, Leone G. E2F1–3 are critical for myeloid development. J Biol Chem 286: 4783–4795, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Z, Sun H, Dai H, Walsh RM, Imakura M, Schelter J, Burchard J, Dai X, Chang AN, Diaz RL, Marszalek JR, Bartz SR, Carleton M, Cleary MA, Linsley PS, Grandori C. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 8: 2756–2768, 2009 [DOI] [PubMed] [Google Scholar]
- 47. Zhou G, Dada LA, Chandel NS, Iwai K, Lecuona E, Ciechanover A, Sznajder JI. Hypoxia-mediated Na-K-ATPase degradation requires von Hippel Lindau protein. FASEB J 22: 1335–1342, 2008 [DOI] [PubMed] [Google Scholar]
- 48. Ziebold U, Reza T, Caron A, Lees JA. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev 15: 386–391, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]